
Fig. 4. Allylic anion model of isomerization for a chromophore containing an electron-withdrawing substituent.
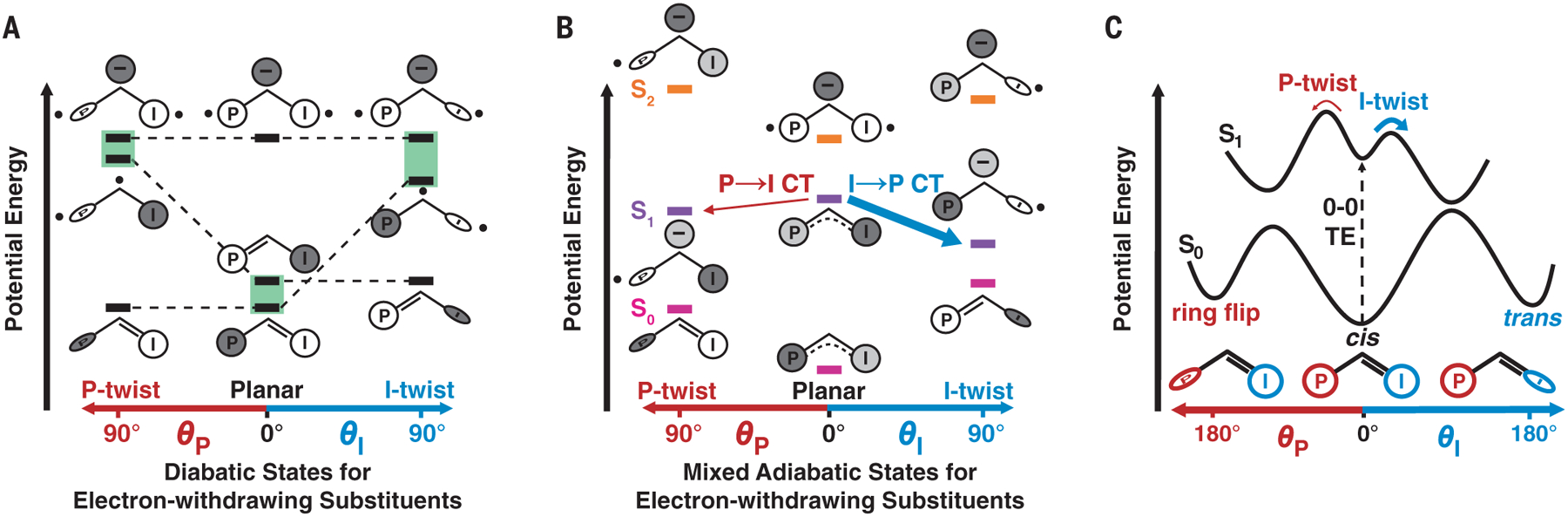
Shades of gray represent relative magnitude of negative charge localized to the methine bridge, P ring, or I ring, and dots represent unpaired electrons. The color scheme is equivalent to that in Fig. 1A. (A) Three diabatic states of the chromophore in planar, I-twisted, and P-twisted geometries, with energetic penalties required for breaking double bonds for rotation. Mixing of the coupled states (highlighted in green) leads to the adiabatic states shown in (B). For variants with electron-withdrawing substituents, the I-twist pathway is more energetically downhill, and thus preferred, compared with the P-twist pathway. Electron-donating substituents would have the opposite energetic effect and favor the P-twist pathway (not shown for clarity). Although the relative energy levels of this allylic anion model are qualitative, they are consistent with high-level calculations on the free chromophore at different bond rotation geometries (15). Negative charge transfer (CT) occurs from I to P for the I-twist pathway and from P to I for the P-twist pathway, which agrees with a Hammett analysis (supplementary text S3) and simulations of the free chromophore (5, 14). (C) Potential energy diagram for FP chromophore isomerization with two competing bond rotation pathways inspired by the mixed adiabatic states in (B). The GS cis chromophore is excited from S0 to S1 and relaxes to an S1 local minimum (relaxation coordinate not shown) (13). From the S1 minimum, the chromophore rotates about either the P or the I bond, depending on the relative ES barrier heights of the competing processes. The diagram represents Dronpa2 variants with electron-withdrawing substituents; variants with electron-donating substituents would have an inverted barrier height ratio between the two competing twisting pathways.