

Abstract
A series of novel compounds 6a–h, 8i–1, 10s–v, and 16a–d were synthesized and evaluated, together with the known analogs 11a–f, for their inhibitory activities towards acetylcholinesterase (AChE) and butyrylcholinesterase (BChE). The inhibitory activities of AChE and BChE were evaluated in vitro by Ellman method. The results show that some compounds have good inhibitory activity against AChE and BChE. Among them, compound 8i showed the strongest inhibitory effect on both AChE (eeAChE IC50 = 0.39 μM) and BChE (eqBChE IC50 = 0.28 μM). Enzyme inhibition kinetics and molecular modeling studies have shown that compound 8i bind simultaneously to the peripheral anionic site (PAS) and the catalytic sites (CAS) of AChE and BChE. In addition, the cytotoxicity of compound 8i is lower than that of Tacrine, indicating its potential safety as anti-Alzheimer’s disease (anti-AD) agents. In summary, these data suggest that compound 8i is a promising multipotent agent for the treatment of AD.
Keywords: Alzheimer’s disease, acetylcholinesterase inhibitor, butyrylcholinesterase inhibitor, cytotoxicity, molecular docking, structural modification, structure-activity relationship
1. Introduction
According to the report of the International Alzheimer’s Association in the last three years, there are more than 36 million AD patients in the world. With the acceleration of the aging process of the population, the amount of AD incidence is increasing year by year, and the number of AD patients in the world will exceed 130 million by 2050 [1,2,3]. The incidence of AD is over 1.9% among people over 60 years old in China [4]. AD is the third most common disease in the elderly after cardio-cerebrovascular disease and cancer. AD can cause dementia, which is one of the six leading causes of death in the United States [5,6].
AD was initially characterized by memory loss, cognitive dysfunction, inability to take care of themselves in daily life, and subsequent exacerbations of mental and behavioral abnormalities [7,8]. AD is not only a seriously threaten of human health and life, but also brings heavy mental burden and economic pressure to the family members and friends of the patients, causing huge fluctuations in the social economy. Therefore, it is an important task of medicinal chemists to develop effective drugs for the treatment of AD [9,10].
AD is a complex neurodegenerative syndrome. Due to the complicated pathogenesis of AD, its etiology is not completely clear. The therapeutic drugs used in the clinical and research stages can only delay the course of AD, but there are no drugs that cure or delay the course of AD [11,12]. A large number of studies have shown that hypotheses of the pathogenesis of AD include the theory of cholinergic damage, tau protein hyperphosphorylation, amyloid β-protein (Aβ) cascade hypothesis, metal ion homeostasis theory, APOE genotype, oxidative stress theory and so on [13,14,15,16,17,18].
Previous studies have identified three pathological features of AD: decreased levels of AChE in the neurotransmitter matrix, deposition of Aβ, and hyperphosphorylation of tau protein. Scientists have been trying to find the etiology and treatment strategy of AD through further study of the above three pathological characteristics [19,20]. At present, acetylcholinesterase inhibitors (AChEIs) are the main treatment for AD. Only five drugs have been approved by the Food and Drug Administration (FDA) to treat AD. Four of them are AChEIs, including Tacrine, Donepezil, Rivastigmine, and Galantamine. Tacrine is the first generation AChEI, but it is limited in clinical usage because of hepatotoxicity [11,21,22].
There are two types of ChEs in the central nervous system, namely AChE and BChE. AChE and BChE are important targets for the development of anti-AD drugs. The physiological function of AChE has been preliminarily understood, but the understanding of BChE is less. Studies have shown that ACh activity in some brain regions of patients with mild to severe AD decreases to 10%–15% of the normal value, and AChEIs have significant effects on patients with mild to moderate AD and can repair their cognitive impairment and other symptoms [23].
When AD develops to the middle and late stage, AChE activity decreases, whereas BChE activity increases, and BChE acts as a metabolic compensation for AChE, partially compensating for the role of AChE in hydrolyzing ACh [11,23,24]. The regulation of AChE is increasingly dependent on BChE, so BChE is gradually accepted as a target of anti-AD drugs. Therefore, the design and development of dual-target inhibitors of AChE and BChE may have the following advantages: it can not only effectively reduce the degradation of AChE and the drug resistance of AChEIs, but also be effective in patients for moderate to severe symptoms of AD. Some people think that appropriate inhibition of AChE and BChE is a more ideal treatment for AD [25,26]. Therefore, we are looking for dual-target ChEs inhibitors with inhibitory activity on both AChE and BChE.
With the study of AChE crystal structure, it was found that the ligand binding pocket of AChE in general is a long and narrow channel extending from the surface to the interior [27]. The channel is dumbbell-shaped. The opening and bottom of the channel are relatively open, and the middle is narrow. The active site of AChE contains two important domains: CAS at the bottom of the channel is the binding site for substrates and inhibitors, consisting of three residues: Ser203, His447, and Glu334; PAS is situated at the opening of the channel, which is the binding site for the enzyme inhibitor. It consists of five residues: Tyr72, Tyr124, Trp286, Tyr34 and Asp74 [11,28,29]. It was found that PAS of AChE could induce the formation of Aβ protein and accelerate its precipitation. Researchers began to develop dual-site AChEIs that act on both CAS and PAS sites simultaneously, which can interfere with the aggregation of Aβ while enhancing the inhibitory activity of AChE, and play a dual role in the treatment of AD [30].
In our previous studies, we found several new ChEIs through virtual screening based on pharmacophore. We have found that G801-0274 AChE (eeAChE IC50 = 2.05 μM), BChE (eqBChE IC50 = 0.03 μM) can inhibit ChEs. In this paper, we used it as the lead compound for structural modification [31]. It has the property of dual-site binding and can bind PAS and CAS sites simultaneously.
In this study, we designed and synthesized a series of new derivatives based on G801-0274 and evaluated their biological activities, including cytotoxicity and AChEs inhibition. By summarizing our data, we found a new type of dual-target inhibitor of AChE and BChE with in vitro activities, hoping to develop anti-AD drugs through further efforts (Figure 1).
Figure 1.

Design of a series of derivatives as dual-target inhibitor of AChE and BChE.
2. Results and Discussion
2.1. Chemistry
The synthesis of the designed compounds 6a–h and 8i–l started from 4-piperidinecarboxamide 1. At the first step, compound 1 was reacted with appropriate substituted Benzylchloride derivatives (2a–d) under potassium carbonate (K2CO3) and potassium iodide (KI) conditions, giving relevant N-Benzylpiperidin-4-carboxamide derivatives (3a–d) with medium yields (67%–81%). In addition, the obtained compounds 3a–d were transformed into relevant substituted (1-Benzylpiperidin-4-yl) methanamine derivatives (4a–d) in the reduction reaction, which was carried out in dry tetrahydrofuran (THF) using lithium aluminum hydride (LiAlH4) under nitrogen atmosphere. The obtained compounds 4a–d were used in further synthesis without purification. The yields of this step were between 74% and 82%. Finally, the commercially available nitrogen-containing heterocyclic aromatic carboxylic acids (5a–d) or 2-Oxoindoline-5-carboxylic acid 7 were activated with Carbonyldiimidazole (CDI) or Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) and reacted with the appropriate amine (4a–d) in dry THF or N,N-dimethylformamide (DMF) to give the target compounds 6a–h and 8i–l with moderate to good yield (35%–80%) (Scheme 1 and Scheme 2). Compounds 10s–v were prepared (73%–88% yield) by reactions of compounds 4a–d with p-Toluenesulfonyl chloride (TsCl, 9) under the presence of Triethylamine (Et3N) in Dichloromethane (DCM) (Scheme 3).
Scheme 1.

Synthesis of compounds 6a–h. Reagents and conditions: (a) compounds 2a–d, KI, K2CO3, acetone, reflux 4 h; (b) LiAlH4, THF, 0 °C, reflux 4 h; (c) 5a–d, CDI, THF, or PyBOP, DIPEA (N,N-Diisopropylethylamine), DMF, r.t.
Scheme 2.

Synthesis of compounds 8i–l. Reagents and conditions: 2-Oxoindoline-5-carboxylic acid 7, PyBOP, DIPEA, DMF, r.t. 24 h.
Scheme 3.

Synthesis of compounds 10s–v. Reagents and conditions: TsCl 9, Et3N, DCM, 0 °C 4 h.
The structures of the new compounds were confirmed by spectral data (1H-NMR, 13C-NMR, and HRMS, see Supplementary Materials).
The synthetic route of compounds 16a–d has been depicted in Scheme 4. Ethyl 2-piperidin-4-ylacetate 11 was reacted with appropriate substituted (2-Bromoethyl) benzene derivatives (12a–b) under potassium carbonate (K2CO3) and potassium iodide (KI) conditions. Then, the obtained compounds 13a–b were used in further synthesis without purification. 4 mol/L potassium hydroxide (KOH) was added to compounds 13a–b in C2H5OH: H2O = 5:1. The reaction mixture was stirred at room temperature for 7 h to give compounds 14a–d. Finally, compounds 14a–d were activated with PyBOP and reacted with compounds 15a–b in DMF to give the target compounds 16a–d with moderate to good yield (30%–80%) (Scheme 4).
Scheme 4.

Synthesis of compounds 16a–d. Reagents and conditions: (a) (2-Bromoethyl) benzene derivatives 12a–b, K2CO3, catalytic amount of KI, acetone, reflux 4 h; (b) 4 mol/L KOH, C2H5OH: H2O = 5:1, r.t. 7 h; (c) compounds 15a–b, PyBOP, DIPEA, DMF, r.t. 24 h.
2.2. AChE and BChE Inhibitory Activity of the Target Molecules
Compounds 6a–h, 8i–l, 11a–f, 10s–v, and 16a–d were evaluated for their anti-ChEs activity. Tacrine and Donepezil were used as reference drugs. According to the method described by Ellman [32], the data were expressed by IC50 values. In vitro experiments showed that some of these compounds could effectively inhibit ChEs in the micromolar range (Table 1).
Table 1.
Structures, eeAChE, and eqBChE inhibitory activities of target compounds.
| Compound | Structure | AChEa (IC50 c, μM or IRd, %) | BChE b (IC50, μM or IR d, %) | SI e |
|---|---|---|---|---|
| 6a |
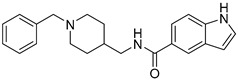
|
52.11 ± 30.25 | 31.37 ± 15.56 | 1.66 |
| 11a f |
|
38.08 ± 12.83 | 10.75 ± 5.55 | 3.54 |
| 6b |
|
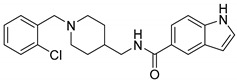
15.02 ± 6.17 | 9.56 ± 4.52 | 1.57 |
| 6c |
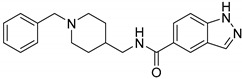
|
9.06 ± 2.57 | 36.64 ± 17.54 | 0.25 |
| 11b f |
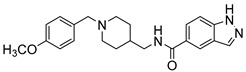
|
48.57 % | 44.57 % | -- |
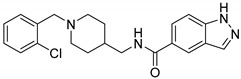
| 11c f |
|
5.44 ± 2.22 | 21.29 ± 5.00 | 0.26 |
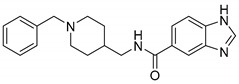
| 6d |
|
4.80 ± 0.68 | 2.15 ± 0.63 | 2.23 |
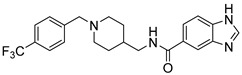
| 6e |
|
48.48 ± 36.19 | 39.88% | -- |
| 11d f |
|
18.81 % | 16.51 % | -- |
| 11e f |
|
7.48 ± 3.10 | 42.12 % | -- |
| 11f f |
|
0.46 ± 0.40 | 43.07 % | -- |
| 6f |
|
43.07 % | 49.63 % | -- |
| 6g |
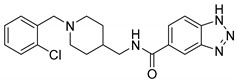
|
29.20 ± 16.38 | 49.87 ± 37.29 | 0.58 |
| 6h |
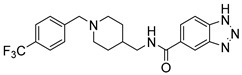
|
40.20 ± 27.64 | 30.71% | -- |
| 8i |
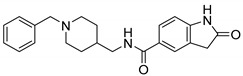
|
0.39 ± 0.04 | 0.28 ± 0.10 | 1.39 |
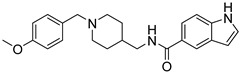
| 8k |
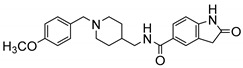
|
no | 22.26% | -- |
| 8j |
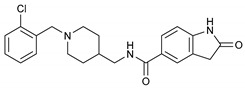
|
14.69% | 22.87% | -- |
| 8l |
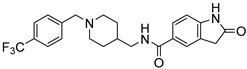
|
21.54 ± 9.37 | 35.20% | -- |
| 10s |
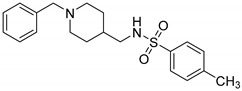
|
4.24 ± 2.44 | 4.10 ± 3.74 | 1.03 |
| 10t |
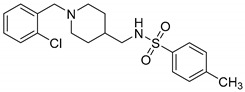
|
5.50 ± 2.56 | 2.01 ± 0.70 | 2.74 |
| 10u |
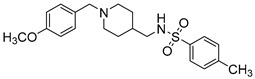
|
11.61% | 18.31% | -- |
| 10v |
|
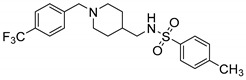
7.20 ± 10.75 | 7.14 ± 5.27 | 1.01 |
| 16a |
|
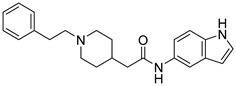
8.61% | 23.49% | -- |
| 16b |
|
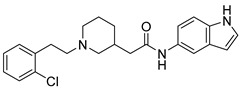
8.95% | 31.21% | -- |
| 16c |
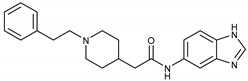
|
no | 30.59% | -- |
| 16d |
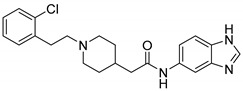
|
8.55% | 38.04% | -- |
| Tacrine | _ | 0.02 ± 0.01 | 0.008 ± 0.004 | 2.50 |
| Donepezil | _ | 0.008 ± 0.002 | 1.734 ± 0.731 | 0.0046 |
a AChE (EC 3.1.1.7) from electric eel. b BChE (EC 3.1.1.8) from horse serum. c Concentration required for 50% inhibition of ChEs, data were shown in mean ± SEM of triplicate independent experiments. d Inhibitory rate of the compounds under 100 μM on ChEs. e Selectivity index (SI) = AChE IC50/BChE IC50. f The known analogs 11a–f [33].
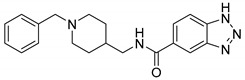
First, we synthesized compounds 6a, 6c, 6d, 8i by maintaining the R as H, X as amide and exploring Y with 1H-Indole, 1H-Indazole, 1H-Benzo[d] imidazole or 2-Oxoindoline, together with the known analog 11f.We found that when Y is substituted by a series of different structures, its activity on AChE are 2-Oxoindoline (8i) > 1H-Benzo[d] [1,2,3] triazole (11f) > 1H-Benzo[d]imidazole (6d) > 1H-Indazole (6c) > 1H-Indole (6a); its activity on BChE are 2-Oxoindoline (8i) > 1H-Benzo[d]imidazole (6d) > 1H-Indole (6a) > 1H-Benzo[d] [1,2,3] triazole (11f). Among them, compound 8i (Figure 2) showed the strongest inhibitory effect on both AChE (eeAChE IC50 = 0.39 μM) and BChE (eqBChE IC50 = 0.28 μM), these results indicated that compound 8i was a potent dual inhibitor against AChE and BChE. It was speculated that 2-Oxoindoline is the key structure for inhibiting two ChEs and is not selective for both ChEs. According to the molecular docking results, for eeAChE, 2-Oxoindoline of compound 8i bound with Trp286 via π-π stacking interaction; for huAChE, 2-Oxoindoline of compound 8i bound with Trp286 and Tyr341 via π-π stacking interaction, and for huBChE, 2-Oxoindoline of compound 8i bound with Phe329 via π-π stacking interaction. These interactions increase the inhibitory activity by enhancing the binding affinity.
Figure 2.

Design of compound 8i for dual-target inhibitor of AChE and BChE.
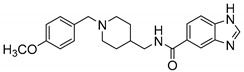
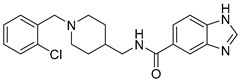
Then, we evaluated the effect of R on ChEs activity. We modified R with different substituent (2-Cl, 4-OCH3, 4-CF3), compared with compound 6d, when R is 4-CF3 (6e), 4-OCH3 (11d), 2-Cl (11e), the ChEs inhibitory activity decreased. In particular, compound 11d showed little inhibitory activity on BChE at 100 μM. In general, with the same Y, when R is H, the compounds have the highest inhibitory activity against ChEs; when Y is substituted by 2-Cl or 4-CF3, the inhibitory activity of the compounds to ChEs were weakened; and when Y is substituted by 4-OCH3, the compounds have the worst inhibitory activity against AChEs. We speculate that enhancing the electron-withdrawing effect or the donor effect on the aromatic ring is not conducive to improving the performance of the analog, and the appropriate space may facilitate the analog to enter the CAS pocket of ChEs.
Next, we investigated the effect of the p-Toluenesulfonamide moiety on the inhibitory activity of ChEs. Not only the amide group was replaced by Sulfonamide moiety based on the principles of bioisosterism, but also the fused nitrogen-containing bicyclic system (Indole, Indazole, Oxoindoline, Benzimidazole) in the previous compounds was replaced by Tosyl moiety. We synthesized compounds 10s–v. Except for compound 10u (eeAChE 11.61% [100 μM], eqBChE 18.31% [100 μM]) has low inhibitory activity against ChEs, the compounds 10s (eeAChE IC50 = 4.24 μM, eqBChE IC50 = 4.10 μM)), 10t (eeAChE IC50 = 5.50 μM, eqBChE IC50 = 2.01 μM), 10v (eeAChE IC50 = 7.20 μM, eqBChE IC50 = 7.14 μM) all can maintain ChEs inhibitory activity at micromolar levels, indicating that the p-Toluenesulfonamide moiety is responsible for maintaining the inhibitory activity of ChEs. On one hand, we speculate that methyl occupies the pocket of the active site and interacts with amino acid residues to increase inhibitory activity. On the other hand, Sulfonamide moiety is very important for maintaining ChEs inhibitory activity, which may be related to the bond angle between Sulfonamide and molecules [33].
In addition, we tested the number of carbon atoms between benzene and piperidine. When the number of carbon atoms becomes two (compounds 16a–d), the compounds had low inhibitory activity against the two kinds of ChEs, in particular, compound 16c has no inhibitory activity against AChE at 100 μM. We speculate that the decrease in the activity of such compounds may be that the molecular volume is too large to enter the active pocket of ChEs, indicating that the residue of N-Benzylpiperidine in the structure is an essential group for inhibiting both ChEs.
Compounds designed with a Piperazine ring instead of a piperidine ring may also have the same or higher inhibitory activity on ChEs. Luca P, Tomás Daniel, Asha H, et al. [34] based on the structure of Donepezil, mainly the conjugation of Benzylpiperidine/ Benzylpiperazine moiety with a biologically active heterocyclic derivative (Benzimidazole or Benzofuran), which gave the compound other relevant properties. It shows good activity (4.0–30.0 μM) for AChE inhibition, and has inhibition of Aβ peptide aggregation, antioxidant activity, and metal chelation.
2.3. Kinetic Studies of AChE and BChE Inhibition
To determine the kinetic types of AChE and BChE inhibition, compounds 8i and 10s were selected for kinetic studies. In each case, the kinetic types of enzyme inhibition were obtained by the modified Ellman’s method and the Lineweaver–Burk secondary plots [35]. The Lineweaver–Burk plots showed both increasing slope (decreased Vmax) and increasing intercept (higher Km) for higher inhibitor concentrations, indicating a mixed-type inhibition, including competitive inhibition and non-competitive inhibition, which possibly was because compound 8i could bind to both CAS and PAS (Figure 3A,B). According to the result of molecular docking study. The same inhibition type between compound 10s and ChEs was found in graphical analysis (Figure 3C,D).
Figure 3.

(A) Lineweaver–Burk plot for the inhibition of eeAChE (A) and eqBChE (B) by compound 8i at different concentrations of substrate. (B) Lineweaver–Burk plot for the inhibition of eeAChE (C) and eqBChE (D) by compound 10s at different concentrations of substrate.
2.4. Docking Studies
To further study the binding mode of compound 8i and ChEs, molecular docking was performed using Discovery Studio software 2016. The predicted binding mode of compound 8i is shown in Figure 4 and Figure 5. Compound 8i could interact with CAS and PAS of AChE simultaneously. For AChE (from Electrophorus electricus (electric eeAChE, Sigma-Aldrich) (Figure 4A), the N-Benzylpiperidine moiety interacted with Trp86 in CAS via aromatic π-π interaction. The Amide group formed hydrogen bond with Phe295. Moreover, 2-Oxoindoline of compound 8i bound with Trp286 via π-π stacking interaction; for human AChE-huAChE (Sigma-Aldrich) (Figure 4B), the N-Benzylpiperidine moiety of compound 8i was bound to CAS, displaying a classic aromatic π-π interaction with Trp86. Moreover, 2-Oxoindoline of compound 8i bound with Trp286 and Tyr341 via π-π stacking interaction. In addition, the Amide group formed hydrogen bond with Phe295. By comparison, it was found that the compounds have similar binding patterns to eeAChE and huAChE. All these facts provide an explanation for the higher inhibitory effects of compound 8i towards AChE.
Figure 4.

(A) Binding mode prediction of compound 8i with eeAChE (PDB ID: 1C2B) (B) Binding mode prediction of compound 8i with huAChE (PDB ID: 4EY7).
Figure 5.

Binding mode prediction of compound 8i with huBChE (PDB ID: 4TPK).
Molecular docking of compound 8i at the active site of human BuChE-huBChE (Sigma-Aldrich, Munich, Germany) has been shown in Figure 5. The N-Benzylpiperidine moiety of compound 8i interacts with Asp70 via electrostatic interaction. Moreover, the N-Benzylpiperidine moiety interacts with Trp82 in CAS by T-shaped π-π interaction, and the N-Benzylpiperidine moiety forms π-alkyl interaction with Ala328. 2-Oxoindoline moiety of compound 8i bound with Phe329 via π-π stacking interaction. These interactions increase the inhibitory activity by enhancing the binding affinity.
From the binding mode prediction of Donepezil with huAChE (Figure 6), we found that the binding pattern of compound 8i is similar to Donepezil in some respects: (i) The N-Benzylpiperidine moiety was bound to CAS, displaying a classic aromatic π-π interaction with Trp86; (ii) The Oxygen atom formed hydrogen bond with Phe295; (iii) Aromatic heterocycle moiety bound with Trp286 and Tyr341 via π-π stacking interaction. In addition, the Indone moiety of Donepezil interacts with Trp286 at the center of the PAS; the piperidine moiety of Donepezil interacts with Tyr337, Phe338, Tyr341.
Figure 6.

(A) Binding mode prediction of Donepezil with eeAChE (PDB ID: 1C2B); (B) Binding mode prediction of Donepezil with huAChE (PDB ID: 4EY7).
From the binding mode prediction of Donepezil with huBChE (Figure 7), we also found that the binding pattern of compound 8i is similar to Donepezil in some respects: (i) The N-Benzylpiperidine moiety interacts with Asp70 via electrostatic interaction. In addition, the N-Benzylpiperidine moiety of Donepezil interacts with Try332 via electrostatic interaction; (ii) the N-Benzylpiperidine moiety interacts with Trp82 in CAS by T-shaped π-π interaction, and the N-Benzylpiperidine moiety form π-alkyl interaction with Ala328. The difference is that 2-Oxoindoline of compound 8i bound with Phe329 via π-π stacking interaction while the 2-Oxoindoline moiety of Donepezil interacts with Gly116 in the center of the PAS.
Figure 7.

Binding mode prediction of Donepezil with huBChE (PDB ID: 4TPK).
Compared with Donepezil, the inhibitory activity of compound 8i on BChE is higher than Donepezil, and has similar inhibitory activity on two ChEs, so that it can exert an anti-ChEs effect in a balanced manner. Studies on compound 8i molecular docking have shown that the Benzylpiperidine moiety of the compound acts on the CAS of the enzyme, while the 2-Oxoindoline moiety binds to the PAS of the enzyme, which is basically consistent with the design idea.
2.5. Cytotoxicity Studies
We focused on the cytotoxicity of the synthetic compounds. The reason for using PC12 is that our compounds act on the central nervous system, so we need to find out whether the compound has a toxic effect on normal nerve cells. Compounds 6b, 6c, 6d, 10s, 8i, and 10v were selected as representative compounds to assess their potential cytotoxic effects. Compounds 6c, 6d, 8i are less toxic than Tacrine; the toxicity of compounds 6b, 10v are similar to that of Tacrine; compound 10s is slightly more toxic than Tacrine. Among them, compound 8i has the lowest toxicity (Figure 8).
Figure 8.

In vitro cell toxicity of compounds 6b, 6c, 6d, 10s, 8i, and 10v on PC-12 cell line. Data were expressed as mean ± SD (n = 3).
3. Materials and Methods
3.1. Chemistry
All reagents were obtained from commercial suppliers and were used without any further purification unless otherwise stated. Flash column chromatography was performed with silica gel (200-300 mesh) purchased from Qingdao Haiyang Chemical Co. Ltd. Thin layer chromatography was performed using silica gel 60 F254 precoated plates (purchased from Qingdao Haiyang Inc., Qingdao, China). Visualization was achieved using Ultraviolet (UV) light (254 nm and 365 nm, Shanghai Yarong Biochemical Instrument Factory, Shanghai, China). Melting points were determined with a Mel-TEMP II melting point apparatus (Beijing Keyi Company, Beijing, China) and was uncorrected. 1H NMR and 13C NMR spectra were recorded with Bruker AV-600, AV-500 or AV-400 MHz instruments (Bruker, Ettlingen, Germany) using DMSO-d6, CD3OD, or CDCl3 as solvent. Chemical shifts were reported as δ values (ppm) from internal reference tetramethylsilane (TMS). All coupling constants were reported in hertz (Hz), All chemical shifts are reported in parts per million (ppm), relative to the internal standard. In addition, proton multiplicities were labeled as br (broad), s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), and m (multiplet). HR-MS were performed on a Waters Vion IMS Q-tof (Waters, MA, USA).
3.2. General Procedure for the Synthesis of Compounds 3a–d
4-Piperidinecarboxamide (1) (3.00 g, 23.4 mmol) and substituted Benzylchloride derivatives (2a–d) (28.1 mmol) were dissolved in 20 mL acetone. Then, anhydrous K2CO3 (6.47 g, 46.8 mmol) and catalytic amount KI were added. The reaction mixture was refluxed for 4 h. After completion of the reaction, acetone was concentrated, and the residue was dissolved in water (60 mL) and extracted with ethyl acetate (60 × 3 mL). The combined organic layers were dried over Na2SO4, filtered, and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph (DCM: methanol = 60:1–5:1) to give target compounds 3a–d.
3.3. General Procedure for the Synthesis of Compounds 4a–d
Compounds 3a–d (3 g) were dissolved in anhydrous THF (17 mL) and then LiAlH4 (5 equiv.) was added to the above cooled solution at 0–5 °C in small portions under stirring. The reaction mixture was further stirred at room temperature for 30 min and finally refluxed for 4 h. After cooling, water and 10% NaOH solution was added at 0–5 °C. Then, the obtained white precipitate was filtered off and washed with THF. The filtrate was extracted with ethyl acetate. The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuum. The obtained compounds 4a–d were used in further synthesis without purification.
3.4. General Procedure for the Synthesis of Compounds 6a–h
CDI (1 equiv.) was added to a solution of the acids (5a–d) ((300 mg, 1 equiv.) in dry THF under nitrogen atmosphere. After 30 min, the solution of substituted 4-Amine-1-benzylpiperidines (4a–d) (1.2 equiv.) in THF were added, and the reaction mixture was stirred at room temperature for 24 h. After the reaction was completed, the solvent was removed under reduced pressure, and then the reaction mixture was quenched with saturated NaCl solution (25 mL). The aqueous phase was extracted with DCM (25 × 3 mL). The DCM layer was combined and washed with brine solution (25 × 3 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph (DCM: methanol = 60:1–5:1) to give target compounds 6a–h.
N-((1-Benzylpiperidin-4-yl)methyl)-1H-indole-5-carboxamide (6a). 1H-Indole-5-carboxylic acid (300 mg, 1.86 mmol), CDI (302 mg, 1.86 mmol), (1-Benzylpiperidin-4-yl)methanamine (457 mg, 2.24 mmol), THF (15 mL) White solid, m.p.: 89–90 °C, yield: 80%, 1H NMR (500 MHz, DMSO-d6) δ 11.50 (s, 1H), 8.47 (s, 1H), 8.17 (s, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H), 7.41 (d, J = 7.1 Hz, 2H), 7.36–7.29 (m, 3H), 6.51 (s, 1H), 3.78 (s, 2H), 3.19 (d, J = 5.5 Hz, 2H), 2.99 (d, J = 10.8 Hz, 2H), 2.31 (d, J = 11.0 Hz, 2H), 1.72 (d, J = 12.0 Hz, 3H), 1.38 (d, J = 11.4 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 168.15, 137.84, 135.07, 130.40, 128.81, 128.39, 127.44, 127.05, 125.94, 120.97, 120.37, 111.37, 102.49, 61.15, 52.37, 44.70, 35.20, 28.63. HRMS (ESI): calcd. For C22H25N3O [M + H]+ 348.2070, found 348.2070.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-1H-Indole-5-carboxamide (6b). 1H-Indole-5-carboxylic acid (300 mg, 1.86 mmol), CDI (302 mg, 1.86 mmol), (1-(2-Chlorobenzyl) piperidin-4-yl)methanamine (532 mg, 2.24 mmol), THF (15 mL) White solid, m.p.: 88–89 °C, yield: 66%, 1H NMR (400 MHz, CD3OD) δ 8.08 (d, J = 1.1 Hz, 1H), 7.58 (dd, J = 8.6, 1.8 Hz, 1H), 7.49 (dd, J = 7.1, 2.2 Hz, 1H), 7.41–7.35 (m, 2H), 7.30–7.24 (m, 3H), 6.51 (d, J = 3.2 Hz, 1H), 3.78 (s, 2H), 3.05 (d, J = 11.7 Hz, 2H), 2.31 (t, J = 11.9 Hz, 2H), 1.77 (t, J = 14.0 Hz, 3H), 1.45–1.33 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 170.60, 138.15, 134.64, 131.71, 129.37, 129.10, 127.70, 126.78, 125.91, 125.17, 120.18, 119.95, 110.67, 102.17, 58.57, 53.06, 44.79, 35.56, 28.93. HRMS (ESI): calcd. For C22H24ClN3O [M + H]+ 382.1681, found 382.1703.
N-((1-Benzylpiperidin-4-yl) methyl)-1H-indazole-5-carboxamide (6c). 1H-Indazole-5-carboxylic acid (300 mg, 1.86 mmol), CDI (302 mg, 1.86 mmol), (1-Benzylpiperidin-4-yl)methanamine (457 mg, 2.24 mmol), THF (15 mL) White solid, m.p.: 111–112 °C, yield: 56%, 1H NMR (500 MHz, DMSO-d6) δ 13.30 (s, 1H), 8.48 (s, 1H), 8.35 (s, 1H), 8.20 (s, 1H), 7.87 (d, J = 8.8 Hz, 1H), 7.57 (d, J = 8.7 Hz, 1H), 7.33–7.27 (m, 3H), 7.24 (d, J = 6.8 Hz, 1H), 3.45 (s, 2H), 3.18 (d, J = 6.0 Hz, 2H), 2.81 (d, J = 11.1 Hz, 2H), 1.92 (t, J = 11.1 Hz, 2H), 1.67 (d, J = 12.4 Hz, 2H), 1.59 (s, 1H), 1.22 (d, J = 9.4 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.07, 141.33, 138.83, 135.11, 129.24, 128.56, 127.67, 127.62, 127.30, 125.79, 122.79, 120.90, 110.16, 62.78, 53.38, 45.31, 36.17, 30.23. HRMS (ESI): calcd. For C21H24N4O [M + H]+ 349.2023, found 349.2019.
N-((1-Benzylpiperidin-4-yl)methyl)-1H-benzo[d]imidazole-5-carboxamide (6d). 1H-Benzo[d]imidazole-5-carboxylic acid (300 mg, 1.85 mmol), CDI (300 mg, 1.85 mmol), (1-Benzylpiperidin-4-yl)methanamine (453 mg, 2.22 mmol), THF (15 mL) Yellow oil, yield: 72%, 1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.27 (d, J = 68.8 Hz, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.70–7.61 (m, 1H), 7.28 (d, J = 6.9 Hz, 3H), 7.21 (d, J = 5.6 Hz, 1H), 7.04 (s, 1H), 3.39 (s, 2H), 3.19 (s, 2H), 2.76 (s, 2H), 1.85 (d, J = 11.8 Hz, 2H), 1.65 (d, J = 11.8 Hz, 3H), 1.20 (d, J = 10.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.40, 144.24, 139.08, 135.64, 129.16, 128.51, 127.18, 121.88, 115.66, 114.85, 62.93, 53.43, 53.35, 45.39, 36.25, 30.32. HRMS (ESI): calcd. For C21H24N4O [M + H]+ 349.2023, found 349.2023.
N-((1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methyl)-1H-benzo[d]imidazole-5-carboxamide (6e). 1H-Benzo[d]imidazole-5-carboxylic acid (300 mg, 1.85 mmol), CDI (300 mg, 1.85 mmol), (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (604 mg, 2.22 mmol), THF (15 mL) White solid, m.p.: 92–93 °C, yield: 77%, 1H NMR (400 MHz, CD3OD) δ 8.29–8.27 (m, 1H), 8.12 (d, J = 1.0 Hz, 1H), 7.87–7.77 (m, 2H), 7.62 (d, J = 7.8 Hz, 1H), 7.56 (t, J = 8.9 Hz, 2H), 7.39 (p, J = 5.5 Hz, 2H), 3.66 (d, J = 6.7 Hz, 2H), 3.30 (s, 1H), 3.27 (s, 1H), 2.90 (d, J = 11.4 Hz, 2H), 2.15–2.06 (m, 2H), 1.79–1.63 (m, 3H), 1.44–1.24 (m, 3H). 13C NMR (126 MHz, CD3OD) δ 169.23, 141.32, 134.81, 131.92, 130.75, 130.34, 128.00, 127.37, 127.30, 125.41, 122.55, 120.67, 114.13, 113.58, 109.69, 58.94, 57.96, 53.34, 53.01, 44.93, 44.83, 35.64, 35.52, 29.26, 29.06. HRMS (ESI): calcd. For C22H23F3N4O [M + H]+ 417.1897, found 417.1895.
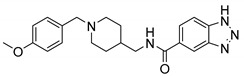
N-((1-(4-Methoxybenzyl)piperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide (6f). 1H-Benzo[d][1,2,3]triazole-5-carboxylic acid (300 mg, 1.84 mmol), CDI (298 mg, 1.84 mmol), (1-(4-Methoxybenzyl)piperidin-4-yl)methanamine (517 mg, 2.21 mmol), THF (15 mL) Yellow oil, yield: 63%, 1H NMR (500 MHz, DMSO-d6) δ 8.53 (s, 1H), 8.32 (s, 1H), 8.16 (s, 1H), 7.75 (d, J = 8.4 Hz, 1H), 7.61 (d, J = 8.3 Hz, 1H), 7.32 (d, J = 8.3 Hz, 2H), 6.91 (d, J = 8.3 Hz, 2H), 3.74 (s, 3H), 3.69 (s, 2H), 3.18 (d, J = 5.9 Hz, 2H), 2.97 (d, J = 11.1 Hz, 2H), 2.26 (s, 2H), 1.73 (d, J = 13.0 Hz, 2H), 1.68 (s, 1H), 1.34 (d, J = 11.2 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 167.31, 159.33, 144.22, 131.57, 128.92, 121.94, 114.16, 60.78, 55.52, 52.34, 44.84, 35.34, 28.86. HRMS (ESI): calcd. For C21H25N5O2 [M + H]+ 380.2081, found 380.2077.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide (6g). 1H-Benzo[d][1,2,3]triazole-5-carboxylic acid (300 mg, 1.84 mmol), CDI (298 mg, 1.84 mmol), (1-(2-Chlorobenzyl)piperidin-4-yl)methanamine (526 mg, 2.21 mmol), THF (15 mL) White solid, m.p.: 77–79 °C, yield: 57%, 1H NMR (500 MHz, DMSO-d6) δ 8.63 (s, 1H), 8.44 (s, 1H), 7.87 (d, J = 2.7 Hz, 2H), 7.47 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 7.8 Hz, 1H), 7.30 (d, J = 6.9 Hz, 1H), 7.27–7.25 (m, 1H), 7.03 (s, 1H), 3.52 (s, 2H), 3.21 (t, J = 6.1 Hz, 2H), 2.82 (d, J = 11.3 Hz, 2H), 2.00 (t, J = 10.4 Hz, 2H), 1.68 (d, J = 12.0 Hz, 2H), 1.61 (s, 1H), 1.23 (d, J = 10.5 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.50, 140.30, 139.38, 138.70, 131.96, 129.27, 128.56, 127.32, 125.46, 115.61, 114.32, 62.73, 53.33, 45.41, 36.08, 30.16. HRMS (ESI): calcd. For C20H22ClN5O [M + H]+ 384.1586, found 384.1584.
N-((1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methyl)-1H-benzo[d][1,2,3]triazole-5-carboxamide (6h). 1H-Benzo[d][1,2,3]triazole-5-carboxylic acid (300 mg, 1.84 mmol), CDI (298 mg, 1.84 mmol), (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (601 mg, 2.21 mmol), THF (15 mL) White solid, m.p.: 90–91 °C, yield: 73%, 1H NMR (400 MHz, CD3OD) δ 8.38 (td, J = 1.5, 1.0 Hz, 1H), 7.92–7.90 (m, 1H), 7.88 (t, J = 1.0 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.61 (dt, J = 7.7, 4.1 Hz, 1H), 7.51–7.42 (m, 2H), 3.89 (d, J = 22.6 Hz, 2H), 3.33 (dd, J = 6.5, 3.7 Hz, 2H), 3.13–3.01 (m, 2H), 2.47–2.31 (m, 2H), 1.90–1.68 (m, 3H), 1.52–1.32 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 166.50, 140.30, 139.38, 138.70, 131.96, 129.27, 128.56, 127.32, 125.46, 115.61, 114.32, 62.73, 53.33, 45.41, 36.08, 30.16. HRMS (ESI): calcd. For C21H22F3N5O [M + H]+ 418.1849, found 418.1850.
3.5. General Procedure for the Synthesis of 8i–l
Intermediates (7) (140 mg, 1.2 equiv.), PyBOP (1.2 equiv.) and DIPEA (1.5 equiv.) were added to DMF and stirred at room temperature for 20 min. Then, intermediates (4a–d) (1.0 equiv.) was added and stirred at room temperature for 4 h. After completion of the reaction, the reaction mixture was quenched with saturated NaCl solution (25 mL). The aqueous phase was extracted with DCM (25 × 3 mL). The DCM layer was combined and washed with brine solution (25 × 3 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph using a methanol in DCM gradient (DCM: methanol= 60:1–5:1) yielded compounds 8i–l.
N-((1-Benzylpiperidin-4-yl)methyl)-2-Oxoindoline-5-carboxamide (8i). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-Benzylpiperidin-4-yl)methanamine (135 mg, 0.66 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.:155–156 °C, yield: 56%, 1H NMR (400 MHz, CD3OD) δ 7.73–7.68 (m, 2H), 7.53–7.42 (m, 5H), 6.91 (d, J = 8.1 Hz, 1H), 4.26 (s, 2H), 3.44 (d, J = 12.5 Hz, 2H), 3.31 (s, 1H), 2.98 (t, J = 12.9 Hz, 2H), 1.97 (d, J = 13.6 Hz, 3H), 1.54 (q, J = 13.1, 12.1 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 178.52, 168.90, 146.69, 130.99, 129.80, 129.40, 128.99, 127.92, 127.60, 125.95, 123.42, 123.40, 108.97, 60.21, 51.86, 43.82, 42.11, 34.01, 26.82. HRMS (ESI): calcd. For C22H25N3O2 [M + H]+ 364.2020, found 364.2032.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-2-Oxoindoline-5-carboxamide (8j). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-(2-Chlorobenzyl)piperidin-4-yl)methanamine (157 mg, 0.66 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.: 151–152 °C, yield: 35%, 1H NMR (600 MHz, CD3OD) δ 7.74–7.69 (m, 2H), 7.50 (d, J = 7.2 Hz, 1H), 7.39 (d, J = 7.6 Hz, 1H), 7.28 (dt, J = 20.1, 7.1 Hz, 2H), 6.93 (d, J = 8.1 Hz, 1H), 3.71 (s, 2H), 3.27 (d, J = 6.8 Hz, 2H), 3.00 (s, 2H), 2.21 (s, 2H), 1.76 (d, J = 12.7 Hz, 2H), 1.69 (s, 1H), 1.37 (q, J = 11.5 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 168.83, 146.63, 135.02, 132.40, 130.20, 129.68, 128.07, 127.58, 127.18, 125.93, 123.41, 108.96, 57.94, 52.76, 44.36, 34.83, 28.01. HRMS (ESI): calcd. For C22H24ClN3O2 [M + H]+ 398.1630, found 398.1652.
N-((1-(4-Methoxybenzyl)piperidin-4-yl)methyl)-2-Oxoindoline-5-carboxamide (8k). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-(4-Methoxybenzyl)piperidin-4-yl)methanamine (154 mg, 0.66 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.: 158–159 °C, yield: 43%, 1H NMR (400 MHz, CD3OD) δ 7.74–7.68 (m, 2H), 7.40 (d, J = 8.7 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 6.91 (d, J = 8.0 Hz, 1H), 4.19 (s, 2H), 3.79 (s, 3H), 3.43 (d, J = 12.0 Hz, 2H), 3.31 (s, 1H), 2.95 (t, J = 11.7 Hz, 2H), 1.97 (d, J = 13.9 Hz, 3H), 1.60–1.45 (m, 2H), 1.26 (s, 1H). 13C NMR (101 MHz, CD3OD) δ 178.54, 168.87, 161.10, 146.66, 132.51, 127.95, 127.62, 125.93, 123.46, 121.09, 114.21, 108.98, 54.55, 51.51, 43.78, 34.03, 27.22, 26.85. HRMS (ESI): calcd. For C23H27N3O3 [M + H]+ 394.2125, found 394.2131.
2-Oxo-N-((1-(4-(trifluoromethyl)benzyl)piperidin-4-yl)methyl)indoline-5-carboxamide (8l). 2-Oxoindoline-5-carboxylic acid (140 mg, 0.79 mmol), (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (179 mg, 0.65 mmol), PyBOP (412 mg, 0.79 mmol), DIPEA(128 mg, 0.99 mmol), DMF (6 mL). White solid, m.p.: 113–115 °C, yield: 40%, 1H NMR (400 MHz, CD3OD) δ 7.79 (d, J = 7.8 Hz, 1H), 7.73–7.69 (m, 2H), 7.62 (d, J = 8.2 Hz, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44–7.32 (m, 2H), 6.95–6.90 (m, 1H), 3.60 (d, J = 17.7 Hz, 2H), 3.24 (dd, J = 9.1, 6.8 Hz, 2H), 2.91–2.77 (m, 2H), 2.09–1.93 (m, 2H), 1.78–1.61 (m, 3H), 1.40–1.21 (m, 3H). 13C NMR (126 MHz, CD3OD)) δ 168.70, 146.44, 131.73, 130.35, 130.30, 128.23, 127.47, 127.30, 126.79, 125.81, 125.26, 123.34, 108.90, 59.79, 59.79, 58.24, 53.43, 53.14, 45.07, 36.04, 36.04, 36.00, 36.00, 29.82, 29.76. HRMS (ESI): calcd. For C23H24F3N3O2 [M + H]+ 432.1893, found 432.1886.
3.6. General Procedure for the Synthesis of 10s–v
To a solution of (1-Benzylpiperidin-4-yl) methanamine derivatives (4a–d) (150 mg, 1 equiv.) in DCM (5 mL) was added Et3N (1.8 equiv.). The mixture was cooled to 0 °C, TsCl (1.3 equiv.) in DCM (5 mL) was added dropwise, and the reaction mixture was stirred for an additional 1 h at 0 °C. After completion of the reaction, the reaction mixture was dissolved in 15 mL saturated sodium bicarbonate (NaHCO3) and extracted with DCM (25 × 3 mL). Organic phases were combined and washed with saturated NaCl solution (25 × 3 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph using a methanol in dichloromethane gradient (dichloromethane: methanol = 60:1–5:1) yielded compounds 10s–v.
N-((1-Benzylpiperidin-4-yl)methyl)-4-methylbenzenesulfonamide (10s). (1-Benzylpiperidin-4-yl) methanamine (150 mg, 0.73 mmol), TsCl (181 mg, 0.95 mmol), Et3N (133 mg, 1.31 mmol), DCM (9 mL) White solid, m.p.:78–79 °C, yield: 80%, 1H NMR (400 MHz, CD3OD) δ 7.68 (d, J = 8.3 Hz, 2H), 7.36–7.24 (m, 7H), 3.60 (s, 2H), 2.93 (d, J = 12.0 Hz, 2H), 2.67 (d, J = 6.8 Hz, 2H), 2.39 (s, 3H), 2.09 (t, J = 12.6 Hz, 2H), 1.68 (d, J = 13.8 Hz, 2H), 1.44 (s, 1H), 1.28–1.13 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 143.22, 137.71, 136.09, 129.69, 129.36, 128.05, 127.39, 126.67, 62.59, 52.72, 35.66, 28.79, 20.11. HRMS (ESI): calcd. For C20H26N2O2S [M + H]+ 359.1788, found 359.1802.
N-((1-(2-Chlorobenzyl)piperidin-4-yl)methyl)-4-methylbenzenesulfonamide (10t). (1-(4-Chlorobenzyl)piperidin-4-yl)methanamine (150 mg,0.63 mmol), TsCl (156 mg, 0.82 mmol), Et3N (114 mg, 1.13 mmol), DCM (9 mL) White solid, m.p.: 111–112 °C, yield: 73%, 1H NMR (400 MHz, CD3OD) δ 7.69 (d, J = 8.3 Hz, 2H), 7.44 (dd, J = 7.4, 2.0 Hz, 1H), 7.37–7.31 (m, 3H), 7.23 (td, J = 7.1, 1.9 Hz, 2H), 3.62 (s, 2H), 2.89 (d, J = 11.8 Hz, 2H), 2.67 (d, J = 6.8 Hz, 2H), 2.39 (s, 3H), 2.06 (t, J = 11.7 Hz, 2H), 1.65 (d, J = 13.1 Hz, 2H), 1.37 (s, 1H), 1.20 (dd, J = 12.3, 3.6 Hz, 1H), 1.15 (s, 1H). 13C NMR (101 MHz, CD3OD) δ 143.42, 137.49, 135.59, 133.53, 131.79, 130.10, 129.48, 127.74, 127.35, 126.68, 126.68, 20.13. HRMS (ESI): calcd. For C20H25ClN2O2S [M + H]+ 393.1398, found 393.1397.
N-((1-(4-Methoxybenzyl)piperidin-4-yl)methyl)-4-methylbenzenesulfonamide (10u). (1-(4-Methoxybenzyl)piperidin-4-yl)methanamine (150 mg, 0.64 mmol), TsCl (158 mg, 0.83 mmol), Et3N (117 mg, 1.15 mmol), DCM (9 mL) White solid, m.p.: 76–77 °C, yield: 87%, 1H NMR (600 MHz, CD3OD) δ 7.61 (d, J = 8.3 Hz, 2H), 7.25 (dd, J = 27.4, 8.3 Hz, 4H), 6.85 (d, J = 8.6 Hz, 2H), 3.80 (s, 2H), 3.70 (s, 3H), 3.10 (d, J = 10.7 Hz, 2H), 2.63 (d, J = 6.7 Hz, 2H), 2.44 (s, 2H), 2.32 (s, 3H), 2.05 (s, 1H), 1.73 (s, 2H), 1.52 (s, 1H), 1.27–1.19 (m, 2H). 13C NMR (101 MHz, CD3OD) δ 160.41, 143.33, 137.60, 131.82, 129.42, 126.66, 113.87, 60.67, 54.47, 51.88, 34.72, 27.47, 20.10. HRMS (ESI): calcd. For C21H28N2O3S [M + H]+ 389.1893, found 389.1894.
4-Methyl-N-((1-(4-(trifluoromethyl)benzyl)piperidin-4-yl)methyl)benzenesulfonamide (10v). (1-(4-(Trifluoromethyl)benzyl)piperidin-4-yl)methanamine (150 mg, 0.55 mmol), TsCl (136 mg, 0.72 mmol), Et3N (100 mg, 0.99 mmol), DCM (9 mL) White solid, m.p.: 87–88 °C, yield: 88%, 1H NMR (400 MHz, CD3OD) δ 7.76 (d, J = 7.8 Hz, 1H), 7.71–7.66 (m, 2H), 7.61 (d, J = 7.9 Hz, 1H), 7.57–7.51 (m, 1H), 7.40–7.31 (m, 4H), 3.58 (d, J = 14.0 Hz, 2H), 2.79 (t, J = 12.2 Hz, 2H), 2.67 (t, J = 6.9 Hz, 2H), 2.40 (s, 3H), 1.97 (t, J = 11.6 Hz, 2H), 1.69–1.59 (m, 2H), 1.40 (dtt, J = 14.5, 7.3, 3.2 Hz, 1H), 1.15 (dqd, J = 24.7, 12.2, 3.7 Hz, 2H). 13C NMR (126 MHz, CD3OD) δ 143.14, 137.67, 133.70, 131.73, 130.36, 130.33, 130.03, 129.28, 127.36, 126.84, 126.60, 125.27, 113.19, 59.65, 58.14, 53.26, 52.97, 48.19, 48.15, 48.12, 48.06, 47.95, 47.89, 47.78, 47.72, 47.61, 47.44, 47.32, 47.27, 47.10, 35.94, 35.86, 29.45, 20.03. HRMS (ESI): calcd. For C21H25F3N2O2S [M + H]+ 427.1662, found 427.1660.
3.7. General Procedure for the Synthesis of 16a–d
Ethyl 2-piperidin-4-ylacetate 11 (1 g, 5.84 mmol, 1.0 equiv.) and substituted (2-romoethyl)benzene derivatives (12a–b) (7.01 mmol, 1.2 equiv.) were dissolved in 20 mL acetone. Then, anhydrous K2CO3 (11.68 mmol, 2 equiv.) and catalytic amount KI were added. The reaction mixture was refluxed for 4 h. After completion of the reaction, acetone was concentrated, and the residue was dissolved in water (60 mL) and extracted with ethyl acetate (60 × 3 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuum. The obtained oil was used in further synthesis without purification yielded compounds 13a–b (Yields were 67% and 72%). Then, 4 mol/L KOH (2.5 equiv.) was added to the solution of compounds 13a–b in C2H5OH: H2O = 5:1(6 mL). The reaction mixture was stirred at room temperature for 7 h. After completion of the reaction, the reaction mixture was evaporated to dryness after neutralization with dilute hydrochloric acid solution. Poured into ethyl acetate to deposit the solid, after cooling off, the mixture was filtered and washed with cold ethyl acetate to give compounds 14a–d.
Finally, intermediates (14a–b) (1.2 equiv.), PyBOP (1.2 equiv.) and DIPEA (1.5 equiv.) were added to 6 mL DMF and stirred at room temperature for 20 min. Then, intermediates (15a–b) (1.0 equiv.) was added and stirred at room temperature for 4 h. After completion of the reaction, the reaction mixture was quenched with saturated NaCl solution. The aqueous phase was extracted with DCM. The DCM layer was combined and washed with brine solution. The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. After concentration, the crude product was purified by silica gel column chromatograph using a methanol in dichloromethane gradient (DCM:methanol = 60:1–5:1) yielded compounds 16a–d.
N-(1H-Indol-5-yl)-2-(1-phenethylpiperidin-4-yl)acetamide (16a). 2-(1-Phenethylpiperidin-4-yl) acetic acid (140 mg, 0.57 mmol), 1H-Indol-5-amine (63 mg, 0.48 mmol), PyBOP (295 mg, 0.57 mmol), DIPEA (93 mg, 0.72 mmol), DMF (6 mL). Yellow solid, m.p.: 165–167 °C, yield: 80.70%, 1H NMR (400 MHz, CD3OD) δ 7.74 (d, J = 2.0 Hz, 1H), 7.28 (tt, J = 13.1, 6.8 Hz, 6H), 7.20 (d, J = 3.1 Hz, 1H), 7.15 (dd, J = 8.6, 2.0 Hz, 1H), 6.37 (d, J = 3.1 Hz, 1H), 3.60 (d, J = 14.9 Hz, 2H), 3.26 (d, J = 5.0 Hz, 1H), 3.08–3.01 (m, 3H), 2.39 (d, J = 7.0 Hz, 2H), 2.21–2.13 (m, 1H), 2.04 (d, J = 15.4 Hz, 2H), 1.74–1.60 (m, 2H), 1.27 (s, 2H). 13C NMR (151 MHz, CD3OD) δ 170.66, 136.38, 133.81, 129.79, 128.59, 128.42, 127.97, 126.86, 125.29, 115.83, 115.76, 112.47, 110.75, 101.07, 72.24, 70.09, 60.80, 57.69, 53.42, 52.37, 41.90, 31.68, 31.23, 29.38, 28.89, 22.34, 13.05. HRMS (ESI): calcd. For C23H27N3O [M + H]+ 362.2227, found 362.2242.
2-(1-(2-Chlorophenethyl)piperidin-4-yl)-N-(1H-indol-5-yl)acetamide (16b). 2-(1-(2-Chlorophenethyl)piperidin-4-yl)acetic acid (140 mg, 0.50 mmol), 1H-Indol-5-amine (55 mg, 0.42 mmol), PyBOP (260 mg, 0.50 mmol), DIPEA(80 mg, 0.62 mmol), DMF (6 mL). Yellow solid, m.p.: 172–173 °C, yield: 30.41%, 1H NMR (400 MHz, CD3OD) δ 7.75 (d, J = 1.9 Hz, 1H), 7.39–7.29 (m, 3H), 7.28–7.20 (m, 2H), 7.19 (d, J = 3.1 Hz, 1H), 7.16 (dd, J = 8.7, 2.0 Hz, 1H), 6.37 (d, J = 4.0 Hz, 1H), 3.45 (d, J = 12.3 Hz, 2H), 3.15–3.09 (m, 2H), 3.08–3.01 (m, 2H), 2.79 (t, J = 13.0 Hz, 2H), 2.36 (d, J = 7.1 Hz, 2H), 2.10 (s, 1H), 2.00–1.92 (m, 2H), 1.60 (q, J = 11.5 Hz, 2H). 13C NMR (151 MHz, CD3OD) δ 171.22, 135.56, 133.78, 133.57, 130.84, 129.85, 129.30, 128.28, 127.96, 127.17, 125.21, 115.80, 112.48, 110.73, 101.06, 56.91, 52.68, 42.55, 32.24, 29.95, 29.32, 29.01. HRMS (ESI): calcd. For C23H26ClN3O [M + H]+ 396.1837, found 396.1838.
N-(1H-Benzo[d]imidazol-5-yl)-2-(1-phenethylpiperidin-4-yl)acetamide (16c). 2-(1-phenethylpiperidin-4-yl)acetic acid (140 mg, 0.57 mmol), 1H-Benzo[d]imidazol-5-amine (64 mg, 0.48 mmol), PyBOP (295 mg, 0.57 mmol), DIPEA(93 mg, 0.72 mmol), DMF (6 mL). Red solid, m.p.: 167–168 °C, yield: 45.92%, 1H NMR (400 MHz, CD3OD) δ 8.09 (s, 1H), 8.03 (s, 1H), 7.52 (d, J = 8.7 Hz, 1H), 7.29–7.12 (m, 6H), 3.07 (d, J = 12.0 Hz, 2H), 2.84–2.78 (m, 2H), 2.64–2.58 (m, 2H), 2.32 (d, J = 7.2 Hz, 2H), 2.17 (t, J = 11.8 Hz, 2H), 1.93 (s, 1H), 1.81 (d, J = 12.9 Hz, 2H), 1.42 (q, J = 12.1, 10.5 Hz, 2H). 13C NMR (101 MHz, CD3OD) δ 171.84, 141.67, 139.67, 133.93, 128.33, 128.21, 128.20, 125.93, 116.32, 60.33, 53.19, 47.89, 43.30, 33.27, 32.51, 31.15. HRMS (ESI): calcd. For C22H26N4O [M + H]+ 363.2179, found 363.2197.
N-(1H-Benzo[d]imidazol-5-yl)-2-(1-(2-chlorophenethyl)piperidin-4-yl)acetamide (16d). 2-(1-(2-Chlorophenethyl)piperidin-4-yl)acetic acid (140 mg, 0.50 mmol), 1H-Benzo[d]imidazol-5-amine (55 mg, 0.42 mmol), PyBOP (259 mg, 0.50 mmol), DIPEA(80 mg, 0.62 mmol), DMF (6 mL). Red solid, m.p.: 176–177 °C, yield: 40.33%, 1H NMR (600 MHz, CDCl3) δ 7.95 (s, 1H), 7.70 (s, 1H), 7.66 (d, J = 8.1 Hz, 1H), 7.48 (d, J = 7.4 Hz, 1H), 7.35 (d, J = 7.8 Hz, 1H), 7.24 (t, J = 7.4 Hz, 1H), 7.19 (t, J = 7.3 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 3.62 (s, 2H), 3.57 (d, J = 11.7 Hz, 1H), 3.37 (d, J = 6.5 Hz, 2H), 2.95 (d, J = 11.2 Hz, 2H), 2.10 (t, J = 11.3 Hz, 2H), 1.75 (d, J = 12.4 Hz, 2H), 1.66 (s, 1H), 1.60 (s, 1H), 1.39 (q, J = 11.5 Hz, 2H). 13C NMR (151 MHz, CD3OD) δ 171.90, 141.65, 137.34, 134.98, 133.95, 133.58, 130.73, 129.20, 127.73, 126.97, 121.27, 116.28, 58.31, 58.27, 53.17, 53.06, 50.62, 43.39, 40.15, 33.39, 32.59, 31.29, 31.15, 30.24. HRMS (ESI): calcd. For C22H25ClN4O [M + H]+ 397.1790, found 397.1797.
3.8. AChE and BChE Inhibition Assay
The inhibitory activity of the target compound against AChE (from Electrophorus electricus (eeAChE), Sigma-Aldrich, Munich, Germany) and horse serum BChE (eqBChE, Sigma-Aldrich, Munich, Germany) were measured by Ellman’s method [32]. In 96-well plates, a mixture of phosphate buffer (0.1 M, pH 8.0, 2 mL), 5,5′-dithiobis-2-nitrobenzoic acid (DTNB, 60 μL), acetylcholinesterase or butyrylcholinesterase (20 μL, 5 IU/mL) and different concentrations of the compounds solution (30 μL) was pre-incubated for 5 min and then substrates (acetylthiocholine iodide or butyrylthiocholine iodide, 20 μL) were added.
Changes in absorbance were measured at 412 nm by using microplate reader (Thermo, Varioskan Flash 3001, Thermo Fisher Scientific, Agawam, MA, USA). The measurement of each concentration for each compound was detected in triplicate. GraphPad Prism 6.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used for data processing. The inhibition curve was fitted by plotting the logarithm of the concentration of the tested compounds with the percentage of enzyme activity (reference to 100%). The (IC50) value was calculated according to the inhibition curve and the data were shown in the layout of mean ± SEM by GraphPad Prism 6.0.
3.9. Kinetics of AChE and BChE Inhibition
Kinetic studies were performed in the same manner as the determination of ChEs inhibition, with substrate (ATC/BTC) concentrations of 90, 150, 226, 452 and 904 μM. The concentration of compound 8i was set to 0, 0.25, 0.5, 0.75, 1 μM, and the concentration of compound 10s was set to 1, 2.5, 5, 7.5, 10 μM. The enzymatic reaction was extended to 7 min for eeAChE and eqBChE before the determination of the absorption. The Vmax and Km values of Michaelis-Menten kinetics were calculated by nonlinear regression from the substrate-velocity curves using GraphPad Prism 6.0. Linear regression was used to fit the Lineweaver–Burk plot.
3.10. Molecular Docking Study
The crystal structures of eeAChE (PDB:1C2B) [36], huAChE (PDB ID: 4EY7) [32] and huBChE (PDB ID: 4TPK) [37] were obtained from the Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). The Discovery Studio software 2016 (DS 2016, BIOVIA, San Diego, CA, USA) was used to study the docking of compound 8i. The three protein structures are pretreated (i.e., protonated, removed water, added Miss sidechains, etc.) by the “prepare protein” module in DS to provide the structures suitable for docking. The “prepare ligand” module in DS is used to test the structural preparation of the compound. The native ligand in the crystal structure was used to define the binding site. The binding site was defined as the site sphere ((in 10 Å radius) around the original ligand in the co-crystal structures. The docking program CDOCKER encoded in DS 2016 was applied to identify the potential binding of compound 8i to eeAChE, huAChE, and huBChE. Other CDOCKER parameters were set to default values. Compound 8i was chosen for molecular modeling as the most active compounds in the series (Table 1). Compound 8i produced 10 poses to eeAChE, huAChE, and huBChE. These postures were visually examined, and the most appropriate docking pose was selected according to the scores and interactions with key residues of the eeAChE, huAChE, and huBChE active sites.
3.11. Cell Studies in Vitro
Pheochromocytoma-derived cell line (PC12 cells) were grown in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS at 37 °C in a humidified atmosphere containing 5% CO2. To carry out the experiment, cells (6 × 103 cells/well) were seeded in 96-well plate in complete medium. After 24 h, the culture medium was removed and the cells were exposed to increasing concentrations of compounds 6b, 6c, 6d, 8i, 10s, 10v or Tacrine (10, 20, 30 and 50 μM) in DMEM for further 24 h. Cell survival was measured by 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) assay [37].
4. Conclusions
In this paper, a series of novel compounds 6a–h, 8i–1, 10s–v, and 16a–d were synthesized and evaluated, together with the known analogs 11a–f, for their inhibitory activities towards AChE and BChE. The results show that most of the compounds have AChE and/or BChE inhibitory activity. Compound 8i showed the strongest inhibitory effect on both AChE (eeAChE IC50 = 0.39 μM) and BChE (eqBChE IC50 = 0.28 μM). Compared with compound G801-0274, compound 8i has comparable inhibitory activity against two ChEs, so that it can exert an anti-ChEs effect in a balanced manner. Kinetic studies indicated a mixed-type inhibition of compound 8i, including competitive inhibition and non-competitive inhibition. Subsequently, molecular docking was performed to evaluate the interaction mechanism between compound 8i and enzymes. Enzyme inhibition kinetics and molecular modeling studies have shown that compound 8i bind simultaneously to the PAS and the CAS of AChE and BChE. Therefore, compounds 8i may be promising scaffold for treatment, and further modifications have been made to obtain novel AChE and BChE dual-target inhibitors.
Supplementary Materials
Copies of the 1H-NMR and 13C-NMR spectra of the compounds are available online.
Author Contributions
Conceptualization, H.S. and Z.L.; Data curation, Y.G., H.Y. and Q.L.; Formal analysis, Y.G., H.Y. and Q.L.; Methodology, Y.G., H.Y., Z.H., S.T., Q.L., C.D., T.C. and Y.L.; Project administration, H.S. and Z.L.; Validation, Y.G., H.Y., Z.H., S.T., Q.L., C.D., T.C. and Y.L.; Writing—original draft, Y.G.; Writing—review and editing, H.S. and Z.L. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the financial support of the National Natural Science Foundation of China (No. 81502983) and the Taishan Scholar Project.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: All samples of the compounds are available from the authors.
References
- 1.Thies W., Bleiler L. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2012;8:131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Paula G., Joseph M., Alma S., Xiangling Y., Jarred R., Dylan G., Katherine D., Debjani T., Jinhua L., Marianne E. A new paradigm for the treatment of Alzheimer’s disease: Targeting vascular activation. J. Alzheimers Dis. 2014;40:619–630. doi: 10.3233/JAD-2014-132057. [DOI] [PubMed] [Google Scholar]
- 3.Gomez-Ramirez J., Wu J. Network-based biomarkers in Alzheimer’s disease: Review and future directions. Front. Aging Neurosci. 2014;6:1–9. doi: 10.3389/fnagi.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geldenhuys W.J., Darvesh A.S. Pharmacotherapy of Alzheimer’s disease: Current and future trends. Expert Rev. Neurother. 2015;15:3–5. doi: 10.1586/14737175.2015.990884. [DOI] [PubMed] [Google Scholar]
- 5.Association A.S. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2014;10:e47–e92. doi: 10.1016/j.jalz.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Alexander A.G., Marfil V., Li C. Use of C. elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet. 2014;5:1–21. doi: 10.3389/fgene.2014.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sperling R.A., Al E. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ávila R., Bottino C.M.C., Carvalho I.A.M., Santos C.B., Seral C., Miotto E.C. Neuropsychological rehabilitation of memory deficits and activities of daily living in patients with Alzheimer’s disease: A pilot study. Braz. J. Med. Biol. Res. 2004;37:1721–1729. doi: 10.1590/S0100-879X2004001100018. [DOI] [PubMed] [Google Scholar]
- 9.Chan K.Y., Wang W., Wu J.J., Liu L., Theodoratou E., Car J., Middleton L., Russ T.C., Deary I.J., Campbell H. Epidemiology of Alzheimer’s disease and other forms of dementia in China, 1990–2010: A systematic review and analysis. Lancet. 2013;381:2016–2023. doi: 10.1016/S0140-6736(13)60221-4. [DOI] [PubMed] [Google Scholar]
- 10.Okuda S., Tetsuka J., Takahashi K., Toda Y., Kubo T., Tokita S. Association between sleep disturbance in Alzheimer’s disease patients and burden on and health status of their caregivers. J. Neurol. 2019;266:1490–1500. doi: 10.1007/s00415-019-09286-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta M., Adem A., Sabbagh M. New Acetylcholinesterase Inhibitors for Alzheimer’s Disease. Int. J. Alzheimers Dis. 2012;5:507–514. doi: 10.1155/2012/728983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domenico F.D., Cenini G., Sultana R., Perluigi M., Butterfield D.A. Glutathionylation of the Pro-apoptotic Protein p53 in Alzheimer’s Disease Brain: Implications for AD Pathogenesis. Neurochem. Res. 2009;34:727–733. doi: 10.1007/s11064-009-9924-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perry E.E., Perry R.H. The cholinergic system in Alzheimer’s disease. Prog. Neurobiol. 1997;5:261–262. doi: 10.1016/0166-2236(82)90165-5. [DOI] [PubMed] [Google Scholar]
- 14.Scarpini E., Scheltens P., Feldman H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003;2:539–547. doi: 10.1016/S1474-4422(03)00502-7. [DOI] [PubMed] [Google Scholar]
- 15.Robert A., Zhang W., Huang D., Huang M., Xingguo L. Preparation of new tetradentate copper chelators as potential anti-Alzheimer agents. Chemmedchem. 2018;13:684–704. doi: 10.1002/cmdc.201700734. [DOI] [PubMed] [Google Scholar]
- 16.Viles J.H. Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord. Chem. Rev. 2012;256:2271–2284. doi: 10.1016/j.ccr.2012.05.003. [DOI] [Google Scholar]
- 17.Huebbe P., Jofre-Monseny L., Ch B.S., Minihane A.M., Rimbach G. Effect of apoE genotype and vitamin E on biomarkers of oxidative stress in cultured neuronal cells and the brain of targeted replacement mice. J. Physiol. Pharmacol. 2007;58:683–698. [PubMed] [Google Scholar]
- 18.Butterfield D.A., Reed T., Newman S.F., Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cummings J.L. Treatment of Alzheimer’s disease: Current and future therapeutic approaches. Rev. Neurol. Dis. 2004;1:60–69. [PubMed] [Google Scholar]
- 20.Gabriel A.J., Almeida M.R., Ribeiro M.H., Durães J., Tábuas-Pereira M., Pinheiro A.C., Rui P., Santana I., Baldeiras I. Association between butyrylcholinesterase and cerebrospinal fluid biomarkers in Alzheimer’s disease patients. Neurosci. Lett. 2017;641:101–106. doi: 10.1016/j.neulet.2017.01.036. [DOI] [PubMed] [Google Scholar]
- 21.Palanimuthu D., Poon R., Sahni S., Anjum R., Hibbs D., Lin H.Y., Bernhardt P.V., Kalinowski D.S., Richardson D.R. A novel class of thiosemicarbazones show multi-functional activity for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2017;139:612–632. doi: 10.1016/j.ejmech.2017.08.021. [DOI] [PubMed] [Google Scholar]
- 22.Reddy E.K., Remya C., Mantosh K., Sajith A.M., Omkumar R.V., Sadasivan C., Anwar S. Novel tacrine derivatives exhibiting improved acetylcholinesterase inhibition: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2017;139:367–377. doi: 10.1016/j.ejmech.2017.08.013. [DOI] [PubMed] [Google Scholar]
- 23.Giacobini E. Cholinesterases: New Roles in Brain Function and in Alzheimer’s Disease. Neurochem. Res. 2003;28:515–522. doi: 10.1023/A:1022869222652. [DOI] [PubMed] [Google Scholar]
- 24.Li Q., Yang H., Chen Y., Sun H. Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur. J. Med. Chem. 2017;132:294–309. doi: 10.1016/j.ejmech.2017.03.062. [DOI] [PubMed] [Google Scholar]
- 25.Giacobini E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004;50:433–440. doi: 10.1016/j.phrs.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 26.Kurz A., Farlow M., Lefèvre G. Pharmacokinetics of a novel transdermal rivastigmine patch for the treatment of Alzheimer’s disease: A review. Int. J. Clin. Pract. 2010;63:799–805. doi: 10.1111/j.1742-1241.2009.02052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sugimoto H., Yamanish Y., Iimura Y., Kawakami Y. Donepezil Hydrochloride (E2020) and Other Acetylcholinesterase Inhibitors. Curr. Med. Chem. 2000;7:303–339. doi: 10.2174/0929867003375191. [DOI] [PubMed] [Google Scholar]
- 28.Jiao K., Hong Q., Song L., Lu Z. Studies on interaction of AChE with its substrates by computer simulation. Bull. Acad. Mil. Med. 1998;22:195–199. [Google Scholar]
- 29.Mohamed T., Osman W., Tin G., Rao P.P.N. Selective inhibition of human acetylcholinesterase by xanthine derivatives: In vitro inhibition and molecular modeling investigations. Bioorganic Med. Chem. Lett. 2013;23:4336–4341. doi: 10.1016/j.bmcl.2013.05.092. [DOI] [PubMed] [Google Scholar]
- 30.Belluti F., Rampa A., Piazzi L., Bisi A., Gobbi S., Bartolini M., Andrisano V., Cavalli A., Recanatini M., Valenti P. Cholinesterase inhibitors: Xanthostigmine derivatives blocking the acetylcholinesterase-induced beta-amyloid aggregation. J. Med. Chem. 2005;48:4444–4456. doi: 10.1021/jm049515h. [DOI] [PubMed] [Google Scholar]
- 31.Yao C., Lin H., Yang H., Tan R., Bian Y., Fu T., Wei L., Liang W., Pei Y., Sun H. Discovery of new acetylcholinesterase and butyrylcholinesterase inhibitors through structure-based virtual screening. Rsc Adv. 2017;7:3429–3438. [Google Scholar]
- 32.Ellman G.L., Courtney K.D., Andres V., Jr., Feather-stone R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 33.Mo J., Chen T., Yang H., Guo Y., Li Q., Qiao Y., Lin H., Feng F., Liu W., Chen Y., et al. Design, synthesis, in vitro and in vivo evaluation of benzylpiperidine-linked 1,3-dimethylbenzimidazolinones as cholinesterase inhibitors against Alzheimer’s disease. J. Enzym. Inhib. Med. Chem. 2020;35:330–343. doi: 10.1080/14756366.2019.1699553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piemontese L., Tomás D., Hiremathad A., Capriati V., Candeias E., Cardoso S.M., Chaves S., Santos M.A. Donepezil structure-based hybrids as potential multifunctional anti-Alzheimer’s drug candidates. J. Enzym. Inhib. Med. Chem. 2018;33:1212–1224. doi: 10.1080/14756366.2018.1491564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tamagno E., Bardini P., Obbili A., Vitali A., Borghi R., Zaccheo D., Pronzato M.A., Danni O., Smith M.A., Perry G. Oxidative Stress Increases Expression and Activity of BACE in NT2 Neurons. Neurobiol. Dis. 2002;10:279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 36.Samadi A., Chioua M., Bolea I., Ríos C.D.L., Iriepa I., Moraleda I., Bastida A., Esteban G., Unzeta M., Gálvez E. Synthesis, biological assessment and molecular modeling of new multipotent MAO and cholinesterase inhibitors as potential drugs for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2011;46:4665–4668. doi: 10.1016/j.ejmech.2011.05.048. [DOI] [PubMed] [Google Scholar]
- 37.Brus B., Košak U., Turk S., Pišlar A., Coquelle N., Kos J., Stojan J., Colletier J.P., Gobec S. Discovery, Biological Evaluation, and Crystal Structure of a Novel Nanomolar Selective Butyrylcholinesterase Inhibitor. J. Med. Chem. 2014;57:8167–8179. doi: 10.1021/jm501195e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.