

ABSTRACT
Advanced colorectal cancer (CRC) is a significant cause of cancer mortality, with a poor prognosis. Here, we identified a novel prognostic signature for predicting survival of advanced CRC. Advanced CRC data were used (training set: n = 267 and validation set: n = 264). The survival analyses were investigated. The functional analysis of the prognostic signature was examined. In this study, our 15-gene signature was established and was an independent prognostic factor of advanced CRC. Stratification analyses also showed that this signature was still powerful for survival prediction in each stratum of age, gender, stage, and metastasis status. In mechanism, our signature involved in DNA replication, DNA damage, and cell cycle. Therefore, our findings suggested that this 15-gene signature has prognostic and predictive value in advanced CRC, which could be further used in personalized therapy for advanced CRC.
KEYWORDS: Prognosis, signature, advanced CRC, predictive, therapy
Graphical Abstract
Introduction
Colorectal cancer (CRC) is one of the most common cancers and the second cause of cancer-related mortality in the world [1]. According to the GLOBOCAN estimates, over 1.8 million new cases are clinically diagnosed with CRC, and approximately 881,000 deaths due to CRC are estimated to occur worldwide in 2018 [1]. Although recent advances in the treatment strategies of CRC such as surgery, chemotherapy, radiotherapy, immunotherapy, and targeted therapy have improved, patients with advanced disease (stage 3–4) remain a poor 5-year survival rate (~13.1%) [2–5]. Although many biomarkers have been reported for CRC prognosis, most do not show strong predictive power of CRC patients whose prognosis [6,7]. Thus, identification of novel prognostic signatures is urgently important to effectively predict the prognosis for advanced CRC.
Multiple molecular alterations correlate with the progression and prognosis of human cancers, and molecular biomarkers reveal promise in predicting patients’ survival [8–11]. A number of studies suggest genetic alterations involve in CRC development and progression [12,13]. For example, herpesvirus entry mediator (HVEM) plays a key role in disease progression and may serve a potential prognostic marker in CRC [14]. Activity-dependent neuroprotector homeobox (ADNP) correlates with CRC cell migration, invasion, and proliferation, and the prognosis of CRC [15]. Cyclooxygenase-2 (COX-2) is correlated with shorter survival and is an independent prognostic indicators of CRC [16]. These studies show that single gene expression may have the potential value for predicting prognosis of patients with CRC. Some recent studies have reported the prognostic significance of the gene signature for predicting prognosis of CRC [17,18]. Unfortunately, these recent studies have some limitations, such as relatively small study population, lack of further validation cohort, or analysis of only one or a few genes. Additionally, study on the multi-gene signature is lacking in advanced CRC. Here, using multiple datasets, we constructed a reliable multi-gene signature for prognosis prediction in the training and validation sets of patients with advanced CRC.
In our present work, a novel 15-gene signature was first established in advanced CRC. We found that our signature was significantly correlated with worse survival and was identified as an independent prognostic molecular factor in advanced CRC. Our signature was still significant for predicting survival in each stratum of age, gender, stage, and metastasis status. In mechanism, our signature mediated DNA replication, DNA damage, and cell cycle. Our results demonstrated that this signature could predict survival and provide more effective treatment selection for patients with advanced CRC.
Materials and methods
Sample information
Gene expression data and clinical data for cases with CRC were obtained from The Cancer Genome Atlas (TCGA) database. mRNA expression counts were normalized by using DEseq package. The mean expression levels >1 in all samples were included. Gene expression values were then transformed in the form of log2(x + 1). CRC cases without available survival data were excluded. CRC samples diagnosed with advanced-stage disease (stage 3–4) are included in this study. 267 patients were finally identified from TCGA. CRC expression profiles using Affymetrix U133plus2 chip were downloaded from Gene Expression Omnibus (GEO) GSE39582 dataset, which were normalized by robust multi-array average (RMA) method. The normalized gene expression levels were transformed to log base 2. The advanced-stage CRC patients with sufficient prognostic information were selected. Finally, 264 patients were identified from GSE39582 dataset.
TCGA dataset was used as a training set and GSE39582 dataset was applied as an independent validation set. The clinical data such as age, gender, and distal metastasis status were collected. Overall survival (OS) was evaluated in the study. The detailed clinical characteristics are summarized in Table 1.
Table 1.
Patient characteristics of the TCGA training set and validation set in advanced CRC.
| TCGA training set |
Validation set |
||||
|---|---|---|---|---|---|
| Characteristics | Number of cases | % | Number of cases | % | |
| Age at diagnosis 66 (31–90) | 67 (22–97) | ||||
| Age (years) | |||||
| ≥60 | 175 | 65.5 | 183 | 69.3 | |
| < 60 | 92 | 34.5 | 81 | 30.7 | |
| Gender | |||||
| Male | 139 | 52.1 | 137 | 51.9 | |
| Female | 128 | 47.9 | 127 | 48.1 | |
| Stage | |||||
| 4 | 88 | 33 | 60 | 22.7 | |
| 3 | 179 | 67 | 204 | 77.3 | |
| Distal metastasis | |||||
| Positive | 86 | 36.6 | 60 | 23.6 | |
| Negative | 149 | 63.4 | 194 | 76.4 | |
TCGA: The Cancer Genome Atlas; CRC: colorectal cancer.
Establishment of the prognostic gene signature
Univariate Cox proportional hazard regression analysis and Kaplan-Meier method were conducted to screen for the prognostic genes. The results with P < 0.05 were selected in the training and validation sets (Figure S1). Then, in the training set, 33 survival-related genes were further conducted for variable selection using the least absolute shrinkage and selection operator (LASSO) method, which showed that 23 genes were selected (Figure S2). To gain the final prognostic genes, we further applied multivariate Cox regression analysis in the training set, 15 genes were finally identified in our prognostic model. For every patient, the risk score was calculated by the expression value of a gene with its corresponding regression coefficient. The formula was used as follows: Risk score = expression value of gene1* βgene1 + expression value of gene2* βgene2 + expression value of gene3* βgene3 … + expression value of genei * βgenei. β stood for regression coefficient.
Functional analysis of the prognostic signature
For correlation between the most significant 500 genes and the 15-gene prognostic signature, the results were selected on the basis of the Spearman correlation coefficient with the absolute values of > 0.25. Then, clusterProfiler package was used, functional enrichment analysis of the 15-gene prognostic signature was conducted based on Gene Ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways.
Statistical analysis
The cutoff value was applied based on the median value of the risk scores. The patients were dichotomized into two groups, the high- and low-risk groups. Kaplan-Meier survival curves and the log-rank test were used to evaluate and compare the difference between the high-risk and low-risk groups. To confirm the predictive accuracy of the 15-gene prognostic signature, the receiver operating characteristic (ROC) curve and area under the curve (AUC) were performed. The Hazard ratios (HRs) and 95% confidence intervals (95% CIs) were derived from the Cox proportion hazard model. Univariate Cox hazard regression analysis was performed to assess the correlation between the 15-gene prognostic signature and the survival. The multivariate Cox hazard regression analysis was further conducted to determine whether the 15-gene signature was a potential prognostic factor. The stratified analyses were also carried out in patients with different variables such as age, gender, and distal metastasis status. Statistical analyses were performed using R software (version: 3.5.1; R Foundation for Statistical Computing, Vienna, Austria).
Results
Identification of the 15-gene prognostic signature in advanced CRC
After using Cox proportional hazard regression analysis, Kaplan-Meier method, and LASSO analysis, final 15 genes belonged to protein-coding type and were identified in our prognostic model in the training set (n = 267 patients), which consisted of zinc finger and BTB domain containing 34 (ZBTB34), sosondowah ankyrin repeat domain family member A (SOWAHA), solute carrier family 4 member 2 (SLC4A2), ankyrin repeat domain 16 (ANKRD16), C-type lectin domain containing 16A (CLEC16A), kinesin family member 15 (KIF15), mitochondrial intermediate peptidase (MIPEP), ring finger protein 113A (RNF113A), gap junction protein beta 6 (GJB6), ribonuclease P/MRP subunit p14 (RPP14), hypocretin receptor 1 (HCRTR1), tubulin alpha 1c (TUBA1C), phosphomannomutase 2 (PMM2), jagged canonical Notch ligand 2 (JAG2), and ribophorin II (RPN2). Our 15-gene prognostic signature was established using the expression value of each gene and its corresponding regression coefficient, and we used the following formula: (ZBTB34 * 0.527) + (SOWAHA * (−0.292)) + (SLC4A2 * 0.861) + (ANKRD16 * 0.474) + (CLEC16A * (−1.512)) + (KIF15 * (−0.449)) + (MIPEP * (−0.336)) +(RNF113A * 0.603) + (GJB6 * 0.150) + (RPP14 * (−0.463)) + (HCRTR1 * 0.144) + (TUBA1C * (−0.609)) + (PMM2 * 0.837) + (JAG2 * 0.342) + (RPN2 * (−0.780)). In addition, using our 15-gene prognostic signature, each patient achieved a risk score in the training and validation sets.
A 15-gene signature for survival prediction in advanced CRC
The 15-gene signature was applied to the training and validation sets for estimation of its predictive value. The results demonstrated that the prognostic score can classify advanced CRC patients into either the high-risk group or the low-risk group, including the training set, the validation set, and the whole set (Figure 1(a)). For the 5-year survival prediction, the AUC values of the 15-gene signature were 0.883 in the training set, 0.777 in the validation set, and 0.804 in the whole set (Figure 1(b)). The AUC value was >0.75 in each set, indicating a good performance of our 15-gene signature for predicting survival. Kaplan–Meier survival curves demonstrated that patients in the high-risk group had significantly worse prognosis than patients in the low-risk group in each set (all P values < 0.001) (Figure 1(c)).
Figure 1.
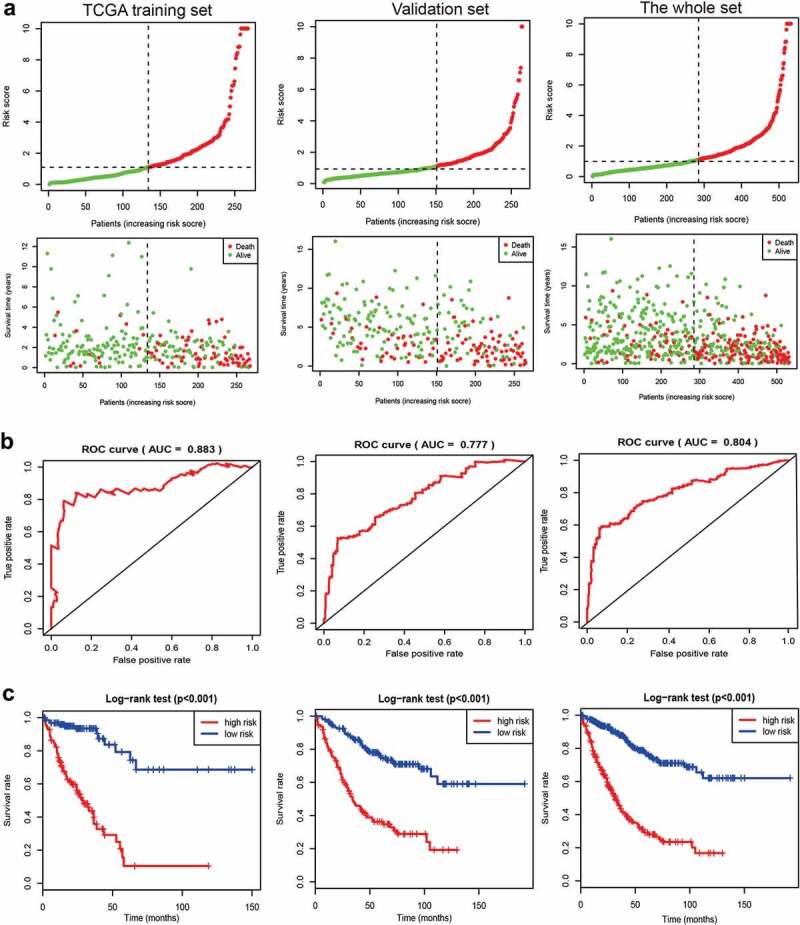
A 15-gene prognostic risk signature in advanced colorectal cancer (CRC) for a TCGA training set, a validation set, and the whole set. (a) Samples sorted by risk score and the corresponding survival status. (b) ROC curve of the 15-gene prognostic risk signature. (c) The Kaplan–Meier curves of the 15-gene prognostic risk signature (the high- and low-risk groups). TCGA: The Cancer Genome Atlas; ROC: the receiver operating characteristic curve; AUC: area under the curve.
A 15-gene signature is an independent prognostic factor
To evaluate the independent prognostic value of the 15-gene signature, univariate and multivariate Cox regression analyses were conducted in the training and validation sets (Table 2). Univariate Cox analysis showed that the high-risk group was significantly correlated with poor prognosis in advanced CRC (the TCGA training set: HR = 8.00, 95% CI = 4.35–14.73, P < 0.001; the validation set: HR = 3.85, 95% CI = 2.58–5.74, P < 0.001). After multivariate adjustment using the clinical variables, the results showed that our 15-gene signature was still an independent prognostic factor (the TCGA training set: HR = 8.19, 95% CI = 4.32–15.54, P < 0.001; the validation set: HR = 3.21, 95% CI = 2.14–4.84, P < 0.001).
Table 2.
Univariate and multivariate Cox regression models of the 15-gene signature in predicting survival of advanced CRC.
| HR (95% CI) |
HR (95% CI) |
|||
|---|---|---|---|---|
| Factors | Univariate Cox analysis | P | Multivariate Cox analysis | P |
| TCGA training set | ||||
| Risk score (high vs. low) | 8.00 (4.35–14.73) | <0.001 | 8.19 (4.32–15.54) | <0.001 |
| Age (≥60 vs. <60 years) | 1.79 (1.08–2.98) | 0.025 | 1.98 (1.14–3.43) | 0.015 |
| Gender (male vs. female) | 0.97 (0.62–1.50) | 0.879 | ||
| Distal metastasis (positive vs. negative) | 2.53 (1.60–4.00) | <0.001 | 2.61 (1.65–4.15) | <0.001 |
| Validation set | ||||
| Risk score (high vs. low) | 3.85 (2.58–5.74) | <0.001 | 3.21 (2.14–4.84) | <0.001 |
| Age (≥60 vs. <60 years) | 1.37 (0.89–2.11) | 0.152 | ||
| Gender (male vs. female) | 1.40 (0.95–2.05) | 0.088 | ||
| Distal metastasis (positive vs. negative) | 4.15 (2.74–6.27) | <0.001 | 3.55 (2.34–5.38) | <0.001 |
TCGA: The Cancer Genome Atlas; CRC: colorectal cancer; HR: hazard ratio; CI: confidence interval.
Stratification analyses
According to available clinical variables such as age (≥60 years and <60 years), gender (male and female), stage (stage 3 and stage 4), and distal metastasis status (yes and no), the stratification analyses of our 15-gene signature were conducted in the entire set. The results demonstrated that the 15-gene signature was closely linked with worse survival in older (HR = 4.931, 95% CI = 3.366–7.223, P < 0.001) or younger patients (HR = 4.727, 95% CI = 2.521–8.861, P < 0.001), male (HR = 3.592, 95% CI = 2.35–5.491, P < 0.001) or female patients (HR = 7.112, 95% CI = 4.262–11.87, P < 0.001), stage 3 (HR = 5.328, 95% CI = 3.475–8.168, P < 0.001) or stage 4 patients (HR = 3.685, 95% CI = 2.229–6.094, P < 0.001), and patients with distal metastasis (HR = 3.685, 95% CI = 2.229–6.094, P < 0.001) or without distal metastasis (HR = 4.928, 95% CI = 3.171–7.659, P < 0.001) (Figures 2–3). Additionally, the AUC value with >0.75 was very good for the 5-year survival prediction in each stratum (Figure S3). These findings indicated that the 15-gene signature was still powerful for predicting survival in each stratum of age, gender, stage, and distal metastasis status.
Figure 2.
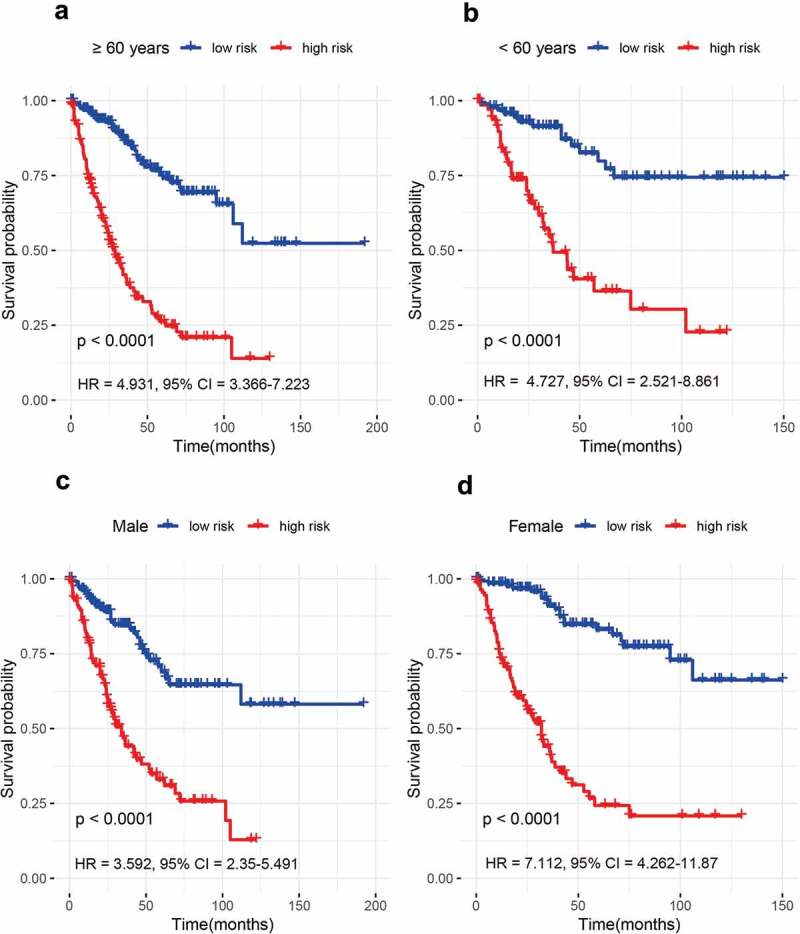
The prognosis of the 15-gene signature for advanced colorectal cancer (CRC) based on age and gender. (a) ≥ 60 years; (b) < 60 years; (c) Male patients; (d) Female patients.
Figure 3.
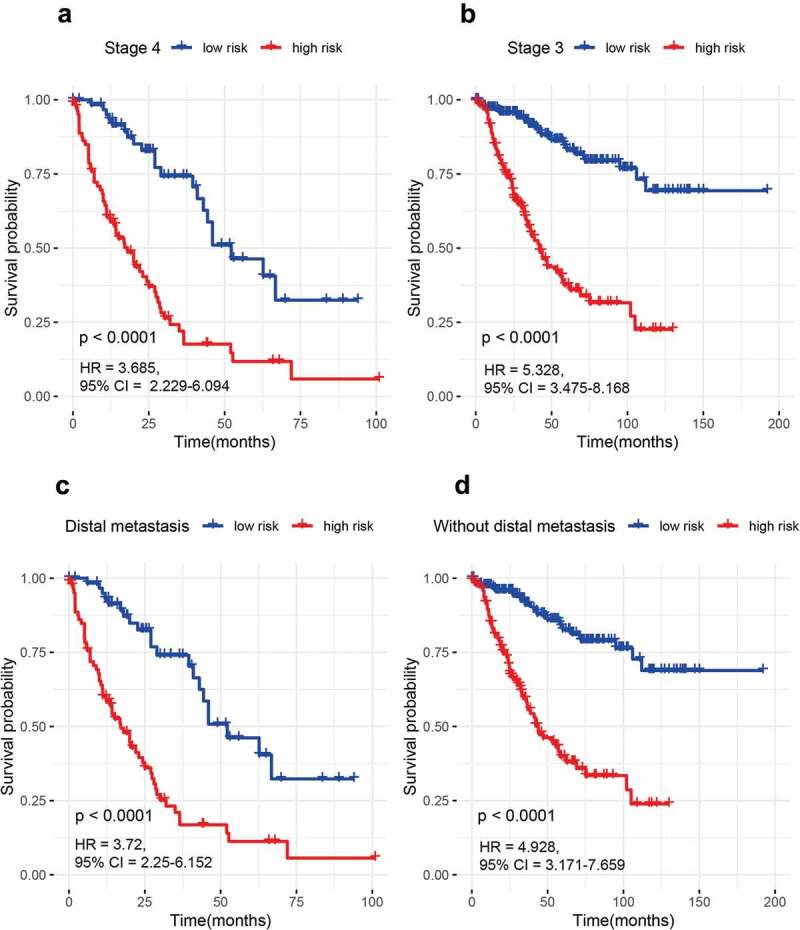
The prognosis of the 15-gene signature for advanced CRC based on stage and distal metastasis status. (a) Stage 4 patients; (b) Stage 3 patients; (c) Patients with distal metastasis; (d) Patients without distal metastasis.
The 15-gene signature-related biological processes and pathways
We further explored the potential biological function and mechanism of 15-gene signature through GO analysis and KEGG pathways. The results showed that signal transduction involved in DNA integrity checkpoint/DNA damage checkpoint/cell cycle checkpoint, regulation of centrosome cycle, chromosome segregation, and DNA replication etc. were enriched (Figure 4), which implied that our 15-gene signature may mainly involve in DNA replication, DNA damage, and cell cycle.
Figure 4.
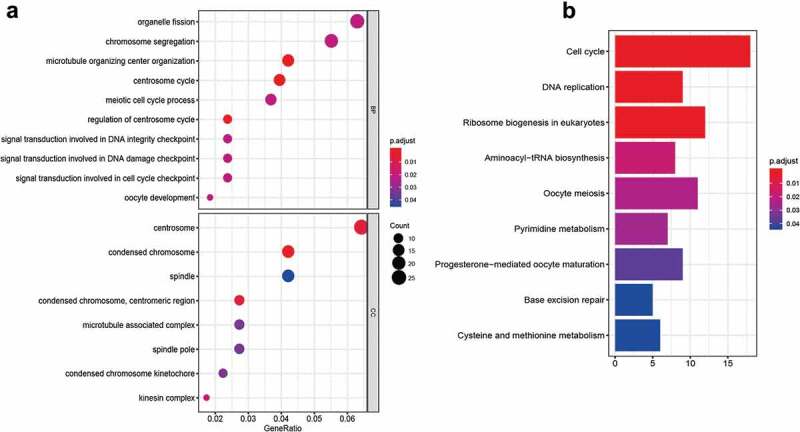
The functional analysis of the 15-gene signature. (a) GO (Gene Ontology) analysis; (b) KEGG (Kyoto Encyclopedia of Genes and Genomes) analysis. BP: biological process; CC: cellular component.
Discussion
CRC is reported to be the second deadliest solid malignancy in the world in 2018 [1]. Despite some significant improvements in the treatment of CRC in recent years, advanced CRC, which is closely linked with spread to distant organs, remains incurable, with an unsatisfactory 5-year survival rate (about 13.1%) [2–4]. Numerous studies reveal genetic alterations play important roles in the development and progression of CRC [12,13]. Some studies have reported that gene alterations could be considered as promising biomarkers for the prediction of the prognosis in CRC. For example, HVEM is associated with CRC progression and could serve an independent prognostic biomarker in CRC [14]. Solute carrier family 12 member 5 (SLC12A5) contributes to disease progression and metastasis, and is an independent prognostic factor in CRC [19]. Additionally, the prognostic value of the gene signature for predicting prognosis of patients with CRC has been reported [17,18]. However, in advanced CRC, the predictive value of the gene signature is still unclear. Thus, we developed and validated a new 15-gene signature for predicting prognosis of patients with advanced CRC.
Natural killer-like signature is shown improved survival in locally advanced CRC [20]. A DNA methylation signature is reported to predict prognosis in locally advanced CRC [21], but the predictive accuracy of this methylation signature is undetermined. In the present work, we constructed a novel 15-gene signature in advanced CRC. Our results found that the 15-gene signature was closely linked with worse prognosis and could classify advanced CRC into high- and low-risk groups in each cohort. In addition, the AUC value of this gene signature was significantly higher in advanced CRC (stage 3–4) than in early CRC (stage 1–2) (0.883 vs. 0.74) (Figure S4), suggesting that our 15-gene signature was very good for the predictive accuracy of prognosis in advanced CRC. These suggested that our 15-gene signature was reliable and effective for survival prediction of advanced CRC.
Our signature included 15 genes. The biological roles of these genes in this prognostic signature are partly explored in tumor. KIF15 mediated pancreatic cancer proliferation through regulating the cell cycle and apoptosis [22]. KIF15 may be a potential prognostic biomarker in bladder cancer [23]. RNF113A expression is correlated with a poor prognosis of esophageal squamous cell carcinoma [24]. GJB6 involves in glioma cell growth and proliferation, and DNA damage. GJB6 expression adversely affected the prognosis of glioma [25]. TUBA1C involves in hepatocellular carcinoma cell migration and proliferation, probability through cell cycle pathway. And TUBA1C is a potential prognostic biomarker of hepatocellular carcinoma [26]. PMM2 expression is significantly correlated with poor prognosis in renal cell carcinoma [27]. JAG2 promotes the motility and invasion of CRC cells [28,29]. RPN2 regulates cell proliferation of CRC cells [30]. RPN2 is a promising prognostic factor in gastric cancer [31]. SLC4A2 (anion exchanger 2, also termed AE2) may contribute to cell cycle progression via mTOR/p70S6K1 pathway. SLC4A2 expression is associated with the shortened survival of ovarian cancer [32,33], which is consistent with our results showed that SLC4A2 upregulation was linked to worse survival of advanced CRC. Here, we pooled 15 genes and found that our 15-gene signature was an independent prognostic molecular marker for patients with advanced CRC. We also showed that our 15-gene signature mainly involved in DNA replication, DNA damage, and cell cycle.
Approximately 20% of patients with CRC present with distant metastasis even at the time of initial diagnosis. Besides, advanced CRC is significantly related to spread to distant organs. The distal metastasis significantly contributes to the high death rate [3,34]. In this work, we showed that our 15-gene signature remained a vigorous tool for the stratification of the risk of patients with distal metastasis or without distal metastasis. In addition, other stratification analyses also demonstrated that the prognostic power of our 15-gene signature was strong in stage 3 or stage 4 patients, male or female patients, and older or younger patients.
Conclusion
In summary, we constructed and validated a 15-gene signature for advanced CRC. Our 15-gene signature contributes to the clinical implications for prognosis prediction and could become an effective prognosis classification for advanced CRC, which could facilitate the management of individualized therapy of patients with advanced CRC. Further investigation and validation are needed in the future.
Article highlights
Our signature was associated with worse survival for advanced colorectal cancer (CRC).
Our signature was an effective prognosis classification for advanced CRC.
Our signature involved in DNA replication, DNA damage, and cell cycle.
Acknowledgements
We gratefully acknowledge the Gene Expression Omnibus and The Cancer Genome Atlas.
Author’s contributions
All authors contributed to the study design; all authors collected the data and performed the data analysis; all authors prepared the manuscript.
Ethics approval and consent to participate
All procedures performed in studies involving human participants were in accordance with the ethical standards of the Gene Expression Omnibus and The Cancer Genome Atlas Human Subjects Protection and Data Access Policies.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. https://portal.gdc.cancer.gov/repository; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39582
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- [2].Han S, Zong S, Shi Q, et al. Is Ep-CAM expression a diagnostic and prognostic biomarker for colorectal cancer? A systematic meta-analysis. EBioMedicine. 2017;20:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Simon K. Colorectal cancer development and advances in screening. Clin Interv Aging. 2016;11:967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Grem JL. Sequencing of treatment in advanced unresectable colorectal cancer. J Natl Compr Canc Network. 2013;11(Suppl 4):S28–37. [DOI] [PubMed] [Google Scholar]
- [5].Poston GJ. Staging of advanced colorectal cancer. Surg Oncol Clin N Am. 2008;17(3):503–17, viii. [DOI] [PubMed] [Google Scholar]
- [6].Shen W, Xu T, Chen D, et al. Targeting SREBP1 chemosensitizes colorectal cancer cells to gemcitabine by caspase-7 upregulation. Bioengineered. 2019;10(1):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lin PS, Semrad TJ. Molecular testing for the treatment of advanced colorectal cancer: an overview. Methods Mol Biol. 2018;1765:281–297. [DOI] [PubMed] [Google Scholar]
- [8].Han S, Huang T, Wu X, et al. Prognostic value of ALDH1 and Nestin in advanced cancer: a systematic meta-analysis with trial sequential analysis. Ther Adv Med Oncol. 2019;11:1758835919830831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bonelli P, Tuccillo FM, Borrelli A, et al. CDK/CCN and CDKI alterations for cancer prognosis and therapeutic predictivity. Biomed Res Int. 2014;2014:361020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cordon-Cardo C. Molecular alterations associated with bladder cancer initiation and progression. Scand J Urol Nephrol Suppl. 2008;42(218):154–165. [DOI] [PubMed] [Google Scholar]
- [11].Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–866. [DOI] [PubMed] [Google Scholar]
- [12].Mehlen P, Tauszig-Delamasure S. Dependence receptors and colorectal cancer. Gut. 2014;63(11):1821–1829. [DOI] [PubMed] [Google Scholar]
- [13].Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135(4):1079–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Inoue T, Sho M, Yasuda S, et al. HVEM expression contributes to tumor progression and prognosis in human colorectal cancer. Anticancer Res. 2015;35(3):1361–1367. [PubMed] [Google Scholar]
- [15].Blaj C, Bringmann A, Schmidt EM, et al. ADNP is a therapeutically inducible repressor of WNT signaling in colorectal cancer. Clin Cancer Res. 2017;23(11):2769–2780. [DOI] [PubMed] [Google Scholar]
- [16].Albasri AM, Elkablawy MA, Hussainy AS, et al. Impact of cyclooxygenase-2 over-expression on the prognosis of colorectal cancer patients. An experience from Western Saudi Arabia. Saudi Med J. 2018;39(8):773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sun G, Li Y, Peng Y, et al. Identification of a five-gene signature with prognostic value in colorectal cancer. J Cell Physiol. 2019;234(4):3829–3836. [DOI] [PubMed] [Google Scholar]
- [18].Schell MJ, Yang M, Missiaglia E, et al. A composite gene expression signature optimizes prediction of colorectal cancer metastasis and outcome. Clin Cancer Res. 2016;22(3):734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Xu L, Li X, Cai M, et al. Increased expression of solute carrier family 12 member 5 via gene amplification contributes to tumour progression and metastasis and associates with poor survival in colorectal cancer. Gut. 2016;65(4):635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Alderdice M, Dunne PD, Cole AJ, et al. Natural killer-like signature observed post therapy in locally advanced rectal cancer is a determinant of pathological response and improved survival. Mod Pathol. 2017;30(9):1287–1298. [DOI] [PubMed] [Google Scholar]
- [21].Gaedcke J, Leha A, Claus R, et al. Identification of a DNA methylation signature to predict disease-free survival in locally advanced rectal cancer. Oncotarget. 2014;5(18):8123–8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wang J, Guo X, Xie C, et al. KIF15 promotes pancreatic cancer proliferation via the MEK-ERK signalling pathway. Br J Cancer. 2017;117(2):245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhao H, Bo Q, Wu Z, et al. KIF15 promotes bladder cancer proliferation via the MEK-ERK signaling pathway. Cancer Manage Res. 2019;11:1857–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang L, Hou Z, Hasim A, et al. RNF113A promotes the proliferation, migration and invasion, and is associated with a poor prognosis of esophageal squamous cell carcinoma. Int J Oncol. 2018;52(3):861–871. [DOI] [PubMed] [Google Scholar]
- [25].Artesi M, Kroonen J, Bredel M, et al. Connexin 30 expression inhibits growth of human malignant gliomas but protects them against radiation therapy. Neuro Oncol. 2015;17(3):392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang J, Chen W, Wei W, et al. Oncogene TUBA1C promotes migration and proliferation in hepatocellular carcinoma and predicts a poor prognosis. Oncotarget. 2017;8(56):96215–96224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yamada Y, Arai T, Sugawara S, et al. Impact of novel oncogenic pathways regulated by antitumor miR-451a in renal cell carcinoma. Cancer Sci. 2018;109(4):1239–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Vaish V, Kim J, Shim M. Jagged-2 (JAG2) enhances tumorigenicity and chemoresistance of colorectal cancer cells. Oncotarget. 2017;8(32):53262–53275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].He W, Chan CM, Wong SC, et al. Jagged 2 silencing inhibits motility and invasiveness of colorectal cancer cell lines. Oncol Lett. 2016;12(6):5193–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li H, Al-Japairai K, Tao Y, et al. RPN2 promotes colorectal cancer cell proliferation through modulating the glycosylation status of EGFR. Oncotarget. 2017;8(42):72633–72651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fujimoto D, Goi T, Hirono Y. Expression of ribophorine II is a promising prognostic factor in human gastric adenocarcinoma. Int J Oncol. 2017;50(2):448–456. [DOI] [PubMed] [Google Scholar]
- [32].Zhang LJ, Lu R, Song YN, et al. Knockdown of anion exchanger 2 suppressed the growth of ovarian cancer cells via mTOR/p70S6K1 signaling. Sci Rep. 2017;7(1):6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Medina JF, Lecanda J, Acin A, et al. Tissue-specific N-terminal isoforms from overlapping alternate promoters of the human AE2 anion exchanger gene. Biochem Biophys Res Commun. 2000;267(1):228–235. [DOI] [PubMed] [Google Scholar]
- [34].Heublein S, Albertsmeier M, Pfeifer D, et al. Association of differential miRNA expression with hepatic vs. peritoneal metastatic spread in colorectal cancer. BMC Cancer. 2018;18(1):201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. https://portal.gdc.cancer.gov/repository; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE39582