
Fig. 1. Mutational paths in ATPα associated with insect specialization on cardiac glycoside-producing plants are constrained.
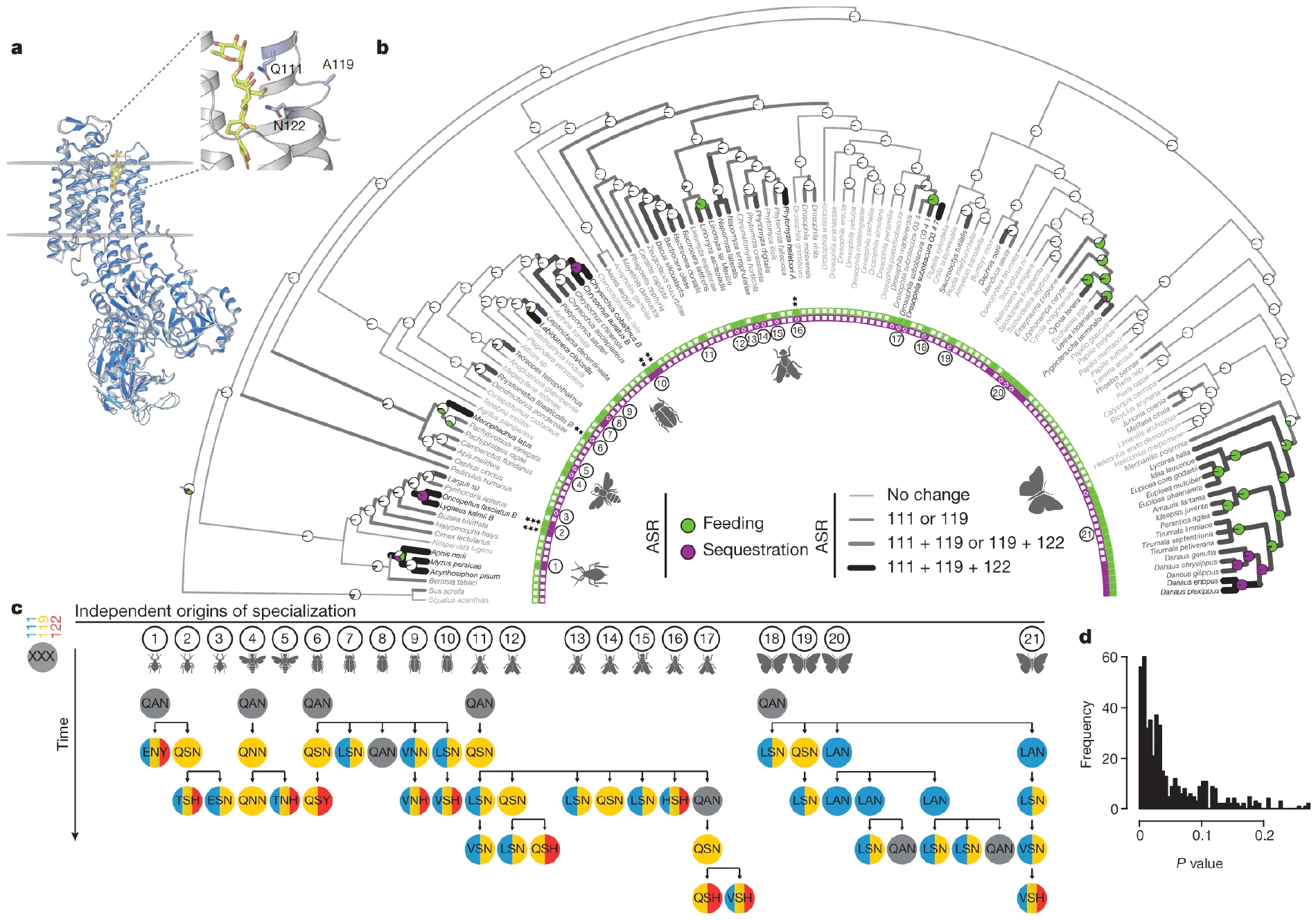
a, Protein homology model of Drosophila melanogaster ATPα (navy) superimposed on a Sus scrofa ATPα crystal structure (light grey) with ouabain (yellow) in the binding pocket. Residues 111, 119 and 122 (sticks) within the H1–H2 extracellular loop are associated with feeding on cardiac glycoside-producing plants and toxin sequestration. b, Maximum likelihood phylogeny based on 4,890 bp from Atpα and coi, with maximum likelihood ancestral state reconstruction (ASR) of feeding and sequestering states, estimated from the states of extant species (inner band of squares). Reconstructions are shown as nodal pie graphs (white, neither feeding nor sequestering; green, feeding; purple, feeding and sequestering), and the number of substituted sites at positions 111, 119 and 122 along branches in grey-scale (light grey 0, medium grey 1, dark grey 2, black 3), based on maximum likelihood ASR of H1–H2 loop amino acid sequences. Black asterisks indicate the Atpα copy number for species with multiple paralogues. c, ATPα substitutions inferred from ASR at positions 111 (blue), 119 (yellow) and 122 (red) in 21 lineages where specialization occurred independently. d, P value distribution from a set of randomized tests to determine the reproducibility of substitutions observed along mutational paths among sub-sampled groups compared to randomly permuted substitutions. On average, 4.9% (considering all mutational steps) of randomly permuted trajectories demonstrate a degree of ordering equal to or greater than observed mutational paths.