

Abstract
Toll-like receptors (TLRs) expressed on both immune cells and hepatocytes recognize microbial danger signals and regulate immune responses. Previous studies showed that TLR9 and TLR2 mediate Propionibacterium acnes–induced sensitization to lipopolysaccharide-triggered acute liver injury in mice. Ligand-specific activation of TLR2 and TLR9 are dependent on the common TLR adaptor, myeloid differentiation factor 88 (MyD88). Here, we dissected the role of MyD88 in parenchymal and bone marrow (BM)–derived cells in liver sensitization. Using chimeric mice with green fluorescent protein–expressing BM cells, we identified that P. acnes–induced liver inflammatory foci are of BM origin. Chimeras with MyD88-deficient BM showed no inflammatory foci after P. acnes or TLR2+TLR9 challenge, suggesting that recruitment of inflammatory cells to the liver required MyD88 expression in BM-derived cells. Further, selective MyD88 deficiency in parenchymal cells in mice with wild-type BM failed to prevent inflammatory cell infiltration. These results demonstrate that MyD88 in immune cells rather than in liver parenchymal cells plays an important role in inflammatory cell recruitment and liver injury.
Multiple conditions can cause sensitization to endotoxin-induced liver injury including drugs, toxins, metabolic factors, and patho-gens.1 Microbial danger signals are recognized by Toll-like receptors (TLRs) resulting in immune activation.2 Of the 10 human TLRs in humans all are expressed in the liver at the messenger RNA level and functional activity of most TLRs was found in the various parenchymal and nonparenchymal liver cell populations.3,4 Ligand recognition of TLRs triggers downstream signaling that is initiated by recruitment of adaptor molecules to the intracellular TIR domain of TLRs. Except for TLR3, all TLRs utilize the myeloid differentiation factor 88 (MyD88) adaptor that, depending on the TLR, results in downstream activation of proinflammatory mediators or Type I interferon (IFN).5,6 MyD88 is expressed in most cell types, and it has been shown to play a role in liver homeostasis both in a TLR-dependent and TLR-independent manner.7,8 Activation of TLRs has been demonstrated in liver diseases such as alcoholic liver disease, ischemia-reperfusion injury, viral hepatitis, biliary disorders, and primary biliary cirrhosis.1–8
Propionibacterium acnes, a Gram-positive bacterium, is a member of the normal skin and intestinal flora that causes inflammatory skin diseases in humans and results in granulomas and sensitization to lipopolysaccharide (LPS)–induced liver failure in mice.9 Previous studies demonstrated that the immunomodulatory effects of P. acnes are mediated by TLR2 and TLR9 activation.10,11 In a previous study, we found that administration of selective TLR9 or TLR2+TLR9 ligands results in both liver granulomas and sensitization to LPS-induced liver injury.12 Further, immune cells account for the majority of the cell population in granulomas, and increased expression of chemokines in the liver contributes to the inflammatory cell recruitment.9,13 However, the cell-type specificity of the TLRs in the process of liver granuloma induction is yet to be delineated. In this study, we aimed first, to determine whether parenchymal or bone marrow (BM)–derived cells participated in the formation of liver granulomas, and second, to evaluate the utilization of the MyD88 TLR adaptor in these cell types.
Materials and Methods
Animals and Experimental Protocol.
Four-week-old to 6-week-old female mice (Table 1) were employed; all animals received proper care in agreement with animal protocols approved by the Institutional Animal Use and Care Committee of the University of Massachusetts Medical School. Mice were injected intraperitoneally with either 0.2 mL 0.9% saline or 0.2 mL 0.9% saline containing combination of 2.5 mg/kg CpG oligonucleotide (ODN1826; InvivoGen, San Diego, CA) and 5 mg/kg lipoteichoic acid (Sigma, St. Louis, MO), or 1 mg heat-killed P. acnes (Van Kampen Group, Hoover, AL). At 3 or 7 days after the above priming stimuli, mice were intraperitoneally administered saline or 0.5 mg/kg lipopolysaccharide (Sigma, St. Louis, MO) and were sacrificed 1.5 hours later.12 There were 3–5 mice in each experimental group.
Table 1.
Annotation of Mouse Chimera Models
| Annotation | WT/WT-GFP+-BM | WT/MyD88−/−-BM | MyD88−/−/WT-BM |
|---|---|---|---|
| Host | WT | WT | MyD88−/− |
| Bone marrow | WT-GFP | MyD88−/− | WT |
Bone Marrow Transplantation.
To induce BM aplasia, mice were irradiated with 900 rads from a 137Cs irradiator. Four hours later, mice were reconstituted with 5 million BM cells via a single tail vein injection. Mice were housed in microisolator cages on conventional racks during the engraftment period, were fed standard mouse chow, and received prophylactic Bactrim in drinking water for days 1–14.
Histopathological Analysis.
Sections of formalin-fixed, paraffin-embedded livers were stained with hematoxylin and eosin (H&E), and assessed for granuloma formation and necroinflammation by a pathologist blinded to the treatment groups.
Serum Liver Enzymes and Cytokines.
Serum alanine aminotransferase (ALT) was determined using a kinetic method (ALT Liquid; Advanced Diagnostics Inc., South Plainfield, NJ). Serum tumor necrosis factor-α(TNFα) and IFNγ levels were determined using specific BD OptEIA enzyme-linked immunosorbent assay kits (BD Biosciences Pharmingen, San Diego, CA).
Immunofluorescence.
The formalin-fixed paraffin-embedded tissue sections were de-waxed and rehydrated, and the antigen retrieval was carried out in citrate buffer pH 6.0 by microwave heat treatment (5 minutes at 750 W). After cooling, washing, and nonspecific blocking with 1% bovine serum albumin–phosphate-buffered saline solution for 10 minutes, the samples were stained with rabbit polyclonal anti-green fluorescent protein (GFP) antibody (Abcam, Cambridge, MA) (1:2000) followed by rhodamine-conjugated goat anti-rabbit immunoglobulin (Chemicon, CA) (1:500) and with 4′−6-diamidino-2-phenylindole (DAPI); the fluorescence (20 fields/sample) was then analyzed.
Western Blot Analysis.
Whole-cell lysates were extracted from liver as previously described.11 The protein of interest was detected by immunostaining with specific primary antibody against GFP (Abcam, Cambridge, MA) followed by horseradish peroxidase–labeled secondary antibody using chemiluminescence as read-out. The immu-noreactive bands were quantified by densitometric analysis using an UVP System (Fuji).
Statistical Analysis.
Statistical significance was determined using the nonparametric Kruskal-Wallis test followed by the Mann-Whitney test. Data are presented as mean ± standard error of the mean, and were considered statistically significant at P < 0.05.
Results
Sensitization of mice with heat-killed P. acnes induces inflammatory cell foci (non-caseating granuloma-like lesions) in the liver and sensitizes to LPS-induced liver injury.12–14 Here, we employed wild-type (WT) chimera mice engrafted with WT GFP-positive BM (WT/WT-GFP+-BM) to differentiate between the impacts of BM-derived and parenchymal cells in a P. acnes–triggered sensitization model for endotoxin-induced liver injury. LPS stimulation resulted in a moderate recruitment of GFP-positive BM-derived cells in the livers of control, saline-primed mice (Fig. 1A), whereas LPS administration in the P. acnes–primed mice leads to massive accumulation of GFP-positive BM-derived cells (Fig. 1B). The distribution of the GFP+ BM-derived cells in the P. acnes–primed mice showed inflammatory foci consistent with liver granulomas.11 Evaluation of the total liver protein confirmed the substantially stronger recruitment of GFP-expressing cells into the liver in P. acnes–challenged mice compared to the saline control (Fig. 1C). These data suggested that BM-derived cells contribute to the inflammatory infiltrates in the liver.
Fig. 1.
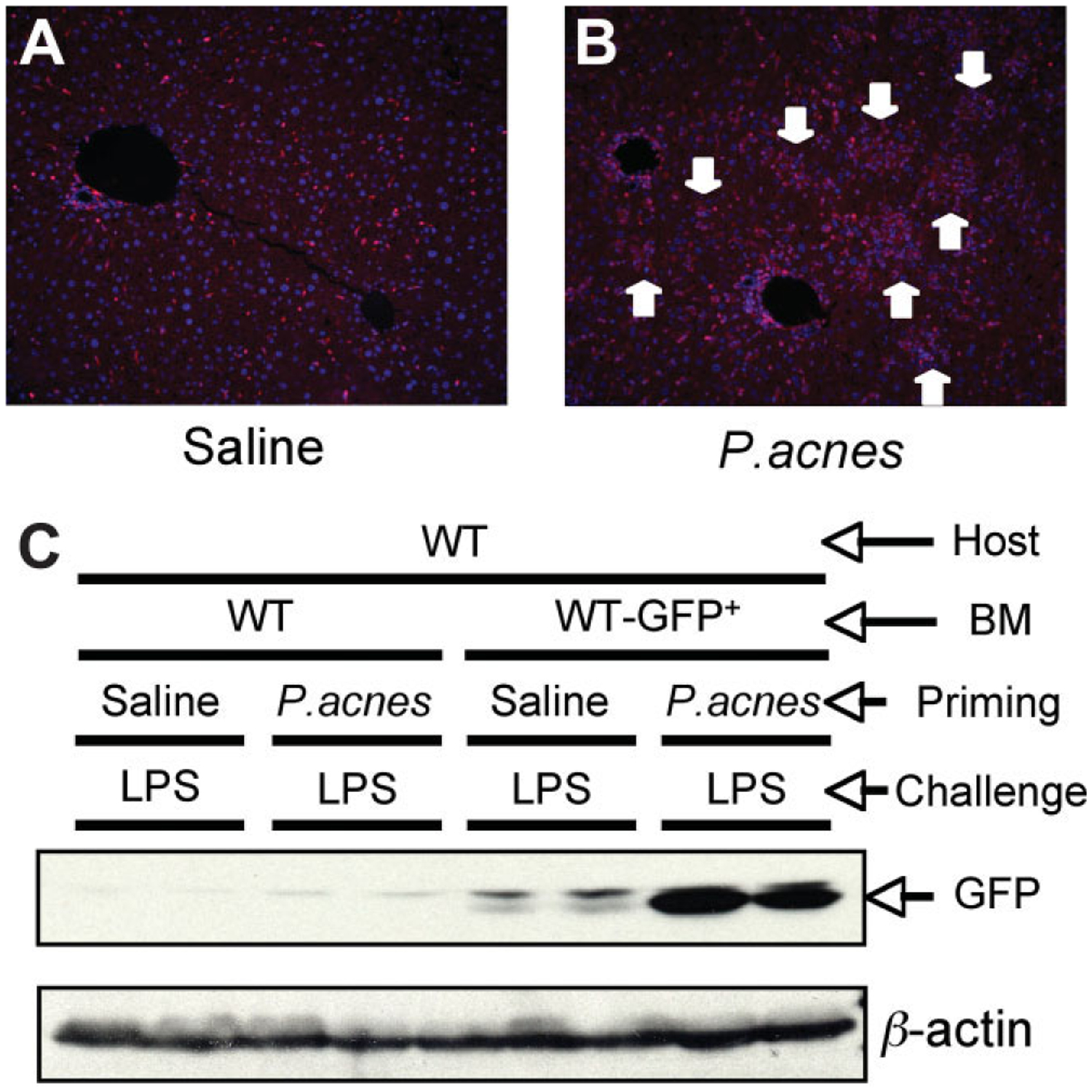
Bone marrow-derived cells are recruited into the liver after P. acnes priming. Wild-type mice transplanted either with wild-type (WT/WT) or wild-type GFP-labeled bone marrow (WT/WT-GFP+-BM) (3–5 per group) were injected intraperitoneally with 0.2 mL saline or 0.2 mL saline containing 1 mg heat-killed P. acnes. Mice were challenged 7 days later with 0.5 mg/kg body weight LPS administered intraperitoneally and mice were sacrificed 1.5 hours later. Sections of formalin-fixed, paraffin-embedded livers were stained with anti-GFP–rhodamine and DAPI for nuclear material and analyzed for immunofluorescence (IF). (A,B). One representative IF section of WT/WT-GFP+-BM after (A) saline plus LPS or (B) P. acnes plus LPS stimulation is shown out of n = 5 with similar results; the nuclei are shown in blue, the GFP is shown in red for color contrast, the arrows point to granulomas. (C) GFP protein expression in liver tissue was analyzed by Western blot analysis (C) and β-actin was used as loading control.
We have recently demonstrated that liver granulomas and sensitization to LPS can be induced in mice by priming with a selective TLR9 ligand and these effects are amplified by coadministration of a selective TLR2 ligand.12 After ligand engagement, both TLR2 and TLR9 utilize the MyD88 adaptor through their intracellular TIR domain to induce downstream signaling that leads to the biological effects of TLR activation.5 Indeed, mice deficient of the MyD88 adaptor, P. acnes, failed to induce granulomas and/or sensitization to LPS-induced liver injury.12 Here, we hypothesized that cell-type specificity of MyD88 expression was required for liver granuloma formation. To address this hypothesis, we employed chimeras with WT parenchymal cells and MyD88-deficient BM (WT/MyD88−/−-BM) and reverse chimeras, with MyD88-deficient mice reconstituted with WT BM (MyD88−/−/WT-BM). As illustrated in Fig. 2, liver granulomas (Fig. 2A,B) and increased serum ALT (Fig. 2C) were induced with administration of TLR2+TLR9 ligands only in chimeras with WT BM, similar to the P. acnes–primed WT positive controls. Selective MyD88 deficiency in the BM-derived cells, but not in liver parenchymal cells, prevented granuloma formation (Fig. 2A,B) as well as induction of liver injury as indicated by increased ALT (Fig. 2C). These results suggested that inflammatory response to the danger signal provided by TLR2+TLR9 ligands occurred at the level of BM-derived cells rather than in liver parencymal cells. Further-more, host cells of non-BM origin, including liver parenchymal cells, appeared to have little if any modulatory effect on the inflammatory cell recruitment and liver injury in this model of liver sensitization.
Fig. 2.
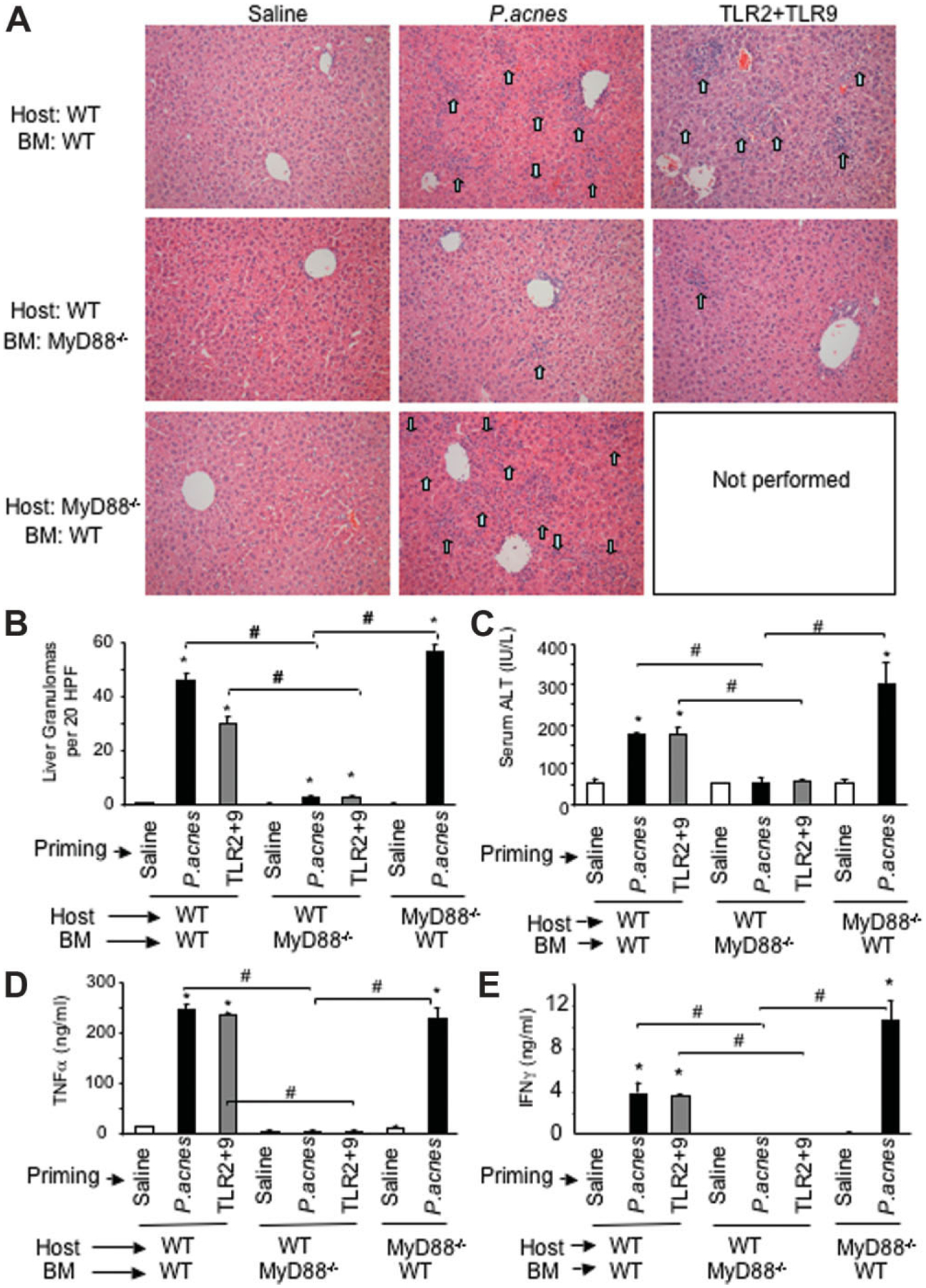
Granuloma formation, liver damage, and cytokine induction are dependent on MyD88 expression in BM-derived cells. Wild-type mice transplanted with wild-type or MyD88-deficient bone marrow (WT/WT-BM and WT/MyD88−/−-BM, respectively) or MyD88-deficient mice transplanted with wild-type bone marrow (MyD88−/−/WT-BM) were injected intraperitoneally with 0.2 mL saline, 0.2 mL saline containing 1 mg heat-killed P. acnes, or combination of a TLR2 (5 mg/kg lipoteichoic acid) and a TLR9 ligand (2.5 mg/kg CpG DNA), as indicated (n = 3–5 mice in each experimental group). Three days after TLR2+TLR9 ligands or 7 days after P. acnes priming, mice were administered intraperitoneally 0.5 mg/kg LPS and were sacrificed 1.5 hours later. (A) Sections of formalin-fixed, paraffin-embedded livers were stained with H&E; representative histology sections are shown for each experimental group; the arrows point to granulomas. (B,C) Average number of liver granulomas per 20 high-power fields (B) and mean serum ALT levels ± standard error of the mean (C) is shown. (D,E) Serum levels of (D) TNFα and (E) IFNγ were analyzed by enzyme-linked immunosorbent assay. In (B) through (E), asterisk (*) indicates P < 0.05 in saline primed versus P. acnes or TLR2+TLR9 ligands–primed groups; the # indicates P < 0.05 as defined.
Accumulation of inflammatory cell foci upon endotoxin-induced liver injury in the P. acnes-primed or TLR2+TLR9-primed liver is associated with increased expression of cytokines and chemokines.11,15 In contrast to the increased serum TNFα levels in WT/WT-BM mice, the WT/MyD88−/−-BM chimeras had negligible TNFα levels after P. acnes or TLR2+TLR9 priming and LPS challenge (Fig. 2D). Importantly, MyD88−/−/WT-BM chimera mice showed TNFα levels similar to the WT/WT-BM controls, suggesting that priming for TNFα production was solely mediated by BM-derived cells (Fig. 2D).
Discussion
Inflammation is a key component of a wide range of liver diseases, including steatohepatitis (SH) of alcoholic (ASH) and nonalcoholic (NASH) origin as well as acute liver failure due to systemic infections. The “two hits” hypothesis was proposed as culprit in the pathogenesis of these diseases, where liver sensitization precedes the endotoxemia, which in turn precipitates the clinical course of the disease. Until recently, the mechanisms of liver sensitization (first hit) and the role of the immune system during endotoxemia (second hit) were largely unknown. The discovery of TLRs allows dissection of the input of different cellular compartments (parenchymal, immune) in liver inflammation.
Here, we report for the first time that expression of the pathogen-sensing machinery in the immune cells is key to liver sensitization (development of the first hit). We employed a chimera mouse system and a liver sensitization model based on administration of P. acnes or TLR2+TLR9 ligands to show that BM-derived immune cells, rather than parenchymal liver cells, contribute to damage of the sensitized liver in response to endotoxin.
Previous studies demonstrated that in P. acnes–primed mice, LPS challenge results in a massive induction of proinflammatory cytokines.11–15 This priming effect of P. acnes was attributed to activation of a Th1-type immune response with predominant IFNγ, interleukin-12 (IL-12), and IL-18 induction.13,16 Consistent with this, we found no induction of IFNγ (Fig. 2E) and IL-12 (data not shown) in chimeras with MyD88-deficient BM (WT/Myd88−/−-BM) whereas high serum levels of IFNγ and IL-12 were found in WT/WT-BM and in MyD88−/−/WT-BM chimeras. Taken together, these results suggest that BM-derived immune cells have a major role in sensitization to liver injury. This involves recognition of pathogen-derived signals though TLRs and induction of inflammatory mediators that can amplify inflammatory cell recruitment to the liver resulting in sensitization to liver injury caused by subsequent insults, such as LPS. Other studies demonstrated a critical role for MyD88 in liver regeneration,7,8 however, the particular cell population responsible for this effect remains to be defined. Interestingly, inflammatory cytokine induction and liver regeneration after partial hepatectomy was impaired in MyD88-deficient mice but TLR4, CD14, or TLR2 were not required for optimal liver regeneration.8 Another study found that the MyD88-dependence was not linked to TLR2-induced or TLR9-induced activation during liver regeneration.7 Functionally active TLR9 was also found in biliary epithelial cells and sinusoidal endothelial cells in the liver.17,18 Interestingly, in primary biliary cirrhosis, where increased frequency of liver granulomas was noted, TLR9 activation has been shown to play a central role in the maintenance of autoimmune stimulation.19 In the lung, TLR9 activation was also associated with mycobacterial antigen-associated granulomas.20 Although TLR2 and TLR9 are expressed in various parenchymal cells in the liver, our results with the MyD88-deficient chimeras suggest that BM-derived rather than parenchymal cells play a role in liver granuloma formation and injury. Thus, TLR-induced inflammatory cell accumulation and activation can have a major impact on liver injury of various etiologies.
The relevance of the immune cells and MyD88-dependent signaling in sensitization is not limited to endotoxin (LPS)-induced liver injury. Although increased serum endotoxin has been implicated in the pathogenesis of various liver diseases, including alcoholic liver disease and most recently, hepatitis associated with hepatitis C virus and human immunodeficiency virus infection,21–23 sensitization from a first hit in the liver may extend to various second-hit insults from either pathogen or host origin. For example, TLR4, the receptor for LPS, also recognizes host-derived danger signals such as heat shock proteins and high mobility group box-1 (HMGB1) in sterile inflammation such as autoimmunity, cancer trauma, or ischemia-reperfusion injury.24
Our results also point to new possibilities in therapy and/or management of liver diseases. Therapeutic manipulation of TLRs is a common practice in basic research that awaits bench-to-bed translation.4,25 Our findings that immune, rather than parenchymal cells, play a role during the first hit in liver injury may also provide a lever in TLR-based therapeutic interventions due to the shorter life span and higher turnover of the immune cells compared to hepatocytes. Finally, immunomodulation may become a viable option for prevention and/or modulation of the events responsible for the first hit once we gather more specifics of pathogen-derived or host-derived ligands and their role in immune activation to account for the first hit in vivo in humans.
Abbreviations:
- ALT
alanine aminotransferase
- BM
bone marrow
- b.w.
body weight
- IFN
interferon
- IL
interleukin
- KO
knockout
- LPS
lipopolysaccharide
- MyD88
myeloid differentiation factor 88
- TLR
Toll-like receptor
Footnotes
Potential conflict of interest: Nothing to report.
References
- 1.Williams R. Classification, etiology, and considerations of outcome in liver failure. Semin Liver Dis 1996;16:343–348. [DOI] [PubMed] [Google Scholar]
- 2.Pasare C, Medzhitov R. Toll-like receptors: linking innate and adaptive immunity. Adv Exp Med Biol 2005;560:11–18. [DOI] [PubMed] [Google Scholar]
- 3.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 2002; 168:554–561. [DOI] [PubMed] [Google Scholar]
- 4.Szabo G, Dolganiuc A, Mandrekar P. Pattern recognition receptors: a contemporary view on liver diseases. HEPATOLOGY 2006;44:287–298. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. TLR signaling. Semin Immunol 2007;19:24–32. [DOI] [PubMed] [Google Scholar]
- 6.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nature Rev Immunol 2007;7:353–364. [DOI] [PubMed] [Google Scholar]
- 7.Seki E, Tsutsui H, Iimuro Y, Naka T, Son G, Akira S, et al. Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. HEPATOLOGY 2005;41:443–450. [DOI] [PubMed] [Google Scholar]
- 8.Campbell JS, Riehle KJ, Brooling JT, Bauer RL, Mitchell C, Fausto N. Proinflammatory cytokine production in liver regeneration is Myd88-dependent, but independent of CD14, TLR2, and TLR4. J Immunol 2006; 176:2522–2528. [DOI] [PubMed] [Google Scholar]
- 9.Chen YL, Yu CK, Lei HY. Propionibacterium acnes induces acute TNFalpha-mediated apoptosis of hepatocytes followed by inflammatory T-cell-mediated granulomatous hepatitis in mice. J Biomed Sci 1999;6:349–356. [DOI] [PubMed] [Google Scholar]
- 10.Kalis C, Gumenscheimer M, Freudenberg N, Tchaptchet S, Fejer G, Heit A, et al. Requirement for TLR9 in the immunomodulatory activity of Propionibacterium acnes. J Immunol 2005;174:4295–4300. [DOI] [PubMed] [Google Scholar]
- 11.Romics L Jr, Dolganiuc A, Velayudham A, Kodys K, Mandrekar P, Golenbock D, et al. Toll-like receptor 2 mediates inflammatory cytokine induction but not sensitization for liver injury by Propionibacterium acnes. J Leukoc Biol 2005;78:1255–1264. [DOI] [PubMed] [Google Scholar]
- 12.Velayudham A, Hritz I, Dolganiuc A, Mandrekar P, Kurt-Jones E, Szabo G. Critical role of toll-like receptors and the common TLR adaptor, MyD88, in induction of granulomas and liver injury. J Hepatol 2006;45: 813–824. [DOI] [PubMed] [Google Scholar]
- 13.Tsuji H, Mukaida N, Harada A, Kaneko S, Matsushita E, Nakanuma Y, et al. Alleviation of lipopolysaccharide-induced acute liver injury in Propionibacterium acnes-primed IFN-gamma-deficient mice by a concomitant reduction of TNF-alpha, IL-12, and IL-18 production. J Immunol 1999; 162:1049–1055. [PubMed] [Google Scholar]
- 14.Shimizu Y, Margenthaler JA, Landeros K, Otomo N, Doherty G, Flye MW. The resistance of P. acnes–primed interferon gamma-deficient mice to low-dose lipopolysaccharide-induced acute liver injury. HEPATOLOGY 2002;35:805–814. [DOI] [PubMed] [Google Scholar]
- 15.Romics L Jr, Dolganiuc A, Kodys K, Drechsler Y, Oak S, Velayudham A, et al. Selective priming to Toll-like receptor 4 (TLR4), not TLR2, ligands by P. acnes involves up-regulation of MD-2. HEPATOLOGY 2004;40:555–564. [DOI] [PubMed] [Google Scholar]
- 16.Iizasa H, Yoneyama H, Mukaida N, Katakoka Y, Naito M, Yoshida N, et al. Exacerbation of granuloma formation in IL-1 receptor antagonist-deficient mice with impaired dendritic cell maturation associated with Th2 cytokine production. J Immunol 2005;174:3273–3280. [DOI] [PubMed] [Google Scholar]
- 17.Karrar A, Broome U, Sodergren T, Jaksch M, Bergquist A, Bjornstedt M, et al. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology 2007;132: 1504–1514. [DOI] [PubMed] [Google Scholar]
- 18.Martin-Armas M, Simon-Santamaria J, Pettersen I, Moens U, Smedsrod B, Sveinbjornsson B. Toll-like receptor 9 (TLR9) is present in murine liver sinusoidal endothelial cells (LSECs) and mediates the effect of CpG-oligonucleotides. J Hepatol 2006;44:939–946. [DOI] [PubMed] [Google Scholar]
- 19.Moritoki Y, Lian ZX, Wulff H, Yang GX, Chuang YH, Lan RY, et al. AMA production in primary biliary cirrhosis is promoted by the TLR9 ligand CpG and suppressed by potassium channel blockers. HEPATOLOGY 2007;45:314–322. [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Schaller M, Hogaboam CM, Standiford TJ, Chensue SW, Kunkel SL. TLR9 activation is a key event for the maintenance of a mycobacterial antigen-elicited pulmonary granulomatous response. Eur J Immunol 2007;37:2847–2855. [DOI] [PubMed] [Google Scholar]
- 21.Mathurin P, Deng QG, Keshavarzian A, Choudhary S, Holmes EW, Tsukamoto H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. HEPATOLOGY 2000;32:1008–1017. [DOI] [PubMed] [Google Scholar]
- 22.Dolganiuc A, Norkina O, Kodys K, Catalano D, Bakis G, Marshall C, et al. Viral and host factors induce macrophage activation and loss of toll-like receptor tolerance in chronic HCV infection. Gastroenterology 2007;133: 1627–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balagopal A, Philp FH, Astemborski J, Block TM, Mehta A, Long R, et al. Human immunodeficiency virus-related microbial translocation and progression of hepatitis C. Gastroenterology 2008;135:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A. HMGB1: endogenous danger signaling. Mol Med 2008;14:476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barrat FJ, Coffman RL. Development of TLR inhibitors for the treatment of autoimmune diseases. Immunol Rev. 2008;223:271–283. [DOI] [PubMed] [Google Scholar]