


Abstract
The XPA protein functions together with the single-stranded DNA (ssDNA) binding protein RPA as the central scaffold to ensure proper positioning of repair factors in multi-protein nucleotide excision repair (NER) machinery. We previously determined the structure of a short motif in the disordered XPA N-terminus bound to the RPA32C domain. However, a second contact between the XPA DNA-binding domain (XPA DBD) and the RPA70AB tandem ssDNA-binding domains, which is likely to influence the orientation of XPA and RPA on the damaged DNA substrate, remains poorly characterized. NMR was used to map the binding interfaces of XPA DBD and RPA70AB. Combining NMR and X-ray scattering data with comprehensive docking and refinement revealed how XPA DBD and RPA70AB orient on model NER DNA substrates. The structural model enabled design of XPA mutations that inhibit the interaction with RPA70AB. These mutations decreased activity in cell-based NER assays, demonstrating the functional importance of XPA DBD–RPA70AB interaction. Our results inform ongoing controversy about where XPA is bound within the NER bubble, provide structural insights into the molecular basis for malfunction of disease-associated XPA missense mutations, and contribute to understanding of the structure and mechanical action of the NER machinery.
INTRODUCTION
Nucleotide excision repair (NER) is a central DNA repair pathway responsible for removing bulky lesions from the genome (1,2). Defects in NER result in the genetic disorder xeroderma pigmentosum (XP), characterized by extreme hypersensitivity to sunlight and an >2000-fold increase in skin cancer (3,4). NER is a highly dynamic process that operates through the sequential assembly and action of >20 proteins at the DNA lesion (Figure 1A). The core NER reaction occurs only after a lesion has been made accessible in the context of chromatin by the UV-DDB ubiquitin ligase complex and associated factors. The reaction is initiated by XPC-RAD23B, which recognizes destabilization induced in the DNA by a lesion (5). TFIIH is then loaded, likely via the XPB helicase. The second helicase of TFIIH, XPD, is thought to track along the damaged strand, leading to the formation of a DNA bubble structure, until it stalls at the lesion, which serves to verify the presence of the lesion. The xeroderma pigmentosum complementation group A (XPA) protein and replication protein A (RPA) are then recruited to the damage site in synchrony and work together as a scaffold to organize the damaged DNA and NER enzymes (6–8). XPA and RPA are essential for the localization and activation of the two NER endonucleases: ERCC1-XPF and XPG (9,10). RPA binds and protects single-stranded DNA (ssDNA) on the undamaged strand (11,12). XPA is thought to interact with the open bubble without direct contact with the lesion and its position on the bubble has been assigned alternately to either the 3′ or the 5′ ss-dsDNA junction (13–15). Understanding the molecular basis for the function of the XPA-RPA scaffold is of importance given its central role in organizing and orchestrating the action of the NER machinery.
Figure 1.

Human NER factors XPA and RPA. (A) Schematic diagram of the NER pre-incision complex. DNA damage is marked with a star on the substrate. (B) Schematic diagram of the domain structure of human XPA. The XPA DNA binding region, which contains the globular core, is depicted as an oval. DNA- and RPA-binding residues are indicated below the domain, aligned with the XPA residues involved in each interaction. Both previously reported sites for RPA70AB interaction with the XPA globular core are marked for clarity. (C) Schematic diagram of the subunit and domain structure of human RPA heterotrimer. Globular domains are depicted as ovals. All globular domains are OB-folds except for the winged-helix domain RPA32C. (D) Schematic representation of the spatial organization of RPA domains when bound to ssDNA. The RPA DNA binding apparatus consists of RPA70A, RPA70B, RPA70C, RPA32D, and RPA14. A ssDNA molecule of ∼40 nts is depicted as a ribbon. The XPA-binding regions are labeled with arcs.
XPA is composed of a central globular domain (XPA98–219) containing a zinc-binding motif and non-globular N- and C-terminal domains that contain sites for interactions with its many partner proteins (9,10,16–22) (Figure 1B). The DNA-binding domain (DBD) of XPA was initially assigned to a central globular core within the construct spanning residues 98–219, but our subsequent analysis showed that DNA binding requires 20 additional C-terminal residues (23,24). XPA is understood to bind to a ss–ds junction in the NER bubble. Studies using bubble mimics indicated that DNA binding involves a basic cleft in the globular core and basic residues in the C-terminal extension (23,25).
RPA is a heterotrimer of RPA70, RPA32 and RPA14 subunits (Figure 1C). The tandem high affinity DNA-binding domains RPA70AB and the trimer core (composed of the RPA70C, RPA32D and the core RPA14 domains) constitute the DNA-binding apparatus, which interacts with ssDNA in a 5′-3′ orientation (11,26) (Figure 1D). In addition to functioning to protect and organize ssDNA, RPA serves to recruit partner proteins to DNA processing machinery using its RPA70N and RPA32C domains. These domains are linked to the DNA-binding apparatus by flexible tethers of ∼70 and ∼35 residues, respectively. Hence, interactions with these two domains will not significantly influence the orientation of RPA and partner proteins on their DNA substrate. Importantly, in addition to binding to RPA32C or RPA70N, functionally relevant interactions with RPA70AB have been reported for a number of partner proteins (27,28).
The binding of XPA to RPA is one such example of an interaction involving two points of contact. The RPA32C domain binds a short motif (XPA29–46) in the disordered XPA N-terminal domain (19,29). The second contact occurs between RPA70AB and the XPA globular core. While the XPA29–46–RPA32C interaction is of higher affinity, it is likely that the XPA DBD–RPA70AB interaction plays a more significant role in the positioning of XPA and RPA on the NER bubble since they both bind the substrate in addition to each other. Published studies of where RPA70AB contacts XPA are not consistent with each other (30–33). NMR titrations of XPA98–219 with truncated RPA70 constructs (and in the absence of DNA) suggested that the interaction is mediated by the zinc-binding motif in the N-terminal portion of XPA98–219 (18,31). In contrast, biochemical pull-down and cell-free NER assays with XPA mutants suggested that residues at the C-terminal end of XPA98–219 are responsible for interaction with RPA70AB (33,34).
Our long-term objective is to understand the functional relevance and physical basis for how XPA and RPA serve as scaffolds for the NER machinery. We believe that the interaction between XPA DBD and RPA70AB is critical to the positioning of these proteins on the NER bubble and report here a detailed structural analysis of the complexes of XPA DBD and RPA70AB bound to model NER bubble substrates mimicking the 3′ and 5′ ss–ds junctions. NMR spectroscopy was used to map the interaction surfaces on the two proteins. These NMR data along with SAXS data for the complex were used as restraints for docking and refinement of the 3D models of the XPA DBD–DNA–RPA70AB ternary complexes. Structure-based mutations were then designed to enable functional analysis of this XPA–RPA contact. XPA mutants with reduced affinity for RPA70AB were defective in repair of 1,3-cisplatin intrastrand crosslinks. These results show that the XPA DBD–RPA70AB contact is required for the maintenance of NER activity.
MATERIALS AND METHODS
Bacterial expression vectors
We have previously reported the construction and purification of pBG100-XPAFL, pBG100-XPA98–219 and pBG100-XPA98–239 constructs (23), and of pSV281-RPA70AB (RPA70181–422) (35). Site-directed mutagenesis for pBG100-XPA98–239 and pBG100-XPAFL E106K and pBG100-XPA98–239 F112A was performed using the Q5(R) Site-Directed Mutagenesis Kit Quick Protocol (E0554, New England Biolabs) and the following primers:
E106K_F: 5′-TGTAATATGCAAAGAATGTGGG-3′;
E106K_R: 5′-TAATCAAATTCCATAACAGGTC-3′,
F112A_F: 5′-TGGGAAAGAAGCTATGGATTCTTATCTTATG-3′;
F112A_R: 5′-CATTCTTCGCATATTACATAATC-3′.
Vectors for pBG100-XPA98–239 and pBG100-XPAFL M2 (E106K/F112A) and M6 (D101N/E106K/K110E/E111K/F112A/D114N) mutants were prepared by the core facility of the Structural Biology of DNA Repair Machinery Program using the NEB’s Q5 site-directed mutagenesis kit.
-
Primers used were:
XPA-M2_F: 5′-TGGGAAAGAAGCTATGGATTCTTATCTTATG-3′,
XPA-M2_R: 5′- CATTCTTTGCATATTACATAATC-3′,
XPA-FL-M6_F: 5′- AGAATGTGGGGAGGAAGCTATGG-3′,
XPA-FL-M6_R: 5′- TTGCATATTACATAGTTAAATTCC-3′,
XPA-DBD-M6_F: 5′- AGAATGTGGGGAGAAAGCTATGAAC-3′,
XPA-DBD-M6_R: 5′- TTGCATATTACATAGTTAAATTC-3′.
and were shared within the SBDR consortium.
DNA substrates
Supplementary Figure S1 shows the structures of the DNA substrates used in this study. Desalted oligodeoxynucleotides were purchased from Sigma-Aldrich Co. Splayed-arm dsDNA substrates were prepared by mixing an equimolar amount at 1 mM of each strand in the duplex annealing buffer (60 mM KCl, 6 mM Tris pH 7.5, 0.2 mM MgCl2). The mixture was then heated to 95°C in a thermoblock and allowed to cool to room temperature for annealing. Hairpin-containing DNA substrates, including those that were fluorescein-labeled, were dissolved in the same buffer at 2 μM concentration and annealed by heating at 95°C in a thermoblock, followed by immediate cooling on ice.
Protein expression and purification
Unlabeled and uniformly 15N-enriched human XPA98–219 and XPA98–239 were expressed and purified as described previously (23) with some modifications to the protocol. First, 10 μM ZnCl2 was added during inoculation with the overnight small-scale culture of the XPA construct to improve the solubility. A Heparin column (GE Healthcare) purification step was incorporated after the Ni affinity step. After the nickel column, proteins were dialyzed overnight against heparin loading buffer (20 mM Tris pH 7.5, 150 mM NaCl, 10% glycerol, 1 mM DTT), loaded on the heparin column (5 ml; GE Healthcare), washed with 5–10 column volumes (CV) of heparin loading buffer and eluted with a 0–100% gradient of 5 CV of heparin elution buffer (20 mM Tris pH 7.5, 1 M NaCl, 10% glycerol, 1 mM DTT). RPA70AB was produced following the same protocol as that for the XPA constructs, except that no ZnCl2 was added to the overnight preculture and TEV protease was used for His-tag cleavage.
Assessment of formation of the XPA DBD–DNA–RPA70AB ternary complex
In order to test the formation and stability of the ternary complexes, purified XPA98–239 and RPA70AB proteins were mixed in an equimolar ratio with the DNA substrates at a final concentration of 50–100 μM. Analytical size-exclusion chromatography coupled to multi-angle light scattering (DAWN HELEOS, Wyatt Technology) (SEC-MALS) was used to monitor the system. A 24 ml Superdex 75 Increase analytical gel filtration column (GE Healthcare) was used with a flow rate of 0.5 ml/min in a buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, 10% glycerol and 1 mM DTT. ASTRA software (Wyatt Technologies) was used for data processing, average molecular mass calculation and estimation of the peak monodispersity. To assess stoichiometry, fractions were collected and analyzed by SDS-PAGE.
Nuclear magnetic resonance (NMR) spectroscopy
All samples for titrations were concentrated to 50 μM in a buffer containing 20 mM Tris, pH 7.0, 150 mM KCl, 1 mM Tris(2-carboxyethyl) phosphine (TCEP) and 5% 2H2O. Multi-point titrations were performed by preparing two identical solutions of protein, one with no DNA substrate and the other at the highest ratio of DNA substrate to 15N-enriched protein, then collecting spectra for these and intermediate ratios created by mixing of the two solutions. Two-point titrations in the presence of ssDNA were performed by preparing three identical solutions of 15N-enriched protein: (i) 15N-enriched protein alone, (ii) 15N-enriched protein with an equimolar amount of ssDNA, (iii) 15N-enriched protein with an equimolar amount of ssDNA and a 4-fold molar excess of the partner protein. After acquisition of all three individual spectra, intermediate ratios of 15N-enriched and unlabeled proteins were made by mixing samples (ii) and (iii). 15N-1H TROSY-HSQC spectra were recorded in 3-mm tubes at 25°C using a Bruker AVANCE 900 MHz spectrometer equipped with a TCI cryoprobe. Further data processing and analysis were carried out using SPARKY (36). All residues whose cross peaks disappeared due to intermediate exchange line broadening upon binding DNA were placed in the category of significantly perturbed. For residues exhibiting chemical shift perturbations in fast exchange, we calculated the change in chemical shift (Δδ) from the spectra acquired with no substrate and at the highest substrate ratio using the formula: Δδ = √[(Ha - Hb)2 + (0.2*(Na - Nb))2]. The threshold for significant chemical shift perturbation was set to the average Δδ ± 1 or 1.5 standard deviation depending on the relative sensitivity of the spectrum. Resonance assignments for XPA98–219, XPA98–239, RPA70AB and RPA70AB–ssDNA used in the study are available at the BMRB under accession codes 4249, 27131, 5823 and 50121, respectively.
For a comparison of chemical shift perturbations caused by the wild-type and mutant XPA proteins, one-point titrations were performed in the presence of DNA substrate under identical conditions to those described above. Samples were prepared with 15N-enriched RPA70AB protein at 50 μM concentration with an equimolar amount of DNA and in the absence or presence of a 4-fold molar excess of unlabeled XPA (WT, M2 or M6). 15N-1H TROSY-HSQC experiments were acquired with 32 scans, and 2048 and 256 points in direct and indirect dimensions, respectively. Data processing was carried out using Topspin version 3.2 (©Bruker Biospin) or Spectra analysis (Ccpnmr version 2.4) (37).
Small-angle X-ray scattering (SAXS)
The SAXS profiles for the XPA DBD–DNA–RPA70AB complex were collected in SEC-SAXS mode at the ALS beamline 12.3.1 LBNL Berkeley, California (38). The X-ray wavelength λ was 1.03 Å and the sample-to-detector distance was set to 1.5 m resulting in scattering vectors, q, ranging from 0.01 to 0.5 Å−1. The scattering vector is defined as q = 4π sinθ/λ, where 2θ is the scattering angle. All experiments were performed at 20°C (39) and data were processed as described (40). Briefly, the flow through SAXS cell was directly coupled with an online Agilent 1260 Infinity HPLC system using a Shodex KW803 column (Shodex™). The column was equilibrated with running buffer (20 mM Tris pH 7.5, 150 mM NaCl, 2% glycerol, 1 mM DTT) with a flow rate of 0.5 ml/min. A 50 μl sample was run through the SEC and 3.0-s X-ray exposures were collected continuously during a ∼35 min elution. The SAXS frames recorded prior to the protein elution peak were used as buffer blanks to subtract from all other frames. The subtracted frames were examined by radius of gyration (RG) and scattering intensity at q = 0 Å−1 (I(0)), derived using the Guinier approximation I(q) = I(0) exp(-q2RG2/3) with the limits qRG <1.5). I(0) and RG values were compared for each collected SAXS curve (frame) across the entire elution peak (Supplementary Figure S2A). The elution peak was mapped by plotting the scattering intensity at q = 0 Å−1 (I(0)), relative to the recorded frame. Uniform RG values across an XPA DBD–DNA–RPA70AB elution peak represented a homogenous assembly (Supplementary Figure S2A). The merged experimental SAXS data were additionally investigated for aggregation by inspecting Guinier plots (Supplementary Figure S2B). The program SCÅTTER 3.1 was used to compute the distance distribution, P(r) (Supplementary Figure S2E). The distance r where P(r) approaches zero intensity is termed the Dmax of the molecule. P(r) was normalized based on molecular mass (55 kDa) determined by SAXS of assemblies as calculated by SCÅTTER as described (41). The differences in the scattering power of protein and DNA were not taken into account in the determination of molecular mass because the contribution of DNA to the total is much less than that of protein. The SAXS data have been deposited in the SASBDB databank under accession code SASDH44 for the complex with the 3′ junction substrate and SADH54 for the 5′ junction substrate.
Structure calculations
The structural models of XPA98–239 in complex with RPA70AB and the two model NER bubble DNA substrates were generated in four steps. First, a homology model of human XPA102–214 bound to double-stranded DNA was generated using the X-ray crystal structure of Saccharomyces cerevisiae Rad14 (PDB: 5A39) as a template in Modeller 9.19 (42). Five models were created based on the template of one subunit from the dimer present in the Rad14 crystal structure. Model variance was minimal, and the best scoring model was chosen for further work. The C-terminal residues 215–239 were added using the RosettaRemodel application (43). The conformation of these residues was modeled based on analysis of the sequence using the prediction server Jpred (44), which found very high probability of helical structure for residues 215–231 and predicted residues 232–239 to be in a loop conformation. One thousand models were generated and the ten best scoring models analyzed and compared. The model that best matched the predicted secondary structure and had the best Rosetta score was chosen for further calculations. The N-terminal residues 98–101 were added by manual building using the Chimera Build Structure tool. Then the entire set of coordinates was energy minimized using the Relax application in Rosetta 3.9 (45).
The next step was to adjust the DNA in the XPA DBD model to match the experimental substrate, since the sequence was different and the DNA duplex is perturbed by a cisplatin crosslink. This involved docking an ideal 10 basepair B-DNA double helix using the HADDOCK2.2 web server (46). The active residues for XPA were selected based on the NMR chemical shift perturbations reported elsewhere (25) and a solvent accessible surface area >50%. A total of 1000 models were initially generated, and the best scoring 200 were used in the subsequent refinement. Based on the recommended HADDOCK protocol, the refined models were clustered, and the best scoring model of the best scoring cluster was chosen as the representative structure for subsequent refinements.
The third step was docking the XPA DBD–dsDNA structure and the X-ray crystal structure of RPA70AB in complex with d-C8 (PDB: 1JMC). To ensure the results were accurate, this step was performed using HADDOCK (HADDOCK2.2 web server) with the NMR data to drive the generation of the initial models, which were then assessed based on fit to the SAXS data. To ensure the two DNA molecules were within the distance range required for connecting the two sub-complexes with the four-nucleotide linker that was present in the experimental samples, a special distance restraint of 20 ± 3 Å was included in these docking calculations. It was here that the calculations for the two NER substrates diverged. For the 3′ junction substrate, the restraint was set between the 3′ end of the DNA bound to XPA DBD and the 5′ end of the DNA bound to RPA. For the 5′ junction substrate, the restraint was set between the 5′ end of the DNA bound to XPA DBD and the 3′ end of the DNA bound to RPA. Active residues for both proteins were defined as those exhibiting statistically significant NMR CSPs or line broadening and a solvent accessible surface area >50%. Of the total of 1000 models initially generated, the top 200 were refined in HADDOCK with an explicit solvent layer of 8 Å and then clustered based on their structural similarity. Next, a theoretical scattering profile was calculated for best scoring structure in each cluster, then compared to the experimental scattering profile using FoXS (47,48). This confirmed that the best scoring clusters gave reasonable fits to the SAXS data.
To complete the generation of the structural model, the four missing nucleotides in the DNA substrate were built in using the Rosetta stepwise application (49). The theoretical scattering profile was then re-calculated for all structures in the best HADDOCK-scoring clusters (three for the 3′ junction model and two for the 5′ junction model) and compared to the experimental scattering profile (Supplementary Table S1). The fit to the data was qualitatively analyzed by superimposing the structure onto a molecular envelope that was created using DAMMIN in SCÅTTER 3.1. The superimpositions were carried out using SUPCOMB (50). In both cases, it turned out that the best HADDOCK-scoring clusters had the lowest χ fit to the scattering curve. The best HADDOCK-scoring structure was selected as the best representative of the respective RPA70AB-XPA DBD–DNA complex. The structures were analyzed using the PROCHECK server (http://www.ebi.ac.uk/thornton-srv/databases/cgi-bin/pdbsum/GetPage.pl?pdbcode=index.html; Supplementary Materials andSupplementary Table S2). The coordinates for the representative structures have been deposited in the PDB-DEV databank under accession codes PDBDEV_00000039 and PDBDEV_00000040 for the 3′ junction and 5′ junction substrate complexes, respectively.
Circular dichroism (CD) spectroscopy
Samples of XPA DBD were exchanged into a 150 mM K2HPO4 (pH 7.5) buffer, diluted to a concentration of 0.1 mg/ml (5.6 μM), and passed through a 0.2-μm filter. The far-UV CD spectrum over the range 190–260 nm was acquired at room temperature using a Jasco J-810 spectrophotometer. Each spectrum is the average of three scans acquired with a scanning rate of 50 nm/min and data pitch of 1 nm. Prior to generating the overlay, the XPA DBD wild-type and mutant proteins spectra were exported to an Excel spreadsheet, buffer subtracted and plotted.
Microscale thermophoresis (MST)
For measurements of DNA-binding affinity by MST, all proteins were transferred into MST buffer (50 mM Tris-HCl, pH 7.8, 150 mM NaCl, 10 mM MgCl2, 0.05% Tween20 and 1 mM DTT). The experiments were performed with a 5′-FAM DNA substrate (5′-TTTTGCGGCCGCTTTTGCGGCCGC-3′) (25) with a 4 nt hairpin to stabilize the 8 bp duplex. The substrate was annealed by heating to 95°C in a thermoblock, followed by immediate cooling on ice. A fluorescein-labeled DNA stock (2 μM) was diluted in a buffer containing 60 mM KCl, 6 mM Tris pH 7.5 and 0.2 mM MgCl2 to a concentration of 440 nM. A series of 16 dilutions of the protein were prepared at varying concentrations, all with a DNA concentration of 40 nM. All experiments were carried out in standard capillaries at room temperature at 20% LED power and 40% MST power. Final KD values were calculated using MO.Affinity software (NanoTemper, Inc.).
Electrophoretic mobility shift assay (EMSA)
EMSA of full-length XPA binding to DNA three-way junctions was carried out as described previously (9). The labeled 5′-FAM oligonucleotide was annealed with a 2-fold excess of the two unlabeled oligonucleotides
FAM-5′-GTTCGTCAGGATTCCAATTCGTGCAGGCAT
-3′,
5′-ATGCCTGCACGAATTAAGCCATTCGTAATCATGGT-3′,
5′-ACCATGATTACGAATGGCTTGGAATCCTGACGAAC-3′
in 10 mM Tris-HCl (pH 8.0) and 50 mM NaCl. The annealed oligo was incubated with wild-type or mutant XPA (0–40 nM) in a 15 μl mixture containing 25 mM Tris-HCl (pH 8.0), 60 mM NaCl, 0.1 mg/ml BSA, 10% glycerol, 1 mM EDTA and 1 mM DTT at 25°C for 30 min. The reaction mixture was loaded onto a native 8% polyacrylamide gel pre-equilibrated with 0.5 × TBE buffer and run at 4°C for 2 h at 20 mA. Gels were scanned using an Amersham Typhoon RGB imager. Two independent repetitions were performed.
In vitro NER activity assay
In vitro NER assays were carried out using extracts derived from XPA-deficient XP2OS cells and a plasmid containing a site-specific 1,3-intrastrand cisplatin (cis-Pt) lesion as described previously (9,51). For each reaction, 2 μl of repair buffer (200 mM HEPES-KOH, 25 mM MgCl2, 110 mM phosphocreatine (di-Tris salt, Sigma), 10 mM ATP, 2.5 mM DTT and 1.8 mg/ml BSA, adjusted to pH 7.8), 0.2 μl of creatine phosphokinase (2.5 mg/ml, Sigma), 3 μl of XPA-deficient cell extract (about 10 mg/ml), NaCl (to a final concentration of 70 mM) and 50 nM of purified wild-type or mutant XPA in a total volume of 9 μl were pre-warmed at 30°C for 10 min. About 1 μl plasmid containing cis-Pt (50 ng/μl) was added to each reaction, and the samples were incubated at 30°C for different incubation times: 0, 5, 10, 20, 45 and 90 min. After placing the samples on ice, 0.5 μl of 1 μM of a 3′-phophorylated oligonucleotide: (5′-GGGGGAAGAGTGCACAGAAGAAGACCTGGTCGACCp-3′) was added and the mixture heated at 95°C for 5 min. The samples were allowed to cool down to room temperature for 15 min to allow the DNA to anneal. About 1 μl of a Sequenase/[α-32P-dCTP mix (0.25 units of Sequenase and 2.5 μCi of [α-32P]-dCTP per reaction) was added before incubating at 37°C for 3 min, followed by addition of 1.2 μl of dNTP mix (100 μM of each dATP, dTTP, dGTP; 50 μM dCTP) and incubated for another 12 min. The reactions were stopped by adding 12 μl of loading dye (80% formamide/10 mM EDTA) and heating at 95°C for 5 min. The samples were run on a 14% sequencing gel (0.5× TBE) at 45 W for 2.5 h, and the reactions products were visualized using a PhosphorImager (Amersham Typhoon RGB, GE Healthcare Bio-Sciences). Two independent repetitions were performed.
RESULTS
XPA possesses distinct binding motifs for RPA70AB and DNA
As noted above, two different models have been proposed for which region of the XPA DNA-binding domain (DBD) contacts RPA70AB: the zinc-binding motif (18,31) or the C-terminal region (33,34). In order to address this discrepancy, we performed a series of detailed analyses of the XPA and RPA70AB interaction interfaces by 2D heteronuclear 15N-1H NMR. The previously reported studies were performed on XPA98–219, which at the time was understood to be the DBD. However, since we had previously established that the full DNA binding activity required an additional 20 C-terminal residues, we performed the first titration with 15N-enriched XPA98–239. The comparison of the spectra without and with RPA70AB revealed an approximately equal mixture of fast and intermediate exchange effects, reflected in chemical shift perturbations (CSPs) and line broadening (Figure 2A). The effects were observed only for a subset of resonances indicating these arose from a specific binding event. Residues that were perturbed in the titration were mapped on the structure (Figure 2B), which revealed that binding occurs primarily to the zinc-binding motif. These observations are consistent with previous studies that suggested the interaction occurs in this region, but which were not definitive because no control experiments were included to compare the XPA98–219 and XPA98–239 constructs and in one case the RPA construct was truncated in the middle of RPA70B (18,31). In the XPA98–239 titration, no significant effects were observed on residues in the C-terminal extension. We therefore performed a second titration with 15N-enriched XPA98–219 (Figure 2C). Comparison of the effects induced by binding of RPA70AB in the spectra of XPA98–239 versus XPA98–219 revealed a similar pattern of CSPs (cf. Figure 2A–D). This confirms that the contact between XPA DBD and RPA70AB is mediated primarily by residues in the zinc-binding motif. These results also suggest that: (i) since the XPA DBD C-terminal 20 residues required for DNA-binding activity are not required for interaction with RPA70AB, further studies of the interaction with RPA70AB can utilize either construct; (ii) RPA70AB and DNA bind to the same region of XPA, but the sites are sufficiently different to enable formation of the ternary complex.
Figure 2.

NMR mapping of the RPA70AB-binding site in XPA DBD. (A and B) NMR titration of 15N-enriched XPA98–239 with RPA70AB. (A) Overlay of the 900 MHz 15N-1H TROSY-HSQC spectrum of 15N-XPA98–239 in the absence (black) and presence (red) of RPA70AB in a 1:4 molar excess. (B) Residues whose signals are perturbed in (A) mapped in red on the structure of the globular core of XPA. (C–F) NMR titrations of 15N-enriched XPA98–219 with RPA70AB in the absence (C,D) or presence (E,F) of ssDNA. (C) Overlay of the 900 MHz 15N-1H TROSY-HSQC spectrum of 15N-XPA98–219 in the absence (black) and presence (red) of RPA70AB in a 1:4 molar excess. (D) Residues whose signals are perturbed in (C) mapped in red on the structure of the globular core of XPA. (E) Overlay of the 900 MHz 15N-1H TROSY-HSQC spectrum of 15N-XPA98–219 pre-equilibrated with dC8 in the absence (black) and presence (red) of RPA70AB in a 1:4 molar excess. (F) Residues whose signals are perturbed in (E) mapped in red on the structure of the globular core of XPA. The threshold for significance of CSPs and line broadening was set to the average of unperturbed resonances ±1 standard deviation. All spectra were acquired in a buffer containing 20 mM Tris, pH 7.0, 150 mM KCl, 1 mM Tris (2-carboxyethyl) phosphine (TCEP) and 5% 2H2O at 25°C.
XPA DBD can interact with RPA70AB bound to ssDNA
Next, we asked if RPA70AB binding of ssDNA altered how it interacts with XPA? The known RPA70AB high affinity ssDNA substrate dC8 was used for these experiments. A control experiment involving titration of dC8 into a solution of 15N-enriched XPA98–219 revealed no effect on the spectrum (Supplementary Materials andSupplementary Figure S3), as expected based on our previous study showing that the XPA globular core does not bind ssDNA with any appreciable affinity (23). In contrast, further titration of this solution with RPA70AB resulted in a number of CSPs and exchange broadened peaks (Figure 2E), which map primarily to the N-terminal portion of XPA98–219, as we had observed in the titration in the absence of dC8 (cf. Figure 2D and F). Thus, XPA utilizes the same mode of binding with RPA70AB whether or not RPA70AB is engaged on a ssDNA substrate.
To further analyze the interaction of XPA DBD and RPA70AB, a reciprocal 2D heteronuclear 15N-1H NMR titration of 15N-enriched RPA70AB bound to dC8 was performed with unlabeled XPA. Since we had observed that the binding site for RPA70AB did not involve residues 220–229 and the shorter construct is much more soluble and easy to titrate, XPA98–219 was used for this analysis. The titration was performed in two phases, first dC8 was added to the RPA70AB solution, then XPA98–219. The CSPs and line broadening effects on 15N-RPA70AB upon binding ssDNA (Supplementary Materials andSupplementary Figure S4) closely matched those reported previously (51). Further titration of XPA98–219 into 15N-enriched RPA70AB with dC8 bound gives rise to CSPs and line broadening of a subset of signals in the spectrum corresponding to residues in and around the sites where RPA70AB binds ssDNA (Figure 3). The data show that, although the RPA70AB residues perturbed upon binding of dC8 and XPA98–219 are in close proximity and involve the same face of RPA70AB (Figure 3B), a ternary complex with independent binding sites can be formed, i.e. XPA DBD can interact with RPA70AB while RPA is engaged on its ssDNA substrate.
Figure 3.

NMR mapping of XPA-binding site in RPA70AB. NMR titration of 15N-enriched RPA70AB with XPA98–219 in the presence of dC8. (A) Overlay of the 900 MHz 15N-1H TROSY-HSQC spectrum of 15N-RPA70AB pre-incubated with dC8 in the absence (black) and presence (red) of XPA98–219 in a 1:4 molar excess. All spectra were acquired in a buffer containing 20 mM Tris, pH 7.0, 150 mM KCl, 1 mM Tris (2-carboxyethyl) phosphine (TCEP) and 5% 2H2O at 25°C. (B) Residues whose signals are perturbed in C are mapped in red on the structure of the globular core of XPA. The threshold for significance was set to the average of unperturbed resonances ± 1.5 standard deviations.
XPA DBD and RPA70AB form ternary complexes with both 5′ and 3′ model NER junction substrates
To further investigate the molecular basis and function of the XPA DBD–RPA70AB interaction, we set out to determine the structure of a ternary complex with a substrate that mimics the NER DNA bubble. Since this requires both proteins to be bound to their respective portions of the DNA substrate, the experiments were performed with the full XPA DBD construct (XPA98–239). XPA is understood to bind at one of the ss–ds junctions of the NER bubble, but as noted above controversy exists as to which one (15). RPA binds to and protects the undamaged strand of NER substrates.
The first step in the design of the model NER bubble substrate was to accommodate the known 5′-3′ orientation of RPA on ssDNA (11,52); RPA70AB is bound at the 5′ end of the undamaged strand in the NER bubble, which would place XPA DBD at the ss–dsDNA junction 3′ to the lesion. Hence, asymmetric Y-shaped DNA ss–ds junction substrates were constructed with the 5′ arm and duplex serving as the ss–ds junction for XPA DBD and the 3′ ssDNA arm designed for RPA70AB (Supplementary Materials, Supplementary Figure S1A and B). SEC-MALS was used to define an optimal substrate, combining retention on the SEC column and the molecular mass of the complexes/sub-complexes derived from the MALS profile. The specificity and homogeneity of complexes were evaluated using these observations along with the measurements of the extent of monodispersity of the top peak fraction from MALS and evaluation of the stoichiometry of the complex by SDS-PAGE. First, different lengths of the duplex portion were tested, ranging from 4 to 16 bps (Supplementary Materials andSupplementary Figure S1A). We also tested structures causing a kink in the duplex, such as a mismatch or gap, as well as capping the duplex with a 4xT hairpin loop (Supplementary Materials andSupplementary Figure S1A), but none of these modifications to the duplex had any significant effect on formation or stabilization of the ternary complex. The 5′ ssDNA overhang was initially set to 4 nts based on our previous analysis and no benefit was found by making it longer (25). We next tested the length of the 3′ ssDNA overhang for RPA70AB. Assuming that the minimal footprint of RPA70AB is 8 nt as seen in the crystal structure (52,53), we designed substrates with overhangs of 10 or 12 nts (Supplementary Materials andSupplementary Figure S1A) so that they contained a spacer between the RPA70AB-binding site and the ss–ds junction to which XPA98–239 was bound. In the end, the optimal substrate contained 10 bp of duplex with a 4 nt 5′ overhang and a 12 nt 3′ overhang (Supplementary Materials andSupplementary Figure S1B). SDS-PAGE of the complex shows it is homogeneous and has the correct stoichiometric ratio (Supplementary Materials Supplementary Figure S1B). Once formed, this complex remains stable (does not degrade or dissociate) for 1 week.
Having established the ability to form a stable ternary complex with a substrate designed to mimic the 3′ ss–ds DNA junction, we asked whether an equally stable complex could be formed with a substrate that mimics the ss–dsDNA junction 5′ to the lesion. These Y-shaped DNA ss–ds junction substrates were designed with the 3′ arm and the duplex serving as the ss–ds junction for XPA DBD and the 5′ ssDNA arm for RPA70AB (Supplementary Materials andSupplementary Figure S1A and C). The critical factor in thinking about such a substrate is that RPA70AB would have to be inverted to retain its 5′-3′ orientation on ssDNA. We initially assumed that the complex formed would be strained and less stable or that the RPA70AB arm would need to be longer to accommodate its inverted orientation with respect to XPA DBD. In fact, neither of these assumptions proved to be correct as the substrate with a 10-nt 5′ arm (for RPA70AB) was formed cleanly, exhibited the correct stoichiometry (Supplementary Materials andSupplementary Figure S1C) and remained intact for as many days as the substrate with a 12-nt 5′ arm. This observation suggested the possibility that models for XPA binding to both the 3′ and 5′ ss–ds junction in the NER bubble are feasible.
Analysis by SEC-MALS of the ternary complexes formed with the most stable 3′ and 5′ model NER substrates provides only limited insight. To further investigate how XPA DBD and RPA70AB are arranged on the NER bubble and compare these complexes in greater detail, small-angle x-ray scattering (SAXS) data were acquired for both asymmetric Y-shaped 3′ and 5′ model NER substrate complexes. In-line coupled size-exclusion chromatography SAXS (SEC-SAXS) was used to ensure optimal data quality (Supplementary Materials andSupplementary Figure S2A–C). The similarity in the SAXS data for the complexes is remarkable. The SAXS profiles and linearity of the Guinier plots (Supplementary Materials andSupplementary Figure S2B) showed that both complexes were free of aggregation in solution. The Kratky Plots revealed both complexes contain globular domains and some flexible loops and/or linkers (Supplementary Materials andSupplementary Figure S2C). Porod–Debye analysis, including values of 3.8 for the Porod Exponent (Supplementary Materials andSupplementary Figure S2D), indicated the ternary complexes are stable and globular, and suggested that direct analysis of the distance distribution function, P(r), is feasible for both (Supplementary Materials andSupplementary Figure S2E). The primary peak centered at ∼30 Å in both P(r) functions is attributed to distance distributions within the three ∼12 kDa globular domains: XPA98–219, RPA70A and RPA70B. The small feature in P(r) at ∼50 Å is consistent with distances between domains. The extension of the Dmax value out beyond 90 Å is consistent with the DNA duplex extending beyond the portion of the substrate that is bound by the two proteins and with the presence of residues at the XPA DBD N- and C-termini that do not contact DNA.
XPA DBD and RPA70AB form similar compact ternary complexes with model NER junction substrates
Structural models of the two different ternary complexes were generated using a integrated structural approach that combines a model of XPA DBD bound to a ss–ds DNA junction, the X-ray crystal structure of RPA70AB with dC8 bound (53), NMR identification of the XPA DBD and RPA70AB interaction interfaces, SAXS to define the shape of the complexes, and computational docking and refinement. For RPA, we started with the crystal structure of RPA70AB in complex with dC8 (PDB: 1JMC). Because there was no crystal structure for human XPA when the study was undertaken, a high quality homology model was generated in Modeller using as template the crystal structure of yeast XPA (Rad14 - 32% identity, 56% homology) in complex with a lesion-containing duplex (PDB: 5A39). The Rad14 structure lacks the residues corresponding to XPA 98–101 and 214–239, so these were generated computationally and added onto the XPA homology model. Residues 214–234 were assigned helical conformation based on the consensus of multiple secondary structure prediction programs (23), which has been confirmed by a recent crystal structure (54). After the missing residues were added, the entire structure was energy minimized. The DNA sequence in the Rad14 crystal structure is different from that in our substrates and the DNA structure is perturbed by the presence of the lesion. Consequently, the DNA was removed and an ideal 10-mer duplex was docked to the protein in HADDOCK using our previously published NMR CSPs induced in XPA DBD by the binding of DNA (25) (Table 1).
Table 1.
Active residues identified from NMR data for HADDOCK calculationsa
| XPA DBDb | RPA70AB | |
|---|---|---|
| Contacts to DNA | Contacts to RPA70AB | Contacts to XPA DBD |
| 99 | 101 | 211 |
| 131 | 103 | 218 |
| 142 | 104 | 219 |
| 143 | 106 | 239 |
| 152 | 110 | 264 |
| 153 | 112 | 268 |
| 156 | 115 | 275 |
| 167 | 116 | 290 |
| 168 | 120 | 292 |
| 174 | 127 | 387 |
| 175 | 131 | 388 |
| 176 | 146 | |
| 202 | 174 | |
| 205 | 219 | |
| 206 | ||
| 208 | ||
| 211 | ||
| 212 | ||
| 216 | ||
| 217 | ||
| 218 | ||
| 221 | ||
| 222 | ||
| 225 | ||
| 228 | ||
| 232 | ||
aActive residues have significant CSPs or line broadening plus solvent accessible surface area of >50%.
bData for selecting the active residues for docking of DNA to XPA DBD were reported previously Sugitani et al. (25).
The next step was to dock the XPA-DNA model to the RPA70AB crystal structure. Up to this point, the XPA-DBD model and the RPA70AB crystal structure were separate sub-complexes. To maximize the accuracy and efficiency of docking at this early stage, rather than building in the 4 nts required to attach the two DNA substrates, a restraint was introduced to link the two sub-complexes together. For both models, an optimized restraint of 20 ± 3 Å was the most effective at ensuring the two distinct DNA fragments remain connected while allowing full sampling of configurations for docking. To ensure highest accuracy was obtained, we adopted a strategy in which all experimental data were utilized. Docking of the two structures was performed using HADDOCK based on the NMR data (Table 1). The best scoring HADDOCK structures, which grouped into distinct clusters, were then assessed for their fit to the SAXS data. Next, the four missing nucleotides were built into the structures and the fits to the SAXS data were re-calculated. The final representative structure was the best scoring HADDOCK structure within the best scoring cluster, which also had the best χ fit to the scattering curve (Figure 4A and C). Since the Porod analyses of the SAXS data showed that both 3′ ss–ds junction and 5′ ss–ds junction DNA complexes were well formed and globular, molecular envelopes were calculated. For each complex, SUPCOMB was then used to fit the best scoring structure into the SAXS molecular envelope (Supplementary Materials andSupplementary Figure S5). The excellent fit of the structures into their respective molecular envelopes supports the accuracy of both the 3′ and 5′ ss–ds junction DNA structural models.
Figure 4.

Comparison of structures of the XPA DBD–RPA70AB complexes bound to model NER bubble substrates. (A–C) Ribbon diagrams of the XPA DBD–RPA70AB complex bound to the model 3′ junction DNA substrate with RPA70AB in orange, XPA DBD in blue and DNA in green. (D–F) Ribbon diagrams of the XPA DBD–RPA70AB complex bound to the model 5′ junction DNA substrate with RPA70AB in yellow, XPA DBD in dark blue and DNA in green. The structures in (B) and (E) are aligned to the place RPA70AB in the same position, revealing the reversed orientation of XPA DBD. The structures in (C) and (F) are aligned to place XPA DBD and the DNA in the same position, revealing the reversal in the orientation of RPA70AB.
The XPA DBD–RPA70AB complexes bound to the model 3′ and 5′ ss–ds junction DNA NER substrates are both quite compact and strikingly similar, near mirror images (Figure 4). In both complexes, the XPA–RPA interaction interface involves the same face of RPA70AB adjacent to the ssDNA-binding sites and the zinc-binding domain of XPA DBD (Supplementary Materials; Supplementary Figure S6 and Table S3). Both complexes have substantial interfaces with 24 XPA/20 RPA residues and 783/795 Å2 buried surface areas for the 3′ ss–ds junction substrate, and 29/25 residues and 856/899 Å2 for the 5′ substrate. Both complexes exhibit a mixture of hydrogen bonds (6, 9), salt bridges (4, 3) and non-bonded contacts (69, 102). The similarity extends to the specific residues engaged in the XPA–RPA interface with approximately half being common, and the remainder representing small shifts of only 1–3 residues (Supplementary Materials, Supplementary Figure S6 and Table S3). Thus, these structures lend further support for the possibility that models for XPA binding to the 3′ or the 5′ ss–ds junction in the NER bubble are feasible.
Mutations in XPA DBD inhibit RPA70AB interaction without altering binding of DNA
To test the structural models and generate mutants to probe the functional importance of the interaction between XPA DBD and RPA70AB, four XPA DBD mutants were designed to perturb the interaction with RPA70AB: E106K, F112A, E106K+F112A (M2) and D101N+E106K+K110E+E111K+F112A+D114N (M6) (Figure 5A). E106K and F112A were selected as single mutants because they are in the binding interface in the model and they exhibit the largest perturbations of their NMR signals upon binding to RPA70AB. E106 was just one of the significant number of charged residues in the binding interface and based on initial results for E106K and M2, knowledge that electrostatic forces are long-range, and past experience with multi-site mutations, four more charge reversal mutations were added to the design of M6 with the goal of generating a mutant that would substantially inhibit binding to RPA70AB. The mutations were incorporated into the XPA DBD construct and their effects on the structure, DNA-binding affinity and ability to interact with RPA70AB were determined. The results for the single-site and M2 mutant were similar, most importantly with the respect to the extent of impairment of interaction with RPA70AB, so in the following we describe only the data for the M2 and M6.
Figure 5.

Characterization and effect on NER of XPA mutations in the RPA70AB-binding interface. (A) Close up view of the six mutated sites depicted on the final structural model. (B) MST measurement of DNA binding of XPA DBD wild-type and mutants. Data shown for XPA DBD, M2, M6 from which KD values were determined of 5.7 μM ± 0.4, 6.1 μM ± 0.3, 6.0 μM ± 0.4 and 13.0 μM ± 3.1, respectively. Data are also shown for the negative control XPA K222E. Error bars represent standard deviations from at least three measurements. (C) EMSA analysis of DNA binding of a 5′-FAM-labeled three-way junction substrate with WT or mutant XPA on an 8% native polyacrylamide gel. Protein concentration and the positions of the unbound DNA (free DNA), XPA-bound DNA (protein-DNA) are indicated. (D) In vitro NER activity assay for the wild-type and mutant proteins. A plasmid containing a site-specific Cis-Pt adduct was incubated with either HeLa whole cell extract (M) or XPA-deficient cell extract and recombinant purified WT or mutant XPA. The excision products were detected by annealing to complementary oligonucleotide with 4G overhang, which was used as a template for a fill-in reaction with [α-32P]dCTP. The asterisk indicates background bands resulting from the method used to detect the excised NER fragments. These arise from incubation of the plasmid-containing substrate with Sequenase. As can be seen from the lanes in XPA-deficient cell extracts incubated without XPA protein, they are not dependent on NER. A previous report showed that application of this assay in a quantitative manner is possible even when such non-specific cleavage bands present (63). The NER products were analyzed on 14% denaturing PAGE gel and visualized on a phosphorimager. Reaction times and identity of the proteins and location of NER and non-specific bands are indicated. (E) Close up view of the overlay of 15N-1H HSQC NMR spectra of 15N-enriched XPA DBD and wild-type (WT), M2 and M6 XPA DBD in the presence of a 4-fold molar excess of RPA70AB.
The effect of mutation on the structure of XPA DBD was assessed by circular dichroism (CD) and NMR. The CD spectra of M2 and M6 were very similar to that of the wild-type protein (Supplementary Materials andSupplementary Figure S7A), showing that the two constructs contain the same distribution of secondary structure (55). The 2D 15N-1H HSQC NMR spectra provide insight into the tertiary structure of the protein. Comparison of the spectra of M2, M6 and the wild-type protein reveal the expected trends for multi-mutations that do not perturb the 3D structure: the distribution of peaks is the same in all three spectra but a number of peaks are shifted relative to wild-type as a result of changes in chemical structure of the side chains and small structural adjustments (Supplementary Materials andSupplementary Figure S7B–D). Microscale thermophoresis (MST) was used to measure the DNA-binding affinity of M2 and M6 using a fluorescein-labeled DNA substrate as reported previously (25) (Figure 5B). The K222E mutant, known to have weaker DNA-binding affinity, was used as a negative control. The KD of wild-type, M2 and M6 derived from these data was 5.7 μM ± 0.4, 6.1 μM ± 0.3 and 6.0 μM ± 0.4, respectively, revealing there were no statistically significant differences in DNA-binding affinity induce by the mutations (Figure 5B). The value for wild-type XPA DBD is similar to that reported previously (3.5 μM ± 0.2) (25).
The XPA DBD mutants were then tested for interaction with RPA70AB by comparing NMR CSPs induced by the wild-type and mutant proteins (Supplementary Materials andSupplementary Figure S8). Although the NMR chemical shift perturbations over the course of a titration can be used to extract binding affinities in the micromolar to millimolar range, direct quantitative comparisons of Kd values is not possible in this case because the affinity is weak and cannot be measured accurately. However, because the residues exhibiting CSPs and the nature of the shifts of peaks is the same for M2, M6 and wild-type DBD, a comparison of the relative amount of CSP at a given stoichiometry can be used to generate a qualitative assessment of the relatively affinities of the three proteins. Overlays of two representative peaks showing CSPs induced by identical amounts of wild-type, M2 and M6 reveal that the M2 peak for the RPA70AB bound state is close to but not shifted as far as wild-type, and that the M6 peak is shifted considerably less than M2 (Figure 5E). Thus, M2 and M6 have progressively weaker affinity than wild-type XPA DBD. Together, the structural and biophysical data show that the M2 and M6 mutations achieve our design goal of generating mutants whose sole effect is impairing the binding of XPA DBD to RPA70AB.
Disruption of the XPA DBD–RPA70AB interaction diminishes NER activity
Since the M2 and M6 mutations have been shown to impair the physical interaction of XPA DBD with RPA70AB, we next tested how they influence NER activity. The standard approach used for these studies is to first perform an EMSA assay to determine if there are any effects from the mutations on the DNA-binding properties of the protein (Figure 5C). Interestingly, the M2 mutation appears to have no significant effect on the DNA-binding affinity of full-length XPA, but the addition of the four additional charge reversal mutations in the M6 mutant results in a mild, but discernable reduction in affinity and appearance of a second band in the EMSA. Interestingly, this observation contrasts with the absence of an effect in the quantitative MST analysis. While we are uncertain as to the origin of this difference, we note that the MST analysis was performed with a model NER substrate, whereas the EMSA, which used a fully duplexed three-way DNA junction, was not. Moreover, it is clear that the M6 XPA mutant still binds DNA with appreciable affinity.
The NER assay involved monitoring the excision of a damaged-containing oligonucleotide from a plasmid incubated in cell-free extracts from XPA-deficient cells complemented with XPA wild-type or mutant proteins, as reported previously (9). A plasmid containing a site-specific 1,3-cisplatin intrastrand crosslink was used for these experiments. Repair of the plasmid is evident over time for the wild-type protein (Figure 5D). The mutant E106K, which did not inhibit the interaction with RPA70AB, was used as a positive control and had comparable levels of activity as the wild-type protein. In contrast, M2 exhibited reduced levels of repair and M6 had a more substantial effect on NER activity, proportional to the extent of reduction in their ability to bind to RPA70AB (Figure 5E). Although the effect on NER observed for the M6 mutations may have an additional contribution (63) from the modest reduction in DNA-binding affinity. Overall, these results strongly support the proposal that the interaction of XPA DBD with RPA70AB is functionally relevant and required for effective repair of bulky DNA adducts by NER.
DISCUSSION
Comparison of different models for DNA binding by XPA
Although structures of the globular core of XPA were determined almost 20 years ago, the details of how XPA binds DNA substrates remained unknown until the structure of the globular core of the yeast homolog of XPA, Rad14, was determined in complex with damaged DNA (56). However, concerns arose about whether or not these structures were broadly applicable to DNA binding by XPA, for example in the context of its primary function in NER (25). One concern is that all Rad14–DNA structures were determined for DNA duplexes containing C8-acetylated deoxyguanosine(dG) bulky-adduct lesions [acetylaminophenyl(dG-AAB), acetylamino-naphthyl(dG-AAN), acetylaminoanthryl(dG-AAA) and acetyl-aminopyrenyl(dG-AAP) phosphoramidite], which XPA binds with extraordinary high affinity, substantially higher than undamaged DNA and duplexes containing a large number of other lesions (56–58). A second concern is two molecules of the Rad14 globular core are bound to the end of the lesion-containing duplex. The interpretation of the first structures suggested Rad14 directly recognizes damage, even though the protein does not directly contact the lesion (56). More recently, the high affinity binding of Rad14 was attributed to an intrinsic kink in the duplex induced by the lesion (58). Thus, Rad14 binding appears to correlate with bent DNA duplex structure, as had been found in earlier studies (6).
XPA was discovered and has been extensively studied as a critical factor for NER. Consequently, the relevance of the structure of Rad14 bound to lesion-containing duplex DNA was questioned because XPA is recruited to NER machinery only after damaged duplex DNA is unwound by TFIIH (8). Moreover, since it is generally understood that the NER machinery contains only a single molecule of XPA, simultaneous binding of two Rad14 molecules to the damaged DNA was difficult to fit into a NER model. Nevertheless, the Rad14 structural data provide a basic underpinning for how XPA interacts with DNA.
We have previously shown that full binding affinity for ss–ds junction NER substrates requires not only the XPA globular core but also a helical C-terminal extension from the core (23,25). In our structures, the helical extension binds the duplex while the globular core interacts at the ss/dsDNA junction (Figure 4A and D). Remarkably, even though the helical extension is missing and two Rad14 molecules bind to one duplex in the Rad14 crystal structure, there are substantial similarities in how XPA DBD interacts with the DNA substrate (Supplementary Materials andSupplementary Figure S9). Residues H258 and F262 in Rad14 extend out from the core and are proposed to function as an anchor point for the unwound base pairs of the damaged duplex (56). In the context of NER, the homologous XPA residues, H171 and W175, are seen to form a wedge between the two strands and stabilize the ss–ds junction (Supplementary Materials andSupplementary Figure S9B). However, in contrast to the structures of Rad14 in complex with dG lesion-containing duplexes, our structure shows that XPA requires neither a kink in the DNA nor a lesion to productively interact with the DNA substrate. As we noted previously, these differences in how XPA interacts with different DNA lesion-containing substrates may correlate to XPA functions in repair pathways outside of NER (15).
XPA binding to RPA70AB and the 5′ versus 3′ ss–ds DNA junction
XPA functions in conjunction with RPA as the scaffold for the NER pre-incision complex. The co-localization of the two proteins is mediated by an interaction between RPA32C and a motif in the disordered XPA N-terminal domain (19). However, it is difficult to see how their activity would be coordinated by this interaction. RPA32C is tethered to the DNA substrate binding apparatus by a 33-residue long flexible linker (59). Moreover, the RPA32C-binding motif on XPA is located within the flexible, disordered XPA N-terminal domain, >50 residues from the XPA DBD (19,29). We propose that the critical factor enabling the coordination of XPA and RPA is the direct physical interaction between XPA DBD and RPA70AB, as this interaction is spatially proximate to the binding of both proteins to the NER bubble.
Our results resolve the controversy over which part of XPA DBD mediates the interaction with RPA70AB. Our comprehensive analysis of two XPA constructs and of the effect of DNA substrates shows that RPA70AB binds primarily to the N-terminal zinc-binding motif (residues 98–125) region, confirming the suggestion made from an NMR titration of XPA98–219 with RPA70AB in the absence of DNA (31). The competing proposal, that XPA residues 153–176 are important (33), was based on the loss of binding for a construct in which these residues were deleted. In examining the structure of the XPA DBD, it is clearly evident that this deletion will greatly destabilize the structure. Hence, loss of interaction with RPA was almost certainly due to the inability of the XPA DBD to fold properly, not loss of the critical region for interaction. This issue highlights the inherent risk of interpreting the effects of mutations in the absence of proper controls to test for perturbations of structure and/or stability.
The exact location of XPA within NER complexes has not yet been firmly established. It is commonly agreed that XPA binds at the edge of the NER bubble, i.e. at ss–ds DNA junctions (15,60). However, some models place XPA at the junction 3′ to the lesion whereas others 5′ to the lesion. We began our analysis using a substrate that mimics RPA70AB and XPA binding at the junction 3′ from the lesion, because it readily accommodates the known 5′ to 3′ polarity of RPA (11,51). Studies by Krasikova et al. on binding of RPA to a range of substrates point to a possible preference for XPA binding to the 5′ junction, but their data were not unambiguous and firm conclusions could not be drawn (14,61). A structure has recently been determined of the complex of TFIIH and XPA bound to a Y-shaped DNA substrate mimicking a 5′ junction (62). The primary rationale for models positioning XPA on the ss–dsDNA junction 5′ from the lesion is the need for close proximity to NER nuclease XPF/ERCC1, which incises DNA 5′ from the lesion, and is known to interact with XPA (16). However, these models are drawn from the perspective of linear 2D representations. Upon examination of 3D models as shown schematically in Figure 6, it is clear that XPA bound at the 3′ ss–ds junction would still be able to contact the 5′ junction since its XPF-ERCC1 binding motif resides in the disordered, N-terminal domain (9).
Figure 6.
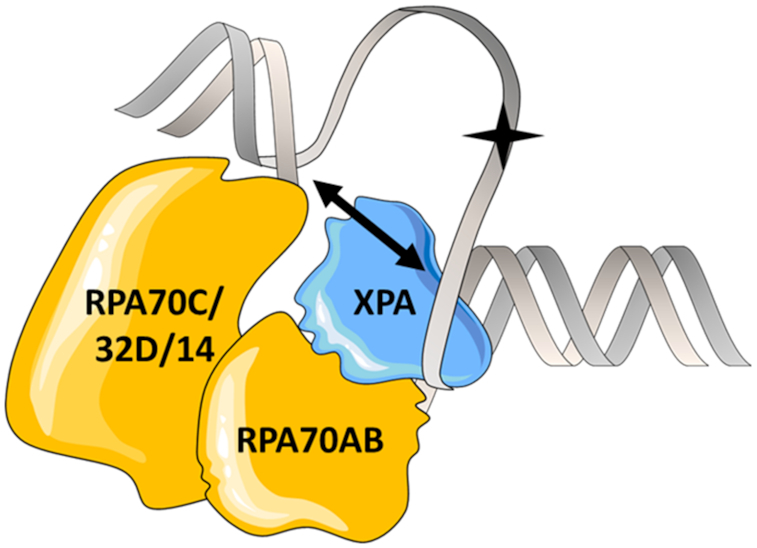
Schematic diagram of XPA and RPA on an NER bubble showing how XPA can be bound to the 3′ ss–ds junction yet still be close to the 5′ ss–ds junction. The lesion is indicated by a star. A two-headed arrow is drawn to show that when the DNA substrate is not extended linearly, the 3′ ss–ds junction can be close enough to the 5′ ss–ds junction for XPA to span the gap.
To test if one, the other or both 3′ and 5′ ss–ds junctions could be accommodated while retaining the critical interaction of XPA DBD with RPA70AB, we obtained experimental data and generated structural models for complexes with substrates mimicking both junctions. Stable complexes were formed using asymmetric Y-shaped ss–ds substrates of the same size that differed only in the 3′ versus 5′ positioning of the short and long arms, and the corresponding complexes were found to be remarkably similar to each other. The two complexes are effectively mirror images, i.e. flipped by 180° relative to one other with the zinc-binding region of XPA DBD rotated with respect to the RPA70AB interface (Figure 4B and E; Supplementary Materials andSupplementary Figure S6). The symmetric relationship is also evident when aligning the structures to the XPA DBD, which shows that the orientation of the RPA70A and RPA70B domains is inverted with respect to the XPA DBD as a result of the 5′-3′ polarity of RPA on ssDNA (Figure 4C and F). The ability to form stable complexes with both substrates suggests the possibility that XPA could bind at either of the two different ss–dsDNA junctions in the bubble. If this were to happen, it would presumably occur at different points in the trajectory of NER.
Implications for coordinated XPA–RPA function in NER
The spatial arrangement of XPA and RPA is critical for the correct positioning of the essential nucleases XPF and XPG on the NER bubble and formation of a properly functioning pre-incision complex. XPA binding to RPA70 was previously shown to be required for NER (34). Our data show that defects even in the weaker of the two XPA–RPA contacts, between XPA DBD and RPA70AB, cause a fundamental dysfunction of nucleotide excision repair in cell extracts (Figure 5D). Structure-based design of mutations enabled us to progressively weaken this interaction, from modest effects for E106K/F112A to nearly eradicating NER by the addition of four more, electrostatic-based mutations. Misalignment of the XPA DBD–RPA70AB interaction may affect the assembly of the entire NER machinery, which can form the basis for XPA-related disease. In this vein, we note the F112C mutation has been reported in the PanCan Atlas as a cancer-associated mutation (TCGA, PanCan Atlas: https://portal.gdc.cancer.gov/genes/ENSG00000136936).
Structural and functional analysis of the XPA DBD binding to RPA70AB has revealed an important function in NER for this interaction, even though the affinity is relatively weak in the micromolar range. Our results have resolved the controversy over exactly where the two domains interact. They also provided strong evidence in support of only a single XPA molecule being involved in binding of DNA substrates and suggest that XPA may in fact bind to both the 3′ or 5′ ss–ds junction in NER complexes. Beyond providing data to predict the molecular basis for dysfunction of disease-associated XPA mutations, the structural characterization of the interaction between XPA DBD and RPA70AB also provides a starting point for small molecule discovery and subsequent evaluation of the therapeutic potential of targeting XPA function.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Dr Miaw-Sheue Tsai of the EMB Core of the Structural Biology of DNA Repair (SBDR) program, who supervised the site-directed mutagenesis and provided the pBG100-XPA E106K/F112A and pBG100-XPA D101N/E106K/K110E/E111K/F112A/D114N plasmids used in this study. We thank Dr Rocco Moretti for assistance with running Rosetta calculations and Dr Swati Balakrishnan for proofreading and editing the manuscript. SAXS experiments were conducted at the SYBILS beamline of the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research.
Author contributions: A.T.-W., N.S., O.D.S. and W.J.C. designed the research; A.T.-W., N.S., J.J.C., R.A.L.M., K.V.O., H.S.K., J.-E.Y., D.R. and M.H. performed experiments; A.T.-W., N.S., R.A.L.M., M.H., O.D.S. and W.J.C. analyzed data; A.T.-W., O.D.S. and W.J.C. wrote the paper; and all authors proofread the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
US National Institutes of Health [R01 CA218315, P01 CA092584 (to O.D.S and W.J.C.)] and Korean Institute for Basic Science [IBS-R022-A1 (to O.D.S)] for operating grants. NIH [T32 GM08320] for stipend to J.J.C. National Science Foundation [0922862], National Institutes of Health [S10 RR025677]; and Vanderbilt University for acquition of NMR instrumentation. National Institutes of Health [S10 OD021483] for for acquition of MST instrumentation. US Department of Energy and National Institute of Health [S10 OD018483, ALS-ENABLE P30 GM124169, SBDR P01 CA092584] for support of the SIBYLS SAXS beamline. Funding for open access charge: Vanderbilt University.
Conflict of interest statement. None declared.
REFERENCES
- 1. Gillet L.C.J., Schärer O.D.. Molecular mechanisms of mammalian global genome nucleotide excision repair. Chem. Rev. 2006; 106:253–276. [DOI] [PubMed] [Google Scholar]
- 2. Schärer O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013; 5:a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. DiGiovanna J.J., Kraemer K.H.. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 2012; 132:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lehmann J., Seebode C., Martens M.C., Emmert S.. Xeroderma pigmentosum - facts and perspectives. Anticancer Res. 2018; 38:1159–1164. [DOI] [PubMed] [Google Scholar]
- 5. Mu H., Geacintov N.E., Broyde S., Yeo J.E., Schärer O.D.. Molecular basis for damage recognition and verification by XPC-RAD23B and TFIIH in nucleotide excision repair. DNA Repair Amst. 2018; 71:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Missoura M., Buterin T., Hindges R., Hübscher U., Kasparkova J., Brabec V., Nägeli H.. Double-check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J. 2001; 20:3554–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riedl T., Hanaoka F., Egly J.. The comings and goings of nucleotide excision repair factors on damaged DNA. EMBO J. 2003; 22:5293–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li C.L., Golebiowski F.M., Onishi Y., Samara N.L., Sugasawa K., Yang W.. Tripartite DNA lesion recognition and verification by XPC, TFIIH, and XPA in nucleotide excision repair. Mol. Cell. 2015; 59:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsodikov O.V., Ivanov D., Orelli B., Staresincic L., Shoshani I., Oberman R., Schärer O.D., Wagner G., Ellenberger T.. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007; 26:4768–4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tripsianes K., Folkers G.E., Zheng C., Das D., Grinstead J.S., Kaptein R., Boelens R.. Analysis of the XPA and ssDNA-binding surfaces on the central domain of human ERCC1 reveals evidence for subfunctionalization. Nucleic Acids Res. 2007; 35:5789–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. de Laat W.L., Appeldoorn E., Sugasawa K., Weterings E., Jaspers N.G., Hoeijmakers J.H.. DNA-binding polarity of human replication protein A positions nucleases in nucleotide excision repair. Genes Dev. 1998; 12:2598–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fanning E., Klimovich V., Nager A.R.. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006; 34:4126–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Z., Roginskaya M., Colis L.C., Basu A.K., Shell S.M., Liu Y., Musich P.R., Harris C.M., Harris T.M., Zou Y.. Specific and efficient binding of xeroderma pigmentosum complementation group A to double-strand/single-strand DNA junctions with 3′- and/or 5′-ssDNA branches. Biochemistry. 2006; 45:15921–15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krasikova Y.S., Rechkunova N.I., Maltseva E.A., Petruseva I.O., Lavrik O.I.. Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic Acids Res. 2010; 38:8083–8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sugitani N., Sivley R.M., Perry K.E., Capra J.A., Chazin W.J.. XPA: A key scaffold for human nucleotide excision repair. DNA Repair Amst. 2016; 44:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li L., Elledge S.J., Peterson C., Bales E.S., Legerski R.J.. Specific association between the human DNA repair proteins XPA and ERCC1. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:5012–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nocentini S., Coin F., Saijo M., Tanaka K., Egly J.-M.. DNA damage recognition by XPA protein promotes efficient recruitment of transcription factor II H. J. Biol. Chem. 1997; 272:22991–22994. [DOI] [PubMed] [Google Scholar]
- 18. Buchko G.W., Daughdrill G.W., de Lorimier R., Rao B.K., Isern N.G., Lingbeck J.M., Taylor J.S., Wold M.S., Gochin M., Spicer L.D. et al.. Interactions of human nucleotide excision repair protein XPA with DNA and RPA70 Delta C327: chemical shift mapping and 15N NMR relaxation studies. Biochemistry. 1999; 38:15116–15128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mer G., Bochkarev A., Gupta R., Bochkareva E., Frappier L., Ingles C.J., Edwards A.M., Chazin W.J.. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 2000; 103:449–456. [DOI] [PubMed] [Google Scholar]
- 20. Neher T.M., Shuck S.C., Liu J.-Y., Zhang J.-T., Turchi J.J.. Biochemical analysis of the damage recognition process in nucleotide excision repair. J. Biol. Chem. 2003; 278:7476–7485. [DOI] [PubMed] [Google Scholar]
- 21. Bernardes de Jesus B.M., Bjørås M., Coin F., Egly J.M.. Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Mol. Cell Biol. 2008; 28:7225–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wakasugi M., Kasashima H., Fukase Y., Imura M., Imai R., Yamada S., Cleaver J.E., Matsunaga T.. Physical and functional interaction between DDB and XPA in nucleotide excision repair. Nucleic Acids Res. 2009; 37:516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugitani N., Shell S.M., Soss S.E., Chazin W.J.. Redefining the DNA binding domain of human XPA. J. Am. Chem. Soc. 2014; 136:10830–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hilton B., Shkriabai N., Musich P.R., Kvaratskhelia M., Shell S.M., Zou Y.. A new structural insight into XPA-DNA interactions. Biosci. Rep. 2014; 34:831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sugitani N., Voehler M.W., Roh M.S., Topolska-Woś A.M., Chazin W.J.. Analysis of DNA binding by human factor xeroderma pigmentosum complementation group A (XPA) provides insight into its interactions with nucleotide excision repair substrates. J. Biol. Chem. 2017; 292:16847–16857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen R., Wold M.S.. Replication protein A: single-stranded DNA’s first responder: dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 2014; 36:1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stauffer M.E., Chazin W.J.. Structural mechanisms of DNA replication, repair, and recombination. J. Biol. Chem. 2004; 279:30915–30918. [DOI] [PubMed] [Google Scholar]
- 28. Jiang X., Klimovich V., Arunkumar A.I., Hysinger E.B., Wang Y., Ott R.D., Guler G.D., Weiner B., Chazin W.J., Fanning E.. Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. EMBO J. 2006; 25:5516–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ali S.I., Shin J.S., Bae S.H., Kim B., Choi B.S.. Replication protein A 32 interacts through a similar binding interface with TIPIN, XPA, and UNG2. Int. J. Biochem. Cell. Biol. 2010; 42:1210–1215. [DOI] [PubMed] [Google Scholar]
- 30. Lee S.-H., Kim D.-K., Drissi R.. Human xeroderma pigmentosum group a protein interacts with human replication protein a and inhibits DNA replication. J. Biol. Chem. 1995; 270:21800–21805. [DOI] [PubMed] [Google Scholar]
- 31. Ikegami T., Kuraoka I., Saijo M., Kodo N., Kyogoku Y., Morikawa K., Tanaka K., Shirakawa M.. Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat. Struct. Biol. 1998; 5:701–706. [DOI] [PubMed] [Google Scholar]
- 32. Daughdrill G.W., Buchko G.W., Botuyan M.V., Arrowsmith C., Wold M.S., Kennedy M.A., Lowry D.F.. Chemical shift changes provide evidence for overlapping single-stranded DNA- and XPA-binding sites on the 70 kDa subunit of human replication protein A. Nucleic Acids Res. 2003; 31:4176–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li L., Lu X., Peterson C.A., Legerski R.J.. An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Mol. Cell. Biol. 1995; 15:5396–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saijo M., Takedachi A., Tanaka K.. Nucleotide excision repair by mutant xeroderma pigmentosum group A (XPA) proteins with deficiency in interaction with RPA. J. Biol. Chem. 2011; 286:5476–5483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arunkumar A.I., Stauffer M.E., Bochkareva E., Bochkarev A., Chazin W.J.. Independent and coordinated functions of replication protein A tandem high affinity single-stranded DNA binding domains. J. Biol. Chem. 2003; 278:41077–41082. [DOI] [PubMed] [Google Scholar]
- 36. Goddard T.D., Kneller D.G.. SPARKY 3. 2008; San Francisco, CA: University of California. [Google Scholar]
- 37. Vranken W.F., Boucher W., Stevens T.J., Fogh R.H., Pajon A., Llinas M., Ulrich E.L., Markley J.L., Ionides J., Laue E.D.. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005; 59:687–696. [DOI] [PubMed] [Google Scholar]
- 38. Classen S., Hura G.L., Holton J.M., Rambo R.P., Rodic I., McGuire P.J., Dyer K., Hammel M., Meigs G., Frankel K.A. et al.. Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. J. Appl. Crystallogr. 2013; 46:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dyer K.N., Hammel M., Rambo R.P., Tsutakawa S.E., Rodic I., Classen S., Tainer J.A., Hura G.L.. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Met. Mol. Biol. 2014; 1091:245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hura G.L., Menon A.L., Hammel M., Rambo R.P., Poole F.L. 2nd, Tsutakawa S.E., Jenney F.E. Jr., Classen S., Frankel K.A., Hopkins R.C. et al.. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Met. 2009; 6:606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rambo R.P., Tainer J.A.. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013; 496:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sali A., Blundell T.L.. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993; 234:779–815. [DOI] [PubMed] [Google Scholar]
- 43. Huang P.S., Ban Y.E., Richter F., Andre I., Vernon R., Schief W.R., Baker D.. RosettaRemodel: a generalized framework for flexible backbone protein design. PLoS One. 2011; 6:e24109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Drozdetskiy A., Cole C., Procter J., Barton G.J.. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015; 43:389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mandell D.J., Coutsias E.A., Kortemme T.. Sub-angstrom accuracy in protein loop reconstruction by robotics-inspired conformational sampling. Nat. Met. 2009; 6:551–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., van Dijk M., de Vries S.J., Bonvin A.M.J.J.. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2015; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 47. Schneidman-Duhovny D., Hammel M., Sali A.. FoXS: A Web server for Rapid Computation and Fitting of SAXS Profiles. Nucleic Acids Res. 2010; 38:W540–W544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A.. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013; 105:962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das R. Atomic-accuracy prediction of protein loop structures enabled by an RNA-inspired Ansatz. PLoS One. 2013; 8:e74830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kozin M., Svergun D.. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001; 34:33–41. [Google Scholar]
- 51. Bhattacharya S., Arunkumar A.I., Sullivan S.L., Botuyan M.V., Arrowsmith C.H., Chazin W.J.. 1H, 13C and 15N assignments of single-stranded DNA binding domains from the 70 kDa subunit of human replication protein A. J. Biomol. NMR. 2004; 28:195–196. [DOI] [PubMed] [Google Scholar]
- 52. Fan J., Pavletich N.P.. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012; 26:2337–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bochkarev A., Pfuetzner R., Edwards A., Frappier L.. Structure of the single-stranded-DNA binding domain of replication protein A bound to DNA. Nature. 1997; 385:176–181. [DOI] [PubMed] [Google Scholar]
- 54. Lian F.M., Yang X., Yang W., Jiang Y.L., Qian C.. The redefined DNA-binding domain of human xeroderma pigmentosum complementation group A: production, crystallization and structure solution. Acta Crystallogr. F Struct. Biol. Commun. 2019; 75:62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Greenfield N.J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2006; 1:2876–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koch S.C., Kuper J., Gasteiger K.L., Simon N., Strasser R., Eisen D., Geiger S., Schneider S., Kisker C., Carell T.. Structural insights into the recognition of cisplatin and AAF-dG lesion by Rad14 (XPA). Proc. Natl. Acad. Sci. U.S.A. 2015; 112:8272–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Simon N., Ebert C., Schneider S.. Structural basis for bulky-adduct DNA-lesion recognition by the nucleotide excision repair protein Rad14. Chemistry. 2016; 22:10782–10785. [DOI] [PubMed] [Google Scholar]
- 58. Ebert C., Simon N., Schneider S., Carell T.. Structural insights into the recognition of N2-Aryl- and C8-Aryl DNA lesions by the repair protein XPA/Rad14. Chembiochem. 2017; 18:1379–1382. [DOI] [PubMed] [Google Scholar]
- 59. Brosey C.A., Chagot M.E., Ehrhardt M., Pretto D.I., Weiner B.E., Chazin W.J.. NMR analysis of the architecture and functional remodeling of a modular multidomain protein, RPA. J. Am. Chem. Soc. 2009; 131:6346–6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fadda E. Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput. Struct. Biotechnol. J. 2016; 14:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Krasikova Y.S., Rechkunova N.I., Maltseva E.A., Lavrik O.I.. RPA and XPA interaction with DNA structures mimicking intermediates of the late stages in nucleotide excision repair. PLoS One. 2018; 13:e0190782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kokic G., Chernev A., Tegunov D., Dienemann C., Urlaub H., Cramer P.. Structural basis of TFIIH activation for nucleotide excision repair. Nat. Commun. 2019; 10:2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yeo J.E., Khoo A., Fagbemi A.F., Schärer O.D.. The efficiencies of damage recognition and excision correlate with duplex destabilization induced by acetylaminofluorene adducts in human nucleotide excision repair. Chem. Res. Toxicol. 2012; 25:2462–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.