

Abstract
This study conducted a comparison of the CaO2-based Fenton (CaO2/Fe(II)) and Fenton-like (CaO2/Fe(III)) systems on their benzene degradation performance. The H2O2, Fe(II), Fe(III), and HO● variations were investigated during the benzene degradation. Although benzene has been totally removed in the two systems, the variation patterns of the investigated parameters were different, leading to the different benzene degradation patterns. In terms of the Fe(II)/Fe(III) conversion, the CaO2/Fe(II) and CaO2/Fe(III) systems were actually inseparable and had the inherent mechanism relationships. For the CaO2/Fe(III) system, the initial Fe(III) must be converted to Fe(II), and then the consequent Fenton reaction could be later developed with the regenerated Fe(II). Moreover, some benzene degradation intermediates could have the ability to facilitate the transformation of the Fe(III) to Fe(II) without the classic H2O2-associated propagation reactions. By varying the Fe(II) dosing method, an effective degradation strategy has been developed to take advantage of the two CaO2-based oxidation systems. The proposed strategy was further successfully tested in TCE degradation, therefore extending the potential for the application of this technique.
Keywords: Calcium Peroxide, Fenton System, Fenton-like System, Hydroxyl Radicals, Application Strategy
1. Introduction
Calcium peroxide (CaO2) is a solid oxidant and has been attracting more and more attention, due to its versatile chemical properties. Based on the Eq. (1), CaO2 usually has been considered as an oxygen releasing compound (ORC) and is therefore applied to oxygen-demanding environmental remediation for many years [1–5]. In terms of the Eq. (2), CaO2 has the potential to act as a solid hydrogen peroxide (H2O2) source, and in recent years a growing number of studies are developing various CaO2 based chemical oxidation systems [6]. It is reported that CaO2 alone can degrade toluene [7] and 2,4-dichlorophenol [8], and can work as an additive in the permeable reactive barrier [9]. Moreover, ozone [10], persulfate [11,12], ultraviolet [13], and other activators have also been studied to promote the CaO2 performance in contaminants remediation [14,15]. Among these emerging oxidation techniques, the CaO2-based Fenton (CaO2/Fe(II)) and Fenton-like (CaO2/Fe(III)) systems are the promising prototypes of the developed CaO2 based oxidation systems, because of their effective decontamination performance [16–23].
| (1) |
| (2) |
The CaO2/Fe(II) and CaO2/Fe(III) systems have been used to treat various contaminated soil and water, and achieved satisfied decontamination performances. Table 1 summarizes the target pollutants which were treated both systems. It is clear that the CaO2/Fe(II) and CaO2/Fe(III) systems were usually considered and studied separately. Most of the testes just concentrated on the decontamination performance under their operational conditions and investigated the influence of some environmental parameters, such as the solution pH, solution matrix, and other common factors [16–23]. Although some studies mentioned the comparison of the CaO2/Fe(II) and CaO2/Fe(III) systems, the aim of these comparisons was not to clarify the relationships among each other. Zhang et al. compared the pollutant degradation performance of the CaO2/Fe(II) and CaO2/Fe(III) systems, but only highlighted the characteristics of their technique and then optimized their experimental conditions [22]. The comparison of the CaO2/Fe(II) and CaO2/Fe(III) systems, so far, are still not thoroughly studied, and the inherent relationships between the two systems are lacking sufficient researches.
Table 1.
Possible reactions involved in the CaO2-based Fenton system
| Type | Reactions | Reference | |
|---|---|---|---|
| Initiation stage | CaO2/Fe(II) system | CaO2 + 2H2O → H2O2 + Ca(OH)2 | [24–26,46,47] |
| Fe2+ + H2O2 → Fe3+ + HO− + HO● | |||
| CaO2/Fe(III) system | CaO2 + 2H2O → H2O2 + Ca(OH)2 | [37,45,46] | |
| Fe3+ + H2O2 → Fe-OOH2+ + H+ | |||
| Fe-OOH2+ → Fe2+ + HO2● | |||
| Fe2+ + H2O2 → Fe3+ + HO− + HO● | |||
| Propagation and termination stage | Fe3+ + HO2● → Fe2+ + H+ + O2 | [23,37,41,45] | |
| HO● + H2O2 → HO2● + H2O | |||
| HO● + Fe2+ → Fe3+ + HO− | |||
| HO● + Ra → ROH● | |||
| ROH● + O2 → ROH + HO2● | |||
| ROH● → R●+ H2O | |||
| R● + O2 → ROO● | |||
| R● + Fe3+ → Fe2+ + R+ | |||
| R● + R● → R2 | |||
| HO● + R2 → R2OH | |||
| Target pollutant | CaO2/Fe(II) system | TCE, PCE, benzene, BTEX, cable insulating oil | [17, 20,21,23,29] |
| CaO2/Fe(III) system | TCE, endocrine-disrupting compounds | [16,19,22] | |
R represents the target compound.
According to the literature reviews on the CaO2/Fe(II) and CaO2/Fe(III) systems, the well-accepted mechanisms for the two systems are concluded (Table 1). Although HO● in the CaO2/Fe(II) and CaO2/Fe(III) systems both originated from the reaction involving Fe(II) and H2O2, the two systems differ in the specific mechanisms. In the CaO2/Fe(II) system, due to the initial Fe(II) presence, HO● can be directly generated from the reaction between Fe(II) and the released H2O2. Previous studies also reported that most of the initial Fe(II) could be quickly converted to Fe(III), and the CaO2/Fe(II) system then actually performed as the CaO2/Fe(III) system for the rest of the experimental time [20,22]. In contrary, for the CaO2/Fe(III) system, the reaction involving Fe(II) and H2O2 is still the dominant HO● source, but the Fe(II) here is from the reduction of the initial Fe(III), and it is the regenerated Fe(II) that causes the Fenton reaction in the CaO2/Fe(III) system. Therefore, the CaO2/Fe(III) system also contains the CaO2/Fe(II) system [26,27]. Although the CaO2/Fe(II) and CaO2/Fe(III) systems were independently studied, the HO● formation is the widely recognized mechanism for both systems [6,24–26]. Moreover, the highlights mentioned above show that that the CaO2/Fe(II) and CaO2/Fe(III) systems are actually related and might have the intrinsic relationships, thus it is necessary to conduct further comparable investigations into the two systems, clarifying the connections between the two systems.
Based on the literature review, trichloroethene (TCE) [17,19,22], BTEX (benzene, toluene, ethylbenzene, and xylenes) [20,23,28], PAHs [13], and other refractory organic pollutants [9–12,16,29–31] have been treated with the CaO2-based oxidation techniques. Among these pollutants, benzene is a typical one in many contaminated sites and also listed as a toxic organic compound [32,33]. Since the reactivity of benzene and HO● is very high (kHO● = 7.8 × 109 M−1 s−1) [34], benzene prefers to react with HO● in the CaO2-based oxidation systems. Thus, benzene, as a target pollutant, was selected in this study, which can provide more convincing explanations of the mechanisms in both systems without other potential debates. Based on the mechanism analysis, an efficient strategy was proposed to optimize the CaO2-based oxidation systems. This strategy was then further tested in TCE treatment, since TCE is another common toxic environmental pollutant that is reported to possess a high reactivity with HO● (kHO● = 3.5 × 108 M−1 s−1) [17,34–36]. This makes TCE suitable to check the proposed strategy, which could further extend the adaptability of our technique in the application of groundwater remediation.
Therefore, the objectives of this study were to (1) compare and evaluate the performances of the CaO2/Fe(II) and CaO2/Fe(III) systems on benzene removal; (2) compare and clarify the intermediates generated in the CaO2/Fe(II) and CaO2/Fe(III) systems; (3) compare and elucidate the benzene degradation mechanisms of the CaO2/Fe(II) and CaO2/Fe(III) systems; and (4) combine the advantages of the CaO2/Fe(II) and CaO2/Fe(III) systems extending the potential application to different pollutants.
2. Experiments
2.1. Experiment procedure
The reactor of 250 mL glass vessel with a water jacket was used in this study and the temperature for all experiments was controlled at 20 ± 2°C. The equilibrated benzene solution was added into the reactor and then diluted to the desired concentration, sealing the reactor and mixing solution with a 600 rpm stirring speed. The initial benzene concentration was set at 1.0 mM, and experiments showed that the volatilization and absorption of benzene caused limited influence in this study (Supplementary Material, Fig. S1). The initial solution pH in this study was adjusted to 3 before dosing Fe(II) or Fe(III) reagent, and the reaction started as soon as adding the predetermined dose of CaO2. At the desired time, 2.5 mL sample was collected in headspace vials containing 1.0 mL methanol solution to stop the reaction. The vial was sealed immediately and then analyzed by headspace-gas chromatography (HS-GC). The tests were conducted duplicate, and the mean values were displayed. The results derived from benzene degradation were further tested in the treatment of TCE, which is another frequently detected toxic pollutant in groundwater and also has a high reactivity with HO● [34–36]. The initial concentration of TCE was set at 0.15 mM, and 1 mL sample was collected in vials containing 1.0 mL hexane at the predetermined time intervals. After the extraction procedure, the extracts were immediately analyzed by GC.
For the HO● measurement, 5,5-Dimethyl-1-Pyrroline N-oxide (DMPO, 8.84 mM) was applied to capture the generated HO● in the CaO2/Fe(II) and CaO2/Fe(III) systems. At the determined time, a 1.0 mL sample was collected from the reactor and then well mixed with 1.0 mL DMPO solution, and then the sample was analyzed using the electron paramagnetic resonance (EPR).
All the chemicals used in this study are listed in Supplementary Material (Text S1). The detailed procedures for the detection of the intermediates could be found in Supplementary Material (Text S2).
2.2. Analytical method
An Agilent HS-GC (Agilent 7890B, Palo Alto, CA, USA) has been used to analyze benzene samples, which coupled with a flame ionization detector (FID), an HP-5 column (30 m length, 0.32 mm I.D., 0.25 μm thickness), and an auto-sampler (Agilent 7967A, Palo Alto, CA, USA). The HO● was captured by DMPO and then tested using the EPR (EMX-8/2.7C, Bruker, Germany) instrument. TCE was measured using a GC (Agilent 7890A, Palo Alto, CA, USA) equipped with an autosampler (Agilent 7693), an electron capture detector (ECD), and a DB-VRX column (250 μm i.d., 1.4 μm thickness, and 60 m length). The intermediates produced in the CaO2/Fe(II) and CaO2/Fe(III) systems were identified by GC/MS (Agilent 6890/5973N, Palo Alto, CA, USA) and high performance liquid chromatography (HPLC, LC-20AT, Shimadzu, Japan). The specific conditions for the analyses can be found in Supplementary Material (Text S2).
For the analysis of H2O2, the filtered sample was analyzed after the reaction with TiSO4 [37]. The concentration of the available Fe(II) and total Fe was determined based on the 1,10-phenanthroline method [38], and the total Fe concentration was determined following Fe(II) procedure after dosing hydroxylamine. The difference between the total Fe and Fe(II) was used to quantify the Fe(III) concentration [39]. The solution pH was measured by a pH meter (Sartorius, PB-10, Germany) equipped with a pH/ATC electrode (Sartorius, Germany).
3. Results and Discussion
3.1. Benzene degradation
The benzene treatment performance of the CaO2/Fe(II) and CaO2/Fe(III) systems were compared and the results have been shown in Fig. 1. Benzene was completely removed in the two systems, but had differing benzene degradation patterns.
Fig. 1.

Benzene degradation in a) the CaO2/Fe(II) system (molar ratio of [CaO2]/[Fe(II)]/[Benzene] were 5/5/1, 10/10/1, 15/15/1, [Benzene]0 = 1.0 mM), and b) the CaO2/Fe(III) system (the molar ratio of [CaO2]/[Fe(III)]/[Benzene] were 5/5/1, 10/10/1, 15/15/1, [Benzene]0 = 1.0 mM). Initial pH = 3.0 ± 0.2.
In the CaO2/Fe(II) system, benzene degradation could be fiinshed in 10 min. When varying the molar ratio of CaO2/Fe(II) from 5/5 to 15/15, benzene removal efficiencies were only enhanced 10% (Fig. 1a), while the observed apparent kinetic reaction rates significantly increased from 0.42 to 4.62 M−1 s−1 according to the second-order kinetic model (Table 2). The results indicating that the molar ratio of CaO2/Fe(II) just slightly influenced the benzene removal efficiency and the benzene degradation rate was regulated by the molar ratio of CaO2/Fe(II). For the CaO2/Fe(III) system, the benzene degradation patterns were different from that in the CaO2/Fe(II) system. When the molar ratio of CaO2/Fe(III) was 5/5, the benzene degradation exhibited a two-stage degradation. During the first 200 min, the benzene removal efficiency was less than 10%, indicating a slow degradation stage; in the following 100 mins, an obvious increase in benzene degradation efficiency leading to a complete removal of benzene by 300 min, indicates a switch to a fast degradation stage. When increasing the molar ratio of CaO2/Fe(III) to 10/10 and 15/15, the benzene degradation finished within 120 and 80 min, respectively (Fig. 1b), while the apparent kinetic reaction rate increased from 5.50 × 10−5 to 1.29× 10−3 M−1 s−1 (Table 2). The results suggested that the benzene degradation performance of the CaO2/Fe(III) system greatly relies on the molar ratio of CaO2/Fe(III) and thus the slow degradation stage could be shortened by increasing the molar ratio CaO2/Fe(III).
Table 2.
The benzene degradation rate constant in the CaO2/Fe(II) and CaO2/Fe(III) systems
| Molar ratio | Rate constant b (M−1 s−1) | Correlation coefficient R2 | |
|---|---|---|---|
| CaO2/Fe(II)/B a | 5/5/1 | 0.42 | 0.91 |
| 10/10/1 | 1.02 | 0.96 | |
| 15/15/1 | 4.62 | 0.99 | |
| CaO2/Fe(III)/B a | 5/5/1 | 5.50 × 10−5 | 0.92 |
| 10/10/1 | 8.63 × 10−4 | 0.99 | |
| 15/15/1 | 1.29 × 10−3 | 0.96 | |
The initial benzene concentration was 1.0 mM.
The presented rate constants were the apparent second-order rate constants, and the benzene degradation stage was fitted by the second-order kinetic model.
The above results indicated that the CaO2/Fe(II) system could achieve a faster benzene degradation than that of the CaO2/Fe(III) system. Although the nano-scale CaO2 alone has the ability to remove some pollutants [6–8], our preliminary study showed that the CaO2 alone only removed little benzene (Supplementary Material, Fig. S1). Hence, it was the Fe(II) or Fe(III) that enhanced benzene degradation in both systems, and the observed different benzene degradation patterns could be ascribed to the different mechanisms of the two systems.
3.2. Mechanisms of the CaO2/Fe(II) and CaO2/Fe(III) systems in benzene degradation
The varied benzene degradation patterns indicated that the benzene degradation mechanisms of the CaO2/Fe(II) and CaO2/Fe(III) were different, so the investigation on the mechanisms would reveal the underlying cause of the disparity between the two systems. The preliminary EPR analysis confirmed that HO● was the dominant reactive oxygen species in the CaO2/Fe(II) and CaO2/Fe(III) systems (Supplementary Material, Fig. S2). Since the dose of H2O2, Fe(II), and Fe(III) could influence HO● generation in both investigated systems, the temporal variations of H2O2, Fe(III), Fe(II), and HO● were studied to clarify the benzene degradation mechanisms of both systems. The mechanism investigation was studied using 1 mM benzene, 10 mM CaO2, and 10 mM Fe(II) or Fe(III).
3.2.1. Fe(II)/Fe(III) conversion in the CaO2/Fe(II) and CaO2/Fe(III) systems
The Fe(II)/Fe(III) conversion is a key factor in the conventional Fenton and Fenton-like systems, thus it is necessary to investigate the Fe(II)/Fe(III) conversion in the CaO2/Fe(II) and CaO2/Fe(III) systems, and the results were presented in Fig. 2.
Fig. 2.

The conversion of Fe(III)/Fe(II) in a) the CaO2/Fe(II) system (control experiment: [CaO2] = [Fe(II)] = 10 mM, [Benzene]0 = 1.0 mM; blank experiment: [CaO2] = [Fe(II)] = 10 mM, [Benzene]0 = 0 mM), and b) the CaO2/Fe(III) system (control experiment: [CaO2] = [Fe(III)] = 10 mM, [Benzene]0 = 1.0 mM; blank experiment: [CaO2] = [Fe(III)] = 10 mM, [Benzene]0 = 0 mM). Initial pH = 3.0 ± 0.2, FeT = [Fe(II)] + [Fe(III)].
The Fe(II)/Fe(III) speciation of the CaO2/Fe(II) system was shown in Fig. 2a. In the blank experiment (without benzene), it was clear that Fe(II) declined to a low concentration in the first few minutes and maintained in this level throughout the experiment, which behaved like a conventional Fenton reaction (Eq. 3). In contrary, for the CaO2/Fe(III) system, it was observed that only trace Fe(II) was detected during the experiment and the Fe(II)/Fe(III) conversion was not clear in this condition. However, the addition of 1 mM benzene led to the different Fe(II)/Fe(III) conversion in both systems. For the CaO2/Fe(II) system, it was still observed a quick decline in the initial Fe(II) concentration, but a clear Fe(II) recovery then appeared in the first few minutes. The recovered Fe(II) obviously declined to a low level at 60 min, accompanied with a slight Fe(II) increase along the rest reaction period. For the CaO2/Fe(III) system, there was still a small amount of Fe(II) in the first 90 min, then the detected Fe(II) concentration increased to the maximum concentration and dropped to a low level accompanied by a gradual increase. This observed Fe(II)/Fe(III) conversion is the typical iron variation in Fenton-like systems and was also reported in previous studies [39,40].
| (3) |
| (4) |
| (5) |
| (6) |
As concluded in Table 1 [41,42], the reaction involved Fe(II) and H2O2 is the main HO● source regardless in Fenton or Fenton-like systems. In the CaO2/Fe(II) system, HO● was directly generated from the reaction between Fe(II) and H2O2, which then resulted in a quick benzene degradation. However, for the CaO2/Fe(III) system, the reaction between Fe(III) and H2O2 cannot directly produce HO●, it is the regenerated Fe(II) which reacted with H2O2 to produce HO●. Since the Fe(II) regeneration reaction rate (Eq. 4) was slower than that of the Eq. 3 (kFe(II), H2O2 = 7.6 M−1 s−1, kFe(III), H2O2 = 2.0 × 10−3 M−1 s−1) [26,27], it was observed a clear lag period before the obvious benzene degradation in the CaO2/Fe(III) system. When increasing the Fe(III) dose, the reaction between Fe(III) and H2O2 would be also facilitated and consequently accelerated the Fe(II) regeneration and HO● formation, leading to a shorter lag period (Fig. 1b). Although many studies reported that the H2O2-associated propagation reactions in classic Fenton or Fenton-like could cause the Fe(III) reduction [41–45], some reports [46–48] also mentioned that the HO2●− could be generated in the CaO2-based systems. This could have the potential to reduce Fe(III) to Fe(II) [48], but the results of the blank experiments indicated that the observed Fe(II) recovery was not owed to these pathways. With the addition of benzene, the clear Fe(II) recovery indicated that the benzene degradation reactions could bring additional reactions participated in Fe(III) reduction in both systems [45]. It is possible that some intermediate products during the benzene degradation could be responsible for Fe(II) recovery (Eq. 6), which were more efficient than the reduction of Fe(III) by H2O2-associated propagation reactions (Eqs. 4 ~ 5). The roles of intermediate products will be discussed in the later section to clarify their effects on the benzene degradation.
3.2.2. H2O2 variation in the CaO2/Fe(II) and CaO2/Fe(III) systems
The variation of H2O2 is an important factor in the studied systems and the temporal H2O2 changes in the CaO2/Fe(II) and CaO2/Fe(III) systems were shown in Fig.3.
Fig. 3.

The decomposition of H2O2 in a) the CaO2/Fe(II) system (control experiment: [CaO2] = [Fe(II)] = 10 mM, [Benzene]0 = 1.0 mM; blank experiment: [CaO2] = [Fe(II)] = 10 mM, [Benzene]0 = 0 mM), and b) the CaO2/Fe(III) system (control experiment: [CaO2] = [Fe(III)] = 10 mM, [Benzene]0 = 1.0 mM; blank experiment: [CaO2] = [Fe(III)] = 10 mM, [Benzene]0 = 0 mM). Initial pH = 3.0 ± 0.2.
As shown in Fig. 3a, for the CaO2/Fe(II) system, in the absence of benzene (the blank experiment), the H2O2 concentration peaked at 5 min and then dropped to a low concentration by 60 min. In the presence of benzene, the detected H2O2 would gradually increase to the maximum concentration by 60 min and then H2O2 would slowly and continuously decline in the remaining test period. As for the CaO2/Fe(III) system, regardless of the presence of benzene, high H2O2 concentrations were measured within the first 120 min, but the H2O2 concentration would steadily decline in the following experiment period in the presence of benzene (Fig. 3b).
The reaction with Fe(II) is the dominant H2O2 sink in the two systems because of the high reaction constant (kFe(II), H2O2 = 7.6 M−1 s−1, kFe(III), H2O2 = 2.0 × 10−3 M−1 s−1) [26,27], and the maximum H2O2 depletion should be observed when the obvious Fe(II) recovery was obtained. Therefore, the H2O2 variation, to some extent, was relevant to Fe(II) variation in the two systems. According to the Fe(II)/Fe(III) variations (Fig. 2), it was clear that the consumption of H2O2 was much faster in the CaO2/Fe(II) system, while H2O2 concentration was maintained at a high level for a much longer time in the CaO2/Fe(III) system. In addition, the presence of benzene could also promote the H2O2 consumption in both systems, which could be mainly ascribed to its influence to the Fe(II)/Fe(III) variations (Fig. 2). Based on the results, it could be concluded that the rapid H2O2 consumption in the CaO2/Fe(II) system could lead to a rapid pollutant degradation; while the CaO2/Fe(III) system could maintain its oxidation capacity for a longer time, which can be an advantage for minimizing the unexpected reagents loss during the injection and distribution process for subsurface remediation application.
3.2.3. HO● generation in the CaO2/Fe(II) and CaO2/Fe(III) systems
Since the Fe(II)/Fe(III) conversion and H2O2 decomposition patterns were different in the CaO2/Fe(II) and CaO2/Fe(III) systems, it could be inferred that the HO● generation pathways could be also different. The HO● variation was analyzed by the EPR instrument and the results were shown in Fig. 4.
Fig. 4.

EPR spectrums at a) the CaO2/Fe(II) system ([CaO2] = [Fe(II)] = 10 mM, [Benzene]0 = 1.0 mM), and b) the CaO2/Fe(III) system ([CaO2] = [Fe(III)] = 10 mM, [Benzene]0 = 1.0 mM). Initial pH = 3.0 ± 0.2.
Fig. 4a showed HO● variation during the benzene degradation in the CaO2/Fe(II) system. At the beginning, the HO● intensity was very high, and then the intensity decreased obviously. For the CaO2/Fe(II) system, due to the adaquate Fe(II), the reaction between Fe(II) and the released H2O2 could quickly occurred and produce excessive HO● in a short time leading to a high HO● intensity at 0.5 and 1 min. Due to the consequent Fe(II) exhaustion and lack of H2O2 (Eqs. 3 ~ 5), a notable decline in the HO● intensity was measured in the following experiment period (Fig. 4a). Conversely, owing to the gradual increase of H2O2 (Fig. 3a), the HO● intensity increased, presenting an enhancement at 30 min (Fig. 4a).
As for the CaO2/Fe(III) system, the reaction between Fe(III) and the released H2O2 could not directly produce HO●: Fe(III) should be reduced to Fe(II) and then it was the regenerated Fe(II) reacted with the released H2O2 producing HO● (Figs. 2b & 4b) [26,27,49]. Hence, in the initial 90 min, due to the low Fe(II) concentration and slow Fe(II) regeneration (Fig. 2b), the measured HO● intensity was weak at 30 and 90 min (Fig. 4b). Then with a notable increase of Fe(II) recovery, the HO● intensity was obviously promoted at 110 min (Figs. 2b & 4b). After the significant intensity enhancement, as a result of Fe(II) and H2O2 exhaustion, the detected HO● intensity at 130 min was similar to that detected before the enhancement. This significant increase of HO● intensity, together with the Fe(II)/Fe(III) conversion and H2O2 variation, reveals that some degradation intermediates could facilitate the HO● production.
3.2.4. Intermediates analysis and benzene degradation pathway in the CaO2/Fe(II) and CaO2/Fe(III) systems
The intermediate products in the CaO2/Fe(II) and CaO2/Fe(III) systems were analyzed with the HPLC and GC/MS. The analytical results showed that phenol, catechol, and benzoquinone were the major intermediates (Supplementary Material, Fig. S3), but their generation patterns were different in the investigated systems. It was clear that phenol was the primary degradation product in the the CaO2/Fe(II) and CaO2/Fe(III) systems, but the observed phenol variations were different. In the CaO2/Fe(II) system, it was observed that phenol sharply increased and declined within the first 10 min (Fig. 5a); in contrary, in the CaO2/Fe(III) system, phenol displayed a gradual increase and reached to its peak around 120 min (Fig. 5b). Along with the increase of phenol, benzoquinone and catechol also reached to their peaks in the two systems. Moreover, all the intermediates peak values were recorded during the fast benzene degradation period, and the benzene degradation pathways are then proposed based on the above experimental results (Fig. 6).
Fig. 5.

Benzene degradation products in a) the CaO2/Fe(II) system ([CaO2] = [Fe(II)] = 10 mM, [Benzene]0 = 1.0 mM; insert figure: peak area variations of benzoquinone and catechol), and b) the CaO2/Fe(III) system ([CaO2] = [Fe(III)] = 10 mM, [Benzene]0 = 1.0 mM; insert figure: peak area variations of benzoquinone and catechol). Initial pH = 3.0 ± 0.2.
Fig. 6.

Proposed benzene degradation pathways in the CaO2/Fe(II) and CaO2/Fe(III) systems ([CaO2] = [Fe(II)] or [Fe(III)] = 10 mM, [Benzene]0 = 1.0 mM). Initial pH = 3.0 ± 0.2.
As for benzene degradation in the CaO2/Fe(II) system, the released H2O2 would quickly react with Fe(II), producing excessive HO● in a short time. The produced HO● then immediately attacked benzene resulting in a rapid, significant decline in benzene while increase in phenol, which then would react with the surrounding HO● generating catechol and hydroquinone. Meanwhile, the generated catechol and hydroquinone could simultaneously reduce Fe(III) to Fe(II) (Eqs. 7 & 8), which could result in the notable Fe(II) increase and benzoquinone formation (Figs. 2a & 5a) [40,50–52]. All the intermediate products would competed for HO● with benzene, presenting clear declines (Fig. 5a). In regards to the CaO2/Fe(III) system, due to the low initial Fe(II) concentration and slow Fe(II) regeneration, the HO● was gradually generated (Figs. 2b & 4b), which limited the benzene degradation rate and resulted in a gentle phenol accumulation (Fig. 5b). With the increas of phenol, in addition to the initial benzene, phenol became another target compound for HO●, and resulted in the increase of catechol and hydroquinone [40,51]. Since catechol and hydroquinone have the ability to reduce the initial Fe(III) to Fe(II), Fe(II) recovery (Fig. 2b) and the subsequent HO● burst (Fig. 4b) along with the intermediates accumulation were then observed [52–54]. When the benzene degradation was completed, the intermediates variations also settled down. In addition, it was found that the HPLC spectrums of the degradation products in the CaO2/Fe(II) system could present the higher levels than that in the CaO2/Fe(III) system. The TOC analysis further confirmed that the CaO2/Fe(II) system produced less benzene mineralization than the CaO2/Fe(III) system (Supplementary Material, Fig. S4).
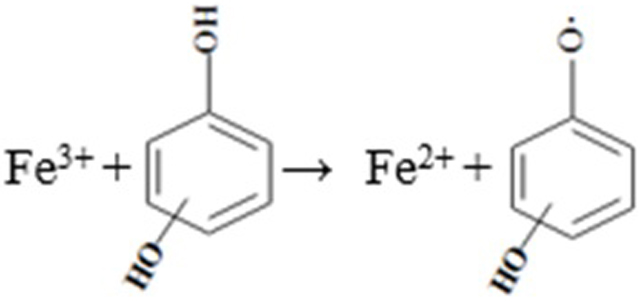 |
(7) |
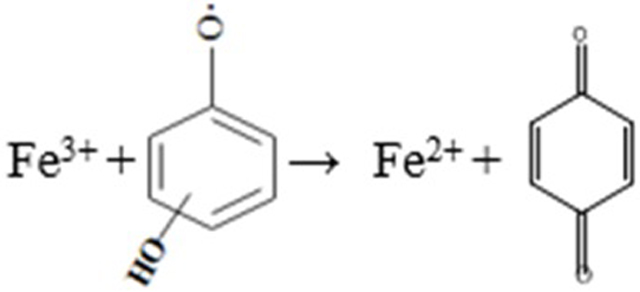 |
(8) |
3.3. An efficient strategy for employing the CaO2-based oxidation system
By comparing the benzene degradation performance in the two systems, it was easy to conclude that the CaO2/Fe(II) system can degrade benzene in a short time, while the CaO2/Fe(III) system can maintain its oxidation capacity for a relatively long time. In comparison to regulate the released H2O2, to manipulate the Fe(II)/Fe(III) conversion could be easier which also has a significant influence on the benzene degradation performance. Thus, we tested different combinations of Fe(II) and Fe(III), attempting to take the advantages of both systems and develop an efficient strategy for applying the CaO2-based oxidation system to groundwater remediation. Fig. 7a showed the benzene degradation results with different combinations of Fe(II) and Fe(III). With Fe(II) addition, it was observed that benzene was completely removed within 5 min regardless of Fe(II) and Fe(III) dosage. According to the previous analysis, the CaO2/Fe(III) system could hold the advantage in maintaining its stable oxidation capacity for a long-term and the lag time before finishing the benzene degradation could be very long. Hence, the addition of Fe(II) could be able to responsible for fast benzene degradation. Moreover, the result suggested that Fe(II) could possibly act as an accelerator to reduce the lag time in the CaO2/Fe(III) system and further experiments were conducted to clarify this hypothesis. 5.0 mM Fe(III) and 5.0 mM CaO2 were added in the reactor, then 1.0 mM Fe(II) was introduced at different times to test whether the benzene degradation could be accelerated, and the results have been shown in Fig. 7b. Before dosing Fe(II), the CaO2/Fe(III) system experienced very slow benzene degradation, however, after the introduction of Fe(II), the benzene degradation was promoted and quickly finished. The significant enhancements clearly indicated that Fe(II) succeeded in accelerating the benzene degradation and Fe(II) can be used as a complementary stimulant in the CaO2/Fe(III) system.
Fig. 7.

a) The influence of Fe(II) and Fe(III) ratio on benzene degradation ([CaO2] = 10 mM, [Benzene]0 = 1 mM), b) regulating benzene degradation with Fe(II) addition at different time ([CaO2] = [Fe(III)] = 5 mM, [Benzene]0 = 1 mM), and c) regulating TCE degradation with Fe(II) addition at different time ([CaO2] = [Fe(III)] = 0.75 mM, [Benzene]0 = 0.15 mM).
The combination of Fe(II) and Fe(III) took the advantages of the CaO2/Fe(II) and CaO2/Fe(III) systems and resulted in a better benzene remediation. TCE is a typical chloride solvent and has been widely used in industry for several decades. Due to its high toxicity and widespread occurrence in groundwater, TCE can bring a significant threat to human health and the natural environment, and has been concerned as priority pollutant in many countries [35,36]. Based on the benzene degradation results, we tested the above strategy in TCE remediation to verify its adaptability. The TCE treatment results with the above strategy was presented in Fig. 7c. TCE degradation was very slow with the CaO2/Fe(III) system alone, however, after dosing Fe(II), it was observed that TCE degradation was greatly enhanced and the degradation finishied as soon as Fe(II) was added. The different TCE degradation patterns indicated that the Fe(II) addition also succeeded in regulating the TCE degradation. The observed TCE decontamination trends were similar to that of the benzene degradation, which demonstrated that the proposed strategy has a good adaptability.
3.4. Implications for the application of the CaO2-based oxidation system
Although the efficiency of the CaO2-based oxidation system in groundwater treatment have been tested with the laboratory-scale experiments, there are still some problems that should be noticed. Unlike the laboratory conditions, the environmental conditions are hard to control and vary significantly in the actual sites. Among the condition parameters, pH is one key factor that should concern us when using the CaO2-based oxidation system. According to the Eqs. 1 ~ 2, the CaO2 dissolution process will unavoidably bring additional alkaline into the solution. Although the natural aquifer has a certain buffer capacity, the CaO2 dissolution can still elevate the solution pH [55,56]. Previous studies reported that the solution pH could be over 10 (even over 11) with the inappropriate molar ratios of CaO2/Fe(II) or CaO2/Fe(III) [20,22,55]. However, it was reported that the Fenton and Fenton-like reactions prefer the pH around 3 [24–27,39], and many documents reported that the elevated solution pH could constrain the performance of the CaO2-based oxidation system in pollutant remediation [17,20,55]. The iron could easily precipitate with the high solution pH, which could inhibit the iron recycle [26,27,39,57]. Particularly, for the CaO2-based oxidation system, the high solution pH would drive the CaO2 dissolution reaction from Eq. 2 to Eq. 1. O2 would replace H2O2 as the dominant product when pH > 10 [55,56], suppressing the subsequent H2O2-based decontamination performance. Besides, recent studies demonstrated that the CaO2-based oxidation system could produce O2●- in neutral pH [48,58], and the HO● yield in the would be greatly inhibited in the alkaline solution [58]. Therefore, the actual remediation process requires some proper conditioning reagents, such as the sulfuric acid [58], chelates [21–23], to attune the site condition, after which the CaO2-based oxidation reagents could be injected into the contaminated sites. Based on the above discussion, a model using the CaO2-based oxidation technique is proposed to remediate subsurface contamination (Supplementary Material, Fig. S5).
Since the CaO2/Fe(III) reagents can maintain their oxidation capacity for a relatively long time, its greater persistence compared with the CaO2/Fe(II) system means that migration distance from the injection well is anticipated to be longer, which results in the extent of larger treatment zones. This, in turn, reduces the number of wells and injection rounds required (thereby reducing costs), thus the CaO2/Fe(III) reagents can be used as an effective injection reagent for the subsurface remediation. Once the CaO2/Fe(III) reagents are injected and well distributed in the contaminated zone, the degradation process can be greatly accelerated by injecting a small amount of Fe(II). During the entire remediation process, the environmental conditions of the contaminated zone are required to be monitored, and the operating parameters need to be adjusted according to the real-time feedback. Moreover, the potential influence of the CaO2-based oxidation system on the subsurface ecosystem is another concern, which needs more investigations when applying this technique to the actual remediation.
4. Conclusions
The results of this study showed the CaO2/Fe(II) and CaO2/Fe(III) systems have the intrinsic relationship. The CaO2/Fe(II) would quickly convert to the CaO2/Fe(III) system during the remediation, while the CaO2/Fe(III) system need convert to the CaO2/Fe(II) system to promote the decontamination. The benzene degradation in the CaO2/Fe(II) system was much faster than that in the CaO2/Fe(III) system, whereas the CaO2/Fe(III) system maintained stable oxidation capacity for a longer time. To take the advantages of both systems, we proposed an effective strategy to treat benzene and TCE using the CaO2-based oxidation system. These encouraging results could provide new knowledge of more effective CaO2-based oxidation technique for subsurface remediation.
Supplementary Material
Acknowledgements
This study was financially supported by a grant from the International Academic Cooperation and Exchange Program of Shanghai Science and Technology Committee (18230722700). The contributions of Mark Brusseau were supported by the NIEHS Superfund Research Program of the United States (PS 42 ES04940).
Abbreviations:
- CaO2
calcium peroxide
- H2O2
hydrogen peroxide
- Fe(II)
ferrous ions
- Fe(III)
ferric ions
- CaO2/Fe(II)
CaO2-based Fenton system
- CaO2/Fe(III)
CaO2-based Fenton-like system
- ORC
oxygen releasing compound
- TCE
trichloroethene
- BTEX
benzene, toluene, ethylbenzene, and xylenes
- HO●
hydroxyl radicals
- HS-GC
headspace-gas chromatography
- EPR
electron paramagnetic resonance
- DMPO
5,5-Dimethyl-1-Pyrroline N-oxide
- FID
flame ionization detector
- GC/MS
gas chromatography/mass spectroscopy
- HPLC
high performance liquid chromatography
- ECD
electron capture detector
- R●
degradation intermediates
References
- [1].P.E.A. Focarino, United States Patent (19) SYS332, (1985).
- [2].Cassidy DP, Irvine RL, Use of calcium peroxide to provide oxygen for contaminant biodegradation in a saturated soil, J. Hazard. Mater 69 (1999) 25–39. doi: 10.1016/S0304-3894(99)00051-5. [DOI] [PubMed] [Google Scholar]
- [3].Liu SJ, Jiang B, Huang GQ, Li XG, Laboratory column study for remediation of MTBE-contaminated groundwater using a biological two-layer permeable barrier, Water Res. 40 (2006) 3401–3408. doi: 10.1016/j.watres.2006.07.015. [DOI] [PubMed] [Google Scholar]
- [4].Thani QA, Schaffer B, Liu G, Vargas AI, Crane JH, Chemical oxygen fertilization reduces stress and increases recovery and survival of flooded papaya (Carica papaya L.) plants, Sci. Hortic. (Amsterdam) 202 (2016) 173–183. doi: 10.1016/j.scienta.2016.03.004. [DOI] [Google Scholar]
- [5].Mosmeri H, Alaie E, Shavandi M, Dastgheib SMM, Tasharrofi S, Bioremediation of benzene from groundwater by calcium peroxide (CaO2) nanoparticles encapsulated in sodium alginate, J. Taiwan Inst. Chem. Eng 78 (2017) 299–306. doi: 10.1016/j.jtice.2017.06.020. [DOI] [Google Scholar]
- [6].Lu S, Zhang X, Xue Y, Application of calcium peroxide in water and soil treatment: A review, J. Hazard. Mater 337 (2017) 163–177. doi: 10.1016/j.jhazmat.2017.04.064. [DOI] [PubMed] [Google Scholar]
- [7].Qian Y, Zhou X, Zhang Y, Zhang W, Chen J, Performance and properties of nanoscale calcium peroxide for toluene removal, Chemosphere. 91 (2013) 717–723. doi: 10.1016/j.chemosphere.2013.01.049. [DOI] [PubMed] [Google Scholar]
- [8].Qian Y, Zhang J, Zhang Y, Chen J, Zhou X, Degradation of 2,4-dichlorophenol by nanoscale calcium peroxide: Implication for groundwater remediation, Sep. Purif. Technol 166 (2016) 222–229. doi: 10.1016/j.seppur.2016.04.010. [DOI] [Google Scholar]
- [9].Gholami F, Shavandi M, Mohammad S, Dastgheib M, Ali M, Chemosphere Naphthalene remediation form groundwater by calcium peroxide (CaO2) nanoparticles in permeable reactive barrier (PRB), Chemosphere. 212 (2018) 105–113. doi: 10.1016/j.chemosphere.2018.08.056. [DOI] [PubMed] [Google Scholar]
- [10].Izadifard M, Achari G, Langford CH, Mineralization of sulfolane in aqueous solutions by Ozone/CaO2 and Ozone/CaO with potential for field application, Chemosphere. 197 (2018) 535–540. doi: 10.1016/j.chemosphere.2018.01.072. [DOI] [PubMed] [Google Scholar]
- [11].Wu H, Sun L, Wang H, Wang X, In situ sodium persulfate/calcium peroxide oxidation in remediation of TPH-contaminated soil in 3D-sand box, Environ. Technol 39 (2018) 91–101. doi: 10.1080/09593330.2017.1296029. [DOI] [PubMed] [Google Scholar]
- [12].Qian Y, Zhou X, Zhang Y, Sun P, Zhang W, Chen J, Guo X, Performance of a methylnaphthalene degradation by dual oxidant of persulfate/calcium peroxide: Implication for ISCO, Chem. Eng. J 279 (2015) 538–546. doi: 10.1016/j.cej.2015.05.053. [DOI] [Google Scholar]
- [13].Kozak J, Makuła MW, Photo-oxidation of PAHs with calcium peroxide as a source of the hydroxyl radicals, E3S Web of Conferences, 02009 (2018) 1–8. [Google Scholar]
- [14].Morikawa CK, Generation of hydroxyl radicals by Fe-polyphenol-activated CaO2 as a potential treatment for soil-borne diseases, Sci. Rep 8 (2018) 9752. doi: 10.1038/s41598-018-28078-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang B, Pignatello JJ, Qu D, Xing B, Activation of hydrogen peroxide and solid peroxide reagents by phosphate ion in alkaline solution 33 (2016) 193–200. doi: 10.1089/ees.2015.0460. [DOI] [Google Scholar]
- [16].Zhang A, Wang J, Li Y, Performance of calcium peroxide for removal of endocrine-disrupting compounds in waste activated sludge and promotion of sludge solubilization, Water Res. 71 (2015) 125–139. doi: 10.1016/j.watres.2015.01.005. [DOI] [PubMed] [Google Scholar]
- [17].Zhang X, Gu X, Lu S, Miao Z, Xu M, Fu X, Qiu Z, Sui Q, Degradation of trichloroethylene in aqueous solution by calcium peroxide activated with ferrous ion, J. Hazard. Mater 284 (2015) 253–260. doi: 10.1016/j.jhazmat.2014.11.030. [DOI] [PubMed] [Google Scholar]
- [18].Chou ML, Jean JS, Yang CM, Hseu ZY, Chen YH, Wang HL, Das S, Chou LS, Inhibition of ethylenediaminetetraacetic acid ferric sodium salt (EDTA-Fe) and calcium peroxide (CaO2) on arsenic uptake by vegetables in arsenic-rich agricultural soil, J. Geochemical Explor. 163 (2016) 19–27. doi: 10.1016/j.gexplo.2016.01.004. [DOI] [Google Scholar]
- [19].Zhang X, Gu X, Lu S, Miao Z, Xu M, Fu X, Danish M, Brusseau ML, Qiu Z, Sui Q, Enhanced degradation of trichloroethene by calcium peroxide activated with Fe(III) in the presence of citric acid, Front. Environ. Sci. Eng 10 (2016) 502–512. doi: 10.1007/s11783-016-0838-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xue Y, Gu X, Lu S, Miao Z, Brusseau ML, Xu M, Fu X, Zhang X, Qiu Z, Sui Q, The destruction of benzene by calcium peroxide activated with Fe(II) in water, Chem. Eng. J 302 (2016) 187–193. doi: 10.1016/j.cej.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang X, Gu X, Lu S, Miao Z, Xu M, Fu X, Qiu Z, Sui Q, Application of calcium peroxide activated with Fe(II)-EDDS complex in trichloroethylene degradation, Chemosphere. 160 (2016) 1–6. doi: 10.1016/j.chemosphere.2016.06.067. [DOI] [PubMed] [Google Scholar]
- [22].Zhang X, Gu X, Lu S, Brusseau ML, Xu M, Fu X, Qiu Z, Sui Q, Application of ascorbic acid to enhance trichloroethene degradation by Fe(III)-activated calcium peroxide, Chem. Eng. J 325 (2017) 188–198. doi: 10.1016/j.cej.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xue Y, Lu S, Fu X, Sharma VK, Mendoza-Sanchez I, Qiu Z, Sui Q, Simultaneous removal of benzene, toluene, ethylbenzene and xylene (BTEX) by CaO2 based Fenton system: Enhanced degradation by chelating agents, Chem. Eng. J 331 (2018) 255–264. doi: 10.1016/j.cej.2017.08.099. [DOI] [Google Scholar]
- [24].Haber F, Weiss J, The catalytic decomposition of hydrogen peroxide by iron salts, Proc. R. Soc. Lond. A 147 (1934) 332–351. doi: 10.1098/rspa.1934.0221. [DOI] [Google Scholar]
- [25].Barb WG, Baxendale JH, George P, Hargrave KR, Reactions of ferrous and ferric ions with hydrogen peroxide, Nature. 163 (1949) 692–694. doi: 10.1038/163692a0. [DOI] [Google Scholar]
- [26].Walling C, Fenton’s reagent revisited, Acc. Chem. Res 8 (1975) 125–131. doi: 10.1021/ar50088a003. [DOI] [Google Scholar]
- [27].Duesterberg CK, Mylon SE, Waite TD, Duesterberg CK, pH effects on iron-catalyzed oxidation using Fenton’s reagent, 42 (2008) 8522–8527. doi: 10.1021/es801720d. [DOI] [PubMed] [Google Scholar]
- [28].Mosmeri H, Alaie E, Shavandi M, Dastghei SMM, Tasharrofi S, Bioremediation of benzene from groundwater by calcium peroxide (CaO2) nanoparticles encapsulated in sodium alginate, J. Taiwan Inst. Chem. E 78 (2017) 299–306. [Google Scholar]
- [29].Goi A, Viisimaa M, Trapido M, Munter R, Polychlorinated biphenyls-containing electrical insulating oil contaminated soil treatment with calcium and magnesium peroxides, Chemosphere. 82 (2011) 1196–1201. doi: 10.1016/j.chemosphere.2010.11.053. [DOI] [PubMed] [Google Scholar]
- [30].Goi A, Viisimaa M, Karpenko O, DDT-contaminated soil treatment with persulfate and hydrogen peroxide utilizing different activation aids and the chemicals combination with biosurfactant, J. Adv. Oxid. Technol 15 (2012) 41. doi: 10.1515/jaots-2012-0105. [DOI] [Google Scholar]
- [31].Xu J, Pancras T, Grotenhuis T, Chemical oxidation of cable insulating oil contaminated soil, Chemosphere. 84 (2011) 272–277. doi: 10.1016/j.chemosphere.2011.03.044. [DOI] [PubMed] [Google Scholar]
- [32].Badham HJ, Winn LM, In vivo exposure to hydroquinone during the early phase of collagen-induced arthritis aggravates the disease, Toxicology 229 (2007) 177–185. doi:org/ 10.1016/j.tox.2006.10.021. [DOI] [PubMed] [Google Scholar]
- [33].ATSDR, Toxicological profile for benzene, Agency Toxic Subst. Dis. Regist (2007). http://www.atsdr.cdc.gov/toxprofiles/tp3.pdf. [PubMed]
- [34].Haag WR, Rate constants for reaction of hydroxyl radicals with several drinking water contaminants, (1992) 1005–1013. doi: 10.1021/es00029a021. [DOI] [Google Scholar]
- [35].Moran MJ, Zogorski JS, Squillace PJ, Chlorinated solvents in groundwater of the United States, Environ. Sci. Technol 41 (2007) 74–81. doi: 10.1021/es061553y. [DOI] [PubMed] [Google Scholar]
- [36].ATSDR, Toxicological profile for trichloroethylene (TCE), Agency Toxic Subst. Dis. Regist (2014). https://www.atsdr.cdc.gov/ToxProfiles/tp19.pdf. [PubMed]
- [37].Eisenberg G, Colorimetric determination of hydrogen peroxide. Ind. Eng. Chem. Anal. Ed 15 (1943) 327–328. doi: 10.1021/i560117a011. [DOI] [Google Scholar]
- [38].Tamura H, Goto K, Yotsuyanagi T, Nagayama M, Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III), Talanta. 21 (1974) 314–318. doi: 10.1016/0039-9140(74)80012-3. [DOI] [PubMed] [Google Scholar]
- [39].Jiang C, Gao Z, Qu H, Li J, Wang X, Li P, Liu H, A new insight into Fenton and Fenton-like processes for water treatment: Part II. Influence of organic compounds on Fe(III)/Fe(II) interconversion and the course of reactions, J. Hazard. Mater 250–251 (2013) 76–81. doi: 10.1016/j.jhazmat.2013.01.055. [DOI] [PubMed] [Google Scholar]
- [40].Chen R, Pignatello JJ, Role of quinone intermediates as electron shuttles in fenton and photoassisted fenton oxidations of aromatic compounds, Environ. Sci. Technol 31 (1997) 2399–2406. doi: 10.1021/es9610646. [DOI] [Google Scholar]
- [41].Bokare AD, Choi W, Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes, J. Hazard. Mater 275 (2014) 121–135. doi: 10.1016/j.jhazmat.2014.04.054. [DOI] [PubMed] [Google Scholar]
- [42].De Laat J, Gallard H, Catalytic decomposition of hydrogen peroxide by Fe(III) in homogeneous aqueous solution: Mechanism and kinetic modeling, Environ. Sci. Technol 33 (1999) 2726–2732. doi: 10.1021/es981171v. [DOI] [Google Scholar]
- [43].Nichela D, Haddou M, Benoit-Marquié F, Maurette MT, Oliveros E, Einschlag FSG, Degradation kinetics of hydroxy and hydroxynitro derivatives of benzoic acid byfenton-like and photo-fenton techniques: A comparative study, Appl. Catal. B 98 (2010) 171–179. doi:org/ 10.1016/j.apcatb.2010.05.026. [DOI] [Google Scholar]
- [44].Tokumura M, Morito R, Hatayama R, Kawase Y, Iron redox cycling in hydroxyl radical generation during the photo-Fenton oxidative degradation: Dynamic change of hydroxyl radical concentration, Appl. Catal. B 106 (2011)565–576. doi:org/ 10.1016/j.apcatb.2011.06.017. [DOI] [Google Scholar]
- [45].Nichela DA, Donadelli JA, Caram BF, Haddou M, Nieto FJR, Oliveros E, Einschlag FSG, Iron cycling during the autocatalytic decomposition of benzoic acid derivatives by Fenton-like and photo-Fenton techniques, Appl. Catal. B 170–171 (2015) 312–321. doi:org/ 10.1016/j.apcatb.2015.01.028. [DOI] [Google Scholar]
- [46].Xue Y, Sui Q, Brusseau ML, Zhang X, Qiu Z, Lyu S, Insight on the generation of reactive oxygen species in the CaO2/Fe(II) Fenton system and the hydroxyl radical advancing strategy, Chem. Eng. J 353 (2018) 657–665. doi:org/ 10.1016/j.cej.2018.07.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Si Amina X, Wu K, Si Y, Yousaf B, Synergistic effects and mechanisms of hydroxyl radical-mediated oxidative degradation of sulfamethoxazole by Fe(II)-EDTA catalyzed calcium peroxide: Implications for remediation of antibiotic-contaminated water, Chem. Eng. J 353 (2018) 80–91. doi:org/ 10.1016/j.cej.2018.07.078. [DOI] [Google Scholar]
- [48].Pan Y, Su H, Zhu Y, Molamahmood HV, Long M, CaO2 based Fenton-like reaction at neutral pH: Accelerated reduction of ferric species and production of superoxide radicals, Water Res. 145 (2018) 731–740. doi:org/ 10.1016/j.watres.2018.09.020. [DOI] [PubMed] [Google Scholar]
- [49].Fu X, Gu X, Lu S, Sharma VK, Brusseau ML, Xue Y, Danish M, Fu GY, Qiu Z, Sui Q, Benzene oxidation by Fe(III)-activated percarbonate: matrix-constituent effects and degradation pathways, Chem. Eng. J 309 (2017) 22–29. doi: 10.1016/j.cej.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Santana-Casiano JM, González-Dávila M, González AG, Millero FJ, Fe(III) reduction in the presence of catechol in seawater, Aquat. Geochemistry 16 (2010) 467–482. doi: 10.1007/s10498-009-9088-x. [DOI] [Google Scholar]
- [51].Liu G, Huang H, Xie R, Feng Q, Fang R, Shu Y, Zhan Y, Ye X, Zhong C, Enhanced degradation of gaseous benzene by a Fenton reaction, RSC Adv. 7 (2017) 71–76. doi: 10.1039/c6ra26016k. [DOI] [Google Scholar]
- [52].Zhou W, Gao J, Zhao H, Meng X, Wu S,The role of quinone cycle in Fe2+-H2O2 system in the regeneration of Fe2+, Environ. Technol 38 (2017) 1887–1896. doi: 10.1080/09593330.2016.1240241. [DOI] [PubMed] [Google Scholar]
- [53].Wang S, A comparative study of Fenton and Fenton-like reaction kinetics in decolourisation of wastewater, Dye. Pigment 76 (2008) 714–720. doi: 10.1016/j.dyepig.2007.01.012. [DOI] [Google Scholar]
- [54].Jiang C, Pang S, Ouyang F, Ma J, Jiang J, A new insight into Fenton and Fenton-like processes for water treatment, J. Hazard. Mater 174 (2010) 813–817. doi: 10.1016/j.jhazmat.2009.09.125. [DOI] [PubMed] [Google Scholar]
- [55].Northup A, Cassidy D, Calcium peroxide (CaO2) for use in modified Fenton chemistry, J. Hazard. Mater 152 (2008) 1164–1170. doi: 10.1016/j.jhazmat.2007.07.096. [DOI] [PubMed] [Google Scholar]
- [56].Wang H, Zhao Y, Li T, Chen Z, Wang Y, Qin C, Properties of calcium peroxide for release of hydrogen peroxide and oxygen: A kinetics study, Chem. Eng. J 303 (2016) 450–457. doi: 10.1016/j.cej.2016.05.123. [DOI] [Google Scholar]
- [57].Valdés-Solís T, Valle-Vigón P, Álvarez S, Marbán G, Fuertes AB, Manganese ferrite nanoparticles synthesized through a nanocasting route as a highly active Fenton catalyst, Catal. Commun 8 (2007) 2037–2042. doi: 10.1016/j.catcom.2007.03.030. [DOI] [Google Scholar]
- [58].Xue Y, Qian S, Brusseau ML, Zhang X, Qiu Z, Lyu S, Insight on the generation of reactive oxygen species in the CaO2/Fe(II) Fenton system and the hydroxyl radical advancing strategy, Chem. Eng. J 353 (2018) 657–665. doi:org/ 10.1016/j.cej.2018.07.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.