

Abstract
Increasing evidence has implicated metabolic pathways as key regulators of cell fate and function. While the metabolism of glucose, amino acids and fatty acids are essential to maintain overall energy homeostasis, the choice of a given metabolic pathways and the levels of particular substrates and intermediates increasingly appears to modulate specific cellular activities. This connection is likely related to the growing appreciation that molecules such as acetyl-CoA act as a shared currency between metabolic flux and chromatin modification. Here, we review recent evidence of a role for metabolism to modulate cellular function in four distinct contexts. These areas include the immune system, the tumor microenvironment, the fibrotic response and the governance of stem cell function. Together, these examples suggest that metabolic pathways do not simply provide the fuel that powers cellular activities but instead, help to shape and determine cellular identity.
Introduction
“Tell me what you eat and I will tell you what you are”
Jean Anthelme Brillat-Savarin, 1826
The internet and magazines are filled with a dazzling array of metabolic intermediates and byproducts whose consumption would appear to cure all that ills us, and in some cases, solve problems we did not even know existed. Indeed, with regards to our own health, perhaps nothing holds the public imagination more than the notion that what we consume determines our well-being. Remarkably, the translation of the notion, ‘you are what you eat’, has been slow to permeate our understanding of intracellular metabolism. For many years, the menu of substrates a cell could consume (e.g. glucose, fatty acids, etc.) were all viewed as essentially indistinguishable. In this framework, the only function of the complex and interconnected web of metabolism was to generate sufficient cellular ATP. That view, however, has substantially changed over the last decade and what has replaced this bioenergtic centered model is a much more nuanced understanding of cellular metabolism. The growing realization that histone modification relies on the by-products of one-carbon metabolism and acetyl-CoA production, and that metabolic intermediaries such as α-ketoglutarate act as regulators of chromatin modifying enzymes, suggested a tight coupling between metabolic flux and cellular fate. Here, we review the recent exploration of this concept in four related but discrete areas. First, we briefly discuss what is perhaps the best-studied example, namely the role that metabolism plays in shaping the immune response. Next, we examine the harsh world of the tumor microenvironment where the competition for substrates limits the body’s defenses and where understanding the metabolic interplay between tumor and stroma might lead to therapeutic advances. We then turn to how metabolism might regulate the processes of fibrosis, a condition linked to the pathology of many chronic disease. Finally, we discuss recent insights into how metabolism can regulate the fate of stem and progenitor cells and their lineage specification. Together, these recent advances demonstrate the recurring theme that metabolism provides not only the energy to run the cell but also the critical determinants of cell fate.
Immunometabolism
There is a growing appreciation that the highly specialized function of various immune cells is intimately linked to their underlying metabolism. This burgeoning field of immunometabolism has been the subject of several excellent reviews1, 2. In brief, perturbations of anaerobic glycolysis, fatty acid synthesis, fatty acid oxidation, glutaminolysis and mitochondrial oxidative phosphorylation has been increasingly used to understand how metabolism is coupled to the function of T cells, macrophages and NK cells. While these approaches initially relied on pharmacological perturbations of metabolism, increasing genetic models are being employed. This genetic approach has sometimes led to a re-evaluation of the precise role that specific metabolic pathways play in immune function3. While the majority of studies have focused on major pathways such as glycolysis and fatty acid oxidation, the influence of metabolism likely extends beyond these core pathways. In particular, the regulation of S-adenosyl methionine (SAM), which serves as the primary methyl donor for histone modification, is likely to be a central node connecting metabolic flux with immune function4. The generation of SAM involves the complicated web of interconnected pathways known as one-carbon metabolism. This involves both the methionine and folate cycles, the former of which is linked to glutathione production and the maintenance of redox homeostasis. One of the major carbon donors to this pathway is the non-essential amino acid serine. In this regard, genes involved in serine metabolism are markedly up-regulated when CD8+ T effector cells are stimulated5. This upregulation appears to be required for T cell expansion, not due solely to bioenergetic needs, but rather, as a requirement to meet a surge in nucleotide synthesis5. Interestingly, mice fed a serine/glycine free diet had impaired in vivo T cell-mediated immunity5. Defects in this pathway have also been recently linked to the inevitable, age-dependent known decline in T cell activation6.
The conversion of serine to glycine is catalyzed by the enzyme serine hydroxymethyltransferase (SHMT), with SHMT1 localized to the cytosol and SHMT2 residing in the mitochondria. Enzymatic activation of these enzymes requires binding of the active form of vitamin B6, pyridoxal-5’-phosphate (PLP) and the formation of a tetramer7. Of note, besides their role in metabolism, SHMT proteins are found in association with BRCC36, a deubiquitinating enzyme (DUB) that regulates immune signaling8. BRCC36 is actually part of a multi-protein complex termed BRISC (the BRCC36 isopeptidase complex), which regulates the ubiquitination and hence levels of interferon receptors8. A recent cryo-electron microscopic structure of the BRISC-SHMT2 complex has been performed9. This demonstrated that inactive dimers of SHMT2 bind and inactivate BRISC activity while active tetramers do not. Levels of PLP regulate the interaction of SHMT2 and BRISC, and this metabolite in turn regulates inflammatory signaling9. As such, SHMT adds to a growing list of metabolic enzymes including pyruvate kinase M2, aldolase and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) that appear to have additional, nonmetabolic and non-cannonical functions10. These additional functions include metabolic enzymes moonlighting to regulate transcription, cytoskeletal dynamics, and, as in the case of SHMT, modulating intracellular signaling. It is likely that additional dual functioning enzymes will be discovered as they provide a structural mechanism to directly link metabolism to energy consuming intracellular functions, much as SHMT ties one-carbon metabolism to interferon signaling. Interestingly, this is unlikely to be the only metabolic control over this pathway, as a recent manuscript demonstrated that oligomerization of the prion-like mitochondrial antiviral-signaling protein (MAVS) was modulated by intracellular lactate and this, in turn, modulated interferon production11.
Recent work has greatly expanded on the myriad roles that metabolism plays in modulating T cell function. For instance, with regard to the mitochondria, Complex III of the electron transport chain appears to play an essential role in Treg suppressive function12. Moreover, in CD4 T cells, a distinct role for Complex I activity versus Complex II activity has recently been delineated13. Disruption of Complex II activity through a conditional deletion of succinate dehydrogenase complex subunit C resulted in TH1 cells that proliferated more than control cells but appeared less capable of differentiation and effector function, as evident by a reduction in interferon gamma (IFNγ) production (Figure 1). The metabolic regulation of IFNγ has also been examined in the setting of T cells which have been engineered to lack lactate dehydrogenase A (LDHA)14. These T cells are unable to reduce pyruvate to lactate, a reaction that also serves to regenerate NAD+. The downstream metabolic consequence of lacking LDHA appears to culminate in a reduction in acetyl-CoA, which in turn leads to reduced histone acetylation and subsequent reduction in IFNγ transcription14. Finally, in Treg, a role for lipid oxidation appears to be important in modulating IFNγ production15. In particular, deletion of liver kinase B (LKB) in Treg cells suppressed lipid metabolism and resulted in augmented IFNγ production thereby triggering an autoimmune type pathology in these mice15. Thus, as the above discussion illustrates, a range of metabolic perturbations, from disrupting mitochondrial electron transport, to manipulating cytosolic glycolysis to modulating fatty acid oxidation can either positively and negatively affect T cell function and IFNγ production (Figure 1).
Figure 1:

Metabolic regulation of T cell function. Manipulation of diverse metabolic pathways can impact T cell activation, as assessed by IFNγ production. This includes genetic disruption of Complex II of the mitochondrial electron transport chain (top), altering cytosolic glycolysis by deletion of LDHA (middle), or altering intracellular lipid metabolism (bottom).
Metabolic modulation of the tumor microenvironment
In all in vitro cell culture models, cells are normally exposed to vast excess of nutrients. As such, neither oxygen nor metabolic substrates are ever truly limiting. This is not, however, the case in vivo, where there is often fierce competition for a limited nutrient supply. This is perhaps best exemplified in the violent, competitive landscape of the tumor microenvironment. Growing evidence suggests that this metabolic competition can alter cell fate and function. Much of this work has been spurred by the desire to better understand why immune cells often fail to effectively neutralize seemingly immunogenic tumors. While the answer to this question is complex, there is growing evidence that at the cellular level, the tumor microenvironment represents a true microcosm of Darwinian survival (Figure 2). Sometimes, this involves the expression within the tumor of enzymes such as indoleamine 2,3-dioxygenase, that avidly consumes tryptophan, thereby depriving surrounding cells of this essential amino acid16. Other times, the hyper-metabolic state of the tumor can produce high levels of byproducts such as lactate, which can modulate the activity and fate of infiltrating T cells17 and macrophages18. This metabolic milieu appears to have important physiological consequences. For instance, high glucose consumption by the tumor appears to limit T cell effector function, with a decrease in interferon gamma (IFN-γ) production and evidence for tumor progression19. Interestingly, checkpoint blockade antibodies, such as CTLA-4, PD-1 and PD-L1, appear to function, in part, by directly altering tumor metabolism, restoring glucose availability to the niche, and thereby rejuvenating infiltrating T cell function19. Other studies have demonstrated that the high glucose consumption of the tumor results in decreased glycolytic flux for infiltrating immune cells, and that the absence of the specific glycolytic metabolite phosphoenolypyruvate (PEP) acts as a T cell metabolic checkpoint20. The consumption of glucose or tryptophan by the tumor is not, however, the only way that the harsh environment of the tumor microenvironment influences the immune response. Within the rapidly expanding tumor, high rates of cell death in turn lead to high levels of extracellular potassium. Emerging evidence suggests that this extracellular potassium dramatically restricts the ability of T cells to take up what limited nutrients are in fact available21. This nutrient deprivation in turn modulates acetyl-CoA levels in T cells, their levels of histone acetylation, and ultimately their function21.
Figure 2:

Metabolic influences in the tumor microenvironment. Numerous metabolites appear to play a role in the tumor microenvironment, and help govern the growth of the tumor and the corresponding strength of the immune response. High rates of tumor glucose and tryptophan consumption can limit the availability of these metabolites for invading immune cells. Moreover, tumor production and subsequent excretion of lactate can have a paracrine influence on a wide range of immune cells. Recent evidence also suggests that the nutrient deprivation tumor create can trigger signaling pathways within immune cells such as activation of the unfolded protein response (e.g. XBP1) or directly trigger growth arrest by reducing the levels of critical nutrients (e.g. PEP).
The tumor milieu can also induce metabolically-driven stress signaling in infiltrating immune cells. A recent report demonstrated that malignant ascites fluid derived from ovarian cancer patients can suppress the expression of glucose transporters such as GLUT1 on the surface of CD4+ T cells22. This metabolic stress, in turn, activated the endoplasmic reticulum stress response, leading to increase activity of the XBP1 arm of the unfolded protein response (UPR)22. Previous work has clearly indicated that ER stress can be induced when nutrients are over-abundant23, however, these recent results would suggest that either an excess or deficit in nutrients could trigger the UPR. The induction of the UPR in T cells exposed to ovarian ascites fluid resulted in reduced expression of multiple glutamine transporters, resulting in a decrease in glutamine influx and hence a decline in overall mitochondrial function. These glutamine-poor T cells appeared to have reduced IFN-γ expression and impaired anti-tumor function22. In contrast, T cells lacking XBP1 were metabolically more robust and appeared to infiltrate tumors better and produce higher levels of IFN-γ. This suggests that in T cells, there is an important connection between nutrient availability, ER homeostasis and anti-tumor activity.
The metabolism of the tumor microenvironment is not just important for the immunological response. Increasingly, the unique metabolism of stromal elements, such as cancer associated fibroblasts (CAFs) are increasingly being dissected. One recent example analyzed 5000 formalin fixed tumor samples in conjunction with a label-free proteomics platform24. This system allowed for an analysis of the proteome of tumors and their associated stroma as disease progressed from an initial in situ lesion, all the way to advanced metastatic disease. Interestingly, this analysis suggested that the tumor proteome was relatively stable over this spectrum of disease, while there were marked differences in the stromal component 24. Notably, the stroma associated with metastatic disease was enriched for the expression of methyltransferase nicotinamide N-methyltransferase (NNMT). This enzyme transfers a methyl group from S-adenosyl methionine (SAM) to nicotinamide. As mentioned previously, within cells, SAM is the universal donor for methylation reactions involving histone and non-histone proteins. Interestingly, within fibroblasts, NNMT was associated with the expression of epithelial-to-mesenchymal transition (EMT) markers, a signature known to be associated with metastatic disease24. Moreover, increased expression of NNMT in normal fibroblasts promoted cancer cell growth, while knockdown of NNMT in CAFs produced the opposite effects, a phenomenon driven by NNMT-dependent alterations in SAM levels24. Interestingly, the interplay of stromal metabolism with the growth of the tumor also involves the ability of CAFs to secrete exosomes that appear to supply the tumor with essential nutrients25. Finally, the metabolism of the tumor can also modulate the niche. In particular, a recent report demonstrated that breast cancers rely on pyruvate metabolism to remodel the extracellular matrix and thereby drive metastatic growth26. Thus, it would appear that the metabolism of the stroma modifies the tumor and the metabolism of the tumor modulates the niche.
Metabolic regulation of fibrosis
Another area where substrate utilization appears to influence and modify cell fate involves the fibrotic response. As we age, alterations in stem and progenitor cell fate occur with both a loss in quiescence and an age-dependent bias in generating differentiated cell types. For instance, older muscle stem cells skew away from generating myofibers and instead preferentially differentiate towards a fibrogenic lineage27. The fact that dietary interventions such as caloric restriction appears to mitigate aspects of stem cell aging has prompted intense investigations regarding the connection between nutrients and progenitor cell fate28. One interesting recent example involves dissecting the metabolic underpinnings for cell fate decisions of beige adipocyte precursor cells. Both brown and beige adipocytes are of interest for their ability to uncouple mitochondrial respiration from ATP production and thereby potentially counteract the harmful effects of nutritional excess and obesity. Beige adipocytes develop in areas of white adipocyte tissue (WAT) in response to external stimuli such as cold exposure or β3-adrenergic stimulation. As mice and humans age, levels of brown and beige fat decline29, 30. The zincfinger transcriptional co-regulator PRD1-BF1-RIZ1 homologous domain-containing protein 16 (PRDM16) is a key determinant for the development and maintenance of brown and beige fat. A new study demonstrated that as mice age, much like muscle stem cells, stimuli that normally would induce precursor cells to generate beige fat cells instead appear to result in a fibrogenic response31. Interestingly, PRDM16+/− mice which had a 50% reduction in the expression of the co-regulator, appeared to mimic the pro-fibrogenic skewing effects of aging, while forced expression of PRDM16 in old mice reduced fibrosis and restored beige adipocyte formation31. This latter effect was demonstrated to be due to a paracrine effect, as PRDM16 overexpressing adipocytes appear to release a factor that suppressed fibrogenesis and stimulated beige adipocyte formation31. RNAseq and metabolomics analysis demonstrated that PRDM16 induced a marked increase in fatty acid oxidation (FAO), as PRDM16-expressing cells incubated with palmitate generating more than 20-fold higher levels of acetyl-CoA than control cells31. Moreover, consistent with this elevated rate of FAO, PRDM16-expresing adipocytes secreted 6-fold higher levels of the ketone body β-hydroxybutyrate (BHB) into the condition media than control cells31. There is a growing appreciation that ketone bodies can act as signaling moieties32, and indeed, in this case, BHB secreted by adipocytes was metabolized in precursor cells and appeared to inhibit the HIF-1α and TGF-β driven fibrogenic program31. It remains unclear how precisely this occurs, however, the metabolism of BHB presumably elevates acetyl CoA and NAD levels in the precursor cells, which may in turn modulate chromatin and cell fate decisions. These results might have practical implications, as this study demonstrated that dietary BHB supplementation appeared to promote beige fat formation31. A similar phenomenon has been previously observed when mice were placed on ketogenic diet33.
The link between FAO and fibrogenesis was also observed in another context, this time involving the conversion of mature endothelial cells into a fibroblast-like state, a process known as endothelial-to-mesenchymal transition (EndoMT). EndoMT is very closely related to the well-studied phenomenon of epithelial-to-mesenchymal transition (EMT) briefly discussed above, and known to be stimulated by TGF-β signaling34. Endothelial cells stimulated to undergo EndoMT with TGF-β developed a marked decrease in FAO, which in turn, led to a marked decline in acetyl-CoA levels35. This TGF-β induced reduction in FAO has been observed in other contexts, including certain disease states like chronic kidney disease characterized by significant fibrosis36. Mitochondrial FAO requires the transport of long-chain fatty acids across the outer mitochondrial membrane, and then yet again across the inner mitochondrial membrane. This transport is catalyzed by the action of two sequential enzymes, namely CPT1 (on the outer mitochondrial membrane) and CPT2 (on the inner mitochondrial membrane). In culture, endothelial knockdown of either CPT1 or CPT2 augmented EndoMT35. This phenomenon could, however, be reversed by supplementing the culture media with acetate, a cell permeant metabolite that can be converted into acetyl-CoA by the action of acetyl-CoA synthetase 2 (ACSS2). Interestingly, in previous studies, it was demonstrated that in neurons, ACSS2 could be found in the nucleus, where it appears to generate a local reservoir of acetyl-CoA required for histone acetylation and for critical functions such as memory formation37. Outside the mitochondria, the generation of acetyl-CoA can also occur by the metabolism of mitochondrial-exported citrate and the subsequent action of the enzyme ATP-citrate lyase (ACLY). Again, the activity of ACLY appears critical for the maintenance of histone acetylation38, as well as for other functions including DNA repair39. Interestingly, in endothelial cells, inhibiting ACLY activity stimulated EndoMT induction suggesting that non-mitochondrial endothelial acetyl-CoA levels were critical determinants of this cell fate change35. The maintenance of acetyl-CoA levels might be cell-type specific with evidence in endothelial cells suggesting it requires FAO, while in other cases; it might derive primarily from pyruvate metabolism40. It is also important to note that the pool of acetyl-CoA in the mitochondria is not directly exchangeable or in equilibrium with the pool of acetyl-CoA in the cytosol and nucleus (Figure 3). Metabolic compartmentation occurs for other metabolites such as NAD/NADH and complicates the interpretation of total cellular metabolomics data. Better tools, including ratiometric fluorescent reporters are needed to allow for real-time and subcellular resolution of metabolites. Some such tools are emerging41. Finally, while the above results highlight a role for endothelial FAO in regulating EndoMT, other reports have shown that endothelial FAO maintains acetyl-CoA levels and is thereby essential for de novo nucleotide synthesis42 and for maintaining redox homeostasis43.
Figure 3:

The compartmentation of acetyl-CoA. Acetyl-CoA can be generated in different compartments of the cell including the mitochondria, cytosol and nucleus. The pool of mitochondrial acetyl-CoA is higher and not in equilibrium with the cytosolic-nuclear pool. Citrate export from the mitochondria can be re-converted to acetyl-CoA by the action of the enzyme ACLY. Moreover, acetate, taken up from the extracellular milieu or produced intracellularly, can also generate acetyl-CoA by the action of the enzyme ACSS2. ACLY and ACSS2 are found in the cytosol and nucleus. In the nucleus, these enzymes are believed to generate locally high concentrations of acetyl-CoA, presumably near sites of active chromatin acetylation.
Interestingly, while the above discussion suggests that a decrease in FAO may push endothelial cells towards a fibroblast lineage; recent reports suggest that the phenotype of mature fibroblasts may be modulated by the balance between FAO and glycolysis44, 45. In particular, as in many other cell types, glycolysis appears to fuel generating biosynthetic mass, which for the case of fibroblasts largely involves production of extracellular matrix (ECM). Earlier studies have demonstrated that human skin maintains a high rate of anaerobic glycolysis, with nearly 70% of consumed glucose diverted towards lactate production46. In contrast, this more recent study suggests that stimulating FAO in fibroblasts appears to stimulate these cells to engage in ECM degradation47. Since FAO can often be stimulated by small molecules that activate the peroxisome proliferator-activated receptor (PPAR) pathway, this hints at a potential metabolic-based therapeutic strategy to modulate excessive fibrosis. Namely by stimulating fibroblast FAO one might potentially impact the excess fibrosis that occurs in a number of skin conditions including scleroderma, graft-versus-host disease or as an unintended consequence of radiation therapy44.
Metabolic regulation of stem and progenitor cells
As discussed in part above, there is an emerging appreciation for the role of metabolism in regulating stem and progenitor cell function. Most of this work has focused on neural stem and progenitor cells48 or in the well-studied hematopoietic stem cell system49. However, the metabolic control of stem and progenitor function extends to other niches. In the gut, intestinal stem cells reside adjacent to differentiated Paneth cells at the bottom of the intestinal crypt. Metabolic analysis has demonstrated that in comparison to Paneth cells, intestinal stem cells demonstrate higher levels of mitochondrial activity50. This increase mitochondrial activity appeared to drive the production of reactive oxygen species (ROS), which functioned as signaling molecules, activating a redox-dependent, p38 MAPK pathway that drives crypt formation50. The beneficial role of ROS here is not unique. For instance, while there are certainly examples where the sustained production of ROS inhibits stem cell function51, 52, there are also contrary examples where the production of ROS appears to provide a necessary signaling function53. For the case of the intestinal stem cell (ISC), the neighboring Paneth cells metabolize glucose and hence secrete lactate that can diffuse out of the Paneth cell and be taken up by the neighboring stem cell. This lactate is then converted into pyruvate within the intestinal stem cell, and used as a mitochondrial substrate in the TCA cycle. This additional example of metabolic compartmentalization, where the Paneth cell supports the mitochondrial activity of the neighboring stem cell, suggests an anatomical-metabolic requirement for optimal stem cell function. In many ways, this is also similar to our previous discussion between the metabolic coupling between mature adipocytes and adipocyte precursor cells31. Interestingly, a similar paradigm exists in the mature brain where astrocytes break down stored glycogen, producing lactate that can be used by adjacent neurons. This process is essential for diverse neuronal functions including memory formation54, as well as excitability and plasticity55.
Other more recent studies have expanded on the role of metabolism on intestinal stem cell biology. Given the presumed benefits of reducing caloric intake on overall health there is substantial interest in understanding how dietary manipulations regulate intestinal stem cell function. In that regard, a recent report demonstrated that acute fasting markedly stimulated the ex vivo organoid-forming capacity of mouse crypts, a measure of ISC function56. Interestingly, this fasting-induced stimulation of ISC function appears to be mediated by an increase in FAO in these cells. In particular, deletion of CPT1A in the ISCs resulted in abrogation of the fasting-induced stimulation of organoid formation56. The potential molecular basis of fad diets like the ketogenic diet has also been recently studied. Interestingly, when compared to their more differentiated progenitor cells, ISCs express significantly higher levels of 3-hydroxy-3-methylglutaryl-CoA synthetase 2 (Hmgcs2), the rate limiting step in ketone body production57. Deletion of Hmgcs2 in ISCs impaired their stem cell regenerative capacity and appeared to cause premature differentiation of these stem cells towards the secretory lineage57. This effect appeared to be mediated through the ability of ketone bodies, such as β-hydroybutyrate, to inhibit histone deacetylases. This study further demonstrated that ketogenic diets could enhance ISC number and function while a glucose-supplemented diet had the opposite effect57. While the above studies have primarily dealt with cell-autonomous metabolism, there is also a growing appreciation that metabolites produced by the gut microbiome can also dramatically affect ISC function58.
There are additional examples where stem cell fate appears to be determined or at least influenced by metabolic determinants. A recent example centers on bone marrow mesenchymal stromal cells (BMSCs) which are normally quiescent, but upon injury can differentiate into either adipocytes, osteoblasts or chondrocytes. As we age, the differentiation potential of BMSCs skews away from osteoblast formation and towards adipogenesis59. This cell fate skewing is believed to contribute to age-related osteoporosis. Interestingly, disruption of glutamine metabolism within BMSCs resulted in a recapitulation of this age-dependent differentiation bias. In particular, conditional deletion of glutaminase (GLS), the rate limiting enzyme in glutamine metabolism, resulted in MSSCs that had increased adipogenic potential and decreased ability to form osteoblasts60. Mice lacking GLS expression in mesenchymal progenitor cells resulted in mice with decreased bone mass and increased marrow adiposity, with the latter phenotype, for unclear reasons, more evident in female mice60. The precise mechanisms for these effects remains elusive but interestingly, some of the defects in GLS-deficient BMSCs could be rescued by supplying α-ketoglutarate (αKG), the metabolite formed following deamination of glutamate60. It is therefore tempting to speculate that some of these effects might be mediated by the Jumonji or TET family of demethylases that use αKG as a cofactor61. Indeed, the TCA metabolite αKG (or alternatively named 2-oxoglutarate, 2OG) has the potential to impact cell fate in multiple ways since this large family of αKG-dependent oxgenases can catalyze a range of enzymatic activities including demethylation, hydroxylation and even halogenation62. This range of enzymatic activities is matched by an even greater set of substrates regulated by of αKG-dependent enzymes (Figure 4). Indeed, the entire central dogma of information, from DNA to RNA to protein falls under some level of αKG-dependent regulation. This includes regulation of transcription through histone and DNA demethylation, regulating translation by demethylation and hydroxylation of mRNA, tRNA and ribosomes and modulating protein stability or function through αKG-dependent hydroxylation (e.g. proline hydroxylation for collagen stabilization and in hypoxia signaling). From a stem cell perspective, a large body of evidence has demonstrated that this family of enzymes appears essential for the maintenance and acquisition of pluripotency61.
Figure 4:
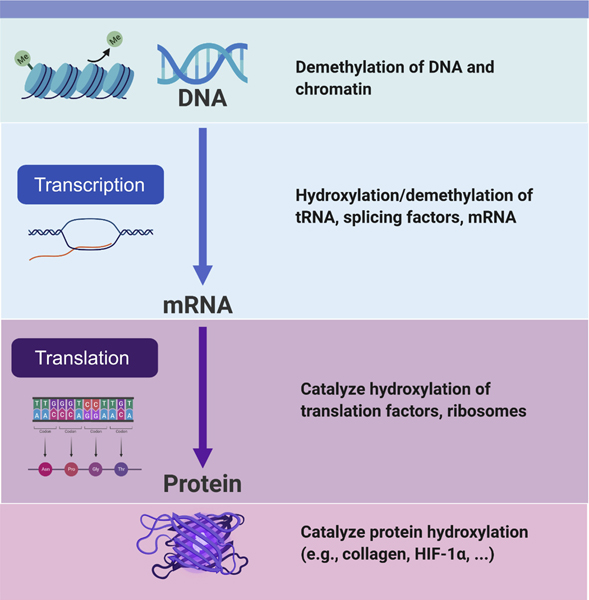
The role of αKG-dependent enzymes. This family of enzymes, all requiring the TCA metabolite αKG, exert influences on all aspects of the central dogma of biology, from DNA to RNA to protein modification. Multiple different enzymes and substrates are involved and reactions include αKG-dependent demethylation and αKG-dependent hydroxylation.
Concluding Remarks
As the work above testifies, the last several years has seen a rapid expansion of our understanding of the coupling between metabolism and cell fate. Much work remains and many outstanding questions remain (see outstanding questions box). This includes a need for better tools and an increase understanding of the specificity of this coupling. Finally, the seemingly important link to age-related diseases and the therapeutic exploitation of these insights is just beginning. For many years, mitochondrial function and metabolic pathways were thought of in purely industrial terms, as Marx would say, ‘the means of production’. In retrospect, this notion of autonomous energy production devoid of any coupling to where that energy was directed and used seems naïve. We are now entering a new phase, one that promises to deliver an ever more sophisticated insight into understanding how what we eat allows us to become what we are.
Outstanding Questions:
Can we develop better tools to easily quantify and dynamically measure the sub-cellular compartmentation of metabolites like acetyl-CoA, NAD+ and SAM in living cells or tissues?
Why are certain chromatin regions preferentially change when metabolic substrates are altered? How is this specificity achieved?
What are the means of regulation for the metabolism-cell fate connection? How does the cell sense a change in acetyl-CoA or SAM? What are the molecular sensors? What are the molecular effectors?
Can the pool of carbon metabolites on chromatin (e.g. methyl groups, acetyl groups) be mobilized to help alleviate nutrient stress? If so, is this an important, perhaps ancient, function for these modifications?
Can cellular substrate utilization be manipulated for therapeutic benefit? Do the changes in cellular metabolism that occur with age act as a primary driver for age-related diseases?
Highlights:
Metabolites, such as acetyl-CoA, directly connect metabolism to the regulation of protein function and to chromatin modification.
The metabolic substrates cells use provides more than bioenergetics and helps shape their identity
Understanding the connection between metabolism and cell fate could usher in a wave of new therapies
Acknowledgments:
We thank members of the Finkel Lab for helpful suggestions. This work was supported by 1R01HL142663 (TF), R01HL142589 (TF) and T32GM008208 (SGC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Chapman NM, Boothby MR and Chi H. Metabolic coordination of T cell quiescence and activation. Nat Rev Immunol. 2019. [DOI] [PubMed] [Google Scholar]
- 2.Buck MD, Sowell RT, Kaech SM and Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169:570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nomura M, Liu J, Rovira, II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ and Finkel T. Fatty acid oxidation in macrophage polarization. Nature immunology. 2016;17:216–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chisolm DA and Weinmann AS. Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu Rev Immunol. 2018;36:221–246. [DOI] [PubMed] [Google Scholar]
- 5.Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, Verway MJ, Raissi TC, Tsui H, Boukhaled G, Henriques da Costa S, Frezza C, Krawczyk CM, Friedman A, Manfredi M, Richer MJ, Hess C and Jones RG. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017;25:482. [DOI] [PubMed] [Google Scholar]
- 6.Ron-Harel N, Notarangelo G, Ghergurovich JM, Paulo JA, Sage PT, Santos D, Satterstrom FK, Gygi SP, Rabinowitz JD, Sharpe AH and Haigis MC. Defective respiration and one-carbon metabolism contribute to impaired naive T cell activation in aged mice. Proc Natl Acad Sci U S A. 2018;115:1334713352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giardina G, Brunotti P, Fiascarelli A, Cicalini A, Costa MG, Buckle AM, di Salvo ML, Giorgi A, Marani M, Paone A, Rinaldo S, Paiardini A, Contestabile R and Cutruzzola F. How pyridoxal 5’-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015;282:1225–41. [DOI] [PubMed] [Google Scholar]
- 8.Zheng H, Gupta V, Patterson-Fortin J, Bhattacharya S, Katlinski K, Wu J, Varghese B, Carbone CJ, Aressy B, Fuchs SY and Greenberg RA. A BRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep. 2013;5:180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walden M, Tian L, Ross RL, Sykora UM, Byrne DP, Hesketh EL, Masandi SK, Cassel J, George R, Ault JR, El Oualid F, Pawlowski K, Salvino JM, Eyers PA, Ranson NA, Del Galdo F, Greenberg RA and Zeqiraj E. Metabolic control of BRISC-SHMT2 assembly regulates immune signalling. Nature. 2019;570:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snaebjornsson MT and Schulze A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp Mol Med. 2018;50:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Wang G, Xu ZG, Tu H, Hu F, Dai J, Chang Y, Chen Y, Lu Y, Zeng H, Cai Z, Han F, Xu C, Jin G, Sun L, Pan BS, Lai SW, Hsu CC, Xu J, Chen ZZ, Li HY, Seth P, Hu J, Zhang X, Li H and Lin HK. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell. 2019;178:176–189 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg SE, Singer BD, Steinert EM, Martinez CA, Mehta MM, Martinez-Reyes I, Gao P, Helmin KA, Abdala-Valencia H, Sena LA, Schumacker PT, Turka LA and Chandel NS. Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature. 2019;565:495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, Kluger Y, Maher LJ, 3rd, Rabinowitz J, Craft J and Flavell RA. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature. 2019;571:403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng M, Yin N, Chhangawala S, Xu K, Leslie CS and Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354:481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Timilshina M, You Z, Lacher SM, Acharya S, Jiang L, Kang Y, Kim JA, Chang HW, Kim KJ, Park B, Song JH, Ko HJ, Park YY, Ma MJ, Nepal MR, Jeong TC, Chung Y, Waisman A and Chang JH. Activation of Mevalonate Pathway via LKB1 Is Essential for Stability of Treg Cells. Cell Rep. 2019;27:2948–2961 e7. [DOI] [PubMed] [Google Scholar]
- 16.Munn DH and Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW and Kreutz M. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9. [DOI] [PubMed] [Google Scholar]
- 18.Stone SC, Rossetti RAM, Alvarez KLF, Carvalho JP, Margarido PFR, Baracat EC, Tacla M, Boccardo E, Yokochi K, Lorenzi NP and Lepique AP. Lactate secreted by cervical cancer cells modulates macrophage phenotype. J Leukoc Biol. 2019;105:1041–1054. [DOI] [PubMed] [Google Scholar]
- 19.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ and Pearce EL. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. 2015;162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC and Kaech SM. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. 2015;162:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, Lee PH, Shin M, Patel SJ, Yu Z, Palmer DC, Kruhlak MJ, Liu X, Locasale JW, Huang J, Roychoudhuri R, Finkel T, Klebanoff CA and Restifo NP. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019;363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song M, Sandoval TA, Chae CS, Chopra S, Tan C, Rutkowski MR, Raundhal M, Chaurio RA, Payne KK, Konrad C, Bettigole SE, Shin HR, Crowley MJP, Cerliani JP, Kossenkov AV, Motorykin I, Zhang S, Manfredi G, Zamarin D, Holcomb K, Rodriguez PC, Rabinovich GA, Conejo-Garcia JR, Glimcher LH and Cubillos-Ruiz JR. IRE1alpha-XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature. 2018;562:423–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH and Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. [DOI] [PubMed] [Google Scholar]
- 24.Eckert MA, Coscia F, Chryplewicz A, Chang JW, Hernandez KM, Pan S, Tienda SM, Nahotko DA, Li G, Blazenovic I, Lastra RR, Curtis M, Yamada SD, Perets R, McGregor SM, Andrade J, Fiehn O, Moellering RE, Mann M and Lengyel E. Proteomics reveals NNMT as a master metabolic regulator of cancerassociated fibroblasts. Nature. 2019;569:723–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA, Alvarez H, Gupta S, Maiti SN, Cooper L, Peehl D, Ram PT, Maitra A and Nagrath D. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife. 2016;5:e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, Boon R, Escalona-Noguero C, Torrekens S, Verfaillie C, Verbeken E, Carmeliet G and Fendt SM. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature. 2019;568:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang AB and Brack AS. Muscle Stem Cells and Aging. Curr Top Dev Biol. 2018;126:299–322. [DOI] [PubMed] [Google Scholar]
- 28.Mana MD, Kuo EY and Yilmaz OH. Dietary Regulation of Adult Stem Cells. Curr Stem Cell Rep. 2017;3:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogers NH, Landa A, Park S and Smith RG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell. 2012;11:1074–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM and Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang W, Ishibashi J, Trefely S, Shao M, Cowan AJ, Sakers A, Lim HW, O’Connor S, Doan MT, Cohen P, Baur JA, King MT, Veech RL, Won KJ, Rabinowitz JD, Snyder NW, Gupta RK and Seale P. A PRDM16-Driven Metabolic Signal from Adipocytes Regulates Precursor Cell Fate. Cell Metab. 2019;30:174–189 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newman JC and Verdin E. Ketone bodies as signaling metabolites. Trends Endocrinol Metab. 2014;25:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srivastava S, Baxa U, Niu G, Chen X and Veech RL. A ketogenic diet increases brown adipose tissue mitochondrial proteins and UCP1 levels in mice. IUBMB Life. 2013;65:58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kovacic JC, Dimmeler S, Harvey RP, Finkel T, Aikawa E, Krenning G and Baker AH. Endothelial to Mesenchymal Transition in Cardiovascular Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73:190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, Fergusson MM, Yu ZX, Rovira, II, Brittain EL, Wolfgang MJ, Jurczak MJ, Fessel JP and Finkel T. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol Cell. 2018;69:689–698 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, Park AS, Tao J, Sharma K, Pullman J, Bottinger EP, Goldberg IJ and Susztak K. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mews P, Donahue G, Drake AM, Luczak V, Abel T and Berger SL. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;546:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR and Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A, Bennett MJ, Minn AJ, Weljie AM, Greenberg RA and Wellen KE. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol Cell. 2017;67:252–265 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Cooper DE, Cluntun AA, Warmoes MO, Zhao S, Reid MA, Liu J, Lund PJ, Lopes M, Garcia BA, Wellen KE, Kirsch DG and Locasale JW. Acetate Production from Glucose and Coupling to Mitochondrial Metabolism in Mammals. Cell. 2018;175:502–513 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hung YP, Albeck JG, Tantama M and Yellen G. Imaging cytosolic NADH-NAD(+) redox state with a genetically encoded fluorescent biosensor. Cell Metab. 2011;14:545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, Heggermont W, Godde L, Vinckier S, Van Veldhoven PP, Eelen G, Schoonjans L, Gerhardt H, Dewerchin M, Baes M, De Bock K, Ghesquiere B, Lunt SY, Fendt SM and Carmeliet P. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kalucka J, Bierhansl L, Conchinha NV, Missiaen R, Elia I, Bruning U, Scheinok S, Treps L, Cantelmo AR, Dubois C, de Zeeuw P, Goveia J, Zecchin A, Taverna F, Morales-Rodriguez F, Brajic A, Conradi LC, Schoors S, Harjes U, Vriens K, Pilz GA, Chen R, Cubbon R, Thienpont B, Cruys B, Wong BW, Ghesquiere B, Dewerchin M, De Bock K, Sagaert X, Jessberger S, Jones EAV, Gallez B, Lambrechts D, Mazzone M, Eelen G, Li X, Fendt SM and Carmeliet P. Quiescent Endothelial Cells Upregulate Fatty Acid beta-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018;28:881–894 e13. [DOI] [PubMed] [Google Scholar]
- 44.Zhao X, Kwan JYY, Yip K, Liu PP and Liu F-F. Targeting metabolic dysregulation for fibrosis therapy. Nature Reviews Drug Discovery. 2019. [DOI] [PubMed] [Google Scholar]
- 45.Rabinowitz JD and Mutlu GM. A metabolic strategy to reverse fibrosis? Nature Metabolism. 2019;1:12–13. [DOI] [PubMed] [Google Scholar]
- 46.Freinkel RK. Metabolism of glucose-C-14 by human skin in vitro. J Invest Dermatol. 1960;34:3742. [PubMed] [Google Scholar]
- 47.Zhao X, Psarianos P, Ghoraie LS, Yip K, Goldstein D, Gilbert R, Witterick I, Pang H, Hussain A, Lee JH, Williams J, Bratman SV, Ailles L, Haibe-Kains B and Liu F-F. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nature Metabolism. 2019;1:147–157. [DOI] [PubMed] [Google Scholar]
- 48.Khacho M, Harris R and Slack RS. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat Rev Neurosci. 2019;20:34–48. [DOI] [PubMed] [Google Scholar]
- 49.Ito K and Ito K. Hematopoietic stem cell fate through metabolic control. Exp Hematol. 2018;64:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, Pras-Raves M, Sacchetti A, Hornsveld M, Oost KC, Snippert HJ, Verhoeven-Duif N, Fodde R and Burgering BM. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature. 2017;543:424–427. [DOI] [PubMed] [Google Scholar]
- 51.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira, II, Xu XL, van Lohuizen M, Motoyama N, Deng CX and Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bigarella CL, Liang R and Ghaffari S. Stem cells and the impact of ROS signaling. Development. 2014;141:4206–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morimoto H, Iwata K, Ogonuki N, Inoue K, Atsuo O, Kanatsu-Shinohara M, Morimoto T, Yabe-Nishimura C and Shinohara T. ROS are required for mouse spermatogonial stem cell self-renewal. Cell Stem Cell. 2013;12:774–86. [DOI] [PubMed] [Google Scholar]
- 54.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ and Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Magistretti PJ and Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018;19:235–249. [DOI] [PubMed] [Google Scholar]
- 56.Mihaylova MM, Cheng CW, Cao AQ, Tripathi S, Mana MD, Bauer-Rowe KE, Abu-Remaileh M, Clavain L, Erdemir A, Lewis CA, Freinkman E, Dickey AS, La Spada AR, Huang Y, Bell GW, Deshpande V, Carmeliet P, Katajisto P, Sabatini DM and Yilmaz OH. Fasting Activates Fatty Acid Oxidation to Enhance Intestinal Stem Cell Function during Homeostasis and Aging. Cell Stem Cell. 2018;22:769–778 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng CW, Biton M, Haber AL, Gunduz N, Eng G, Gaynor LT, Tripathi S, Calibasi-Kocal G, Rickelt S, Butty VL, Moreno-Serrano M, Iqbal AM, Bauer-Rowe KE, Imada S, Ulutas MS, Mylonas C, Whary MT, Levine SS, Basbinar Y, Hynes RO, Mino-Kenudson M, Deshpande V, Boyer LA, Fox JG, Terranova C, Rai K, Piwnica-Worms H, Mihaylova MM, Regev A and Yilmaz OH. Ketone Body Signaling Mediates Intestinal Stem Cell Homeostasis and Adaptation to Diet. Cell. 2019;178:1115–1131 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee YS, Kim TY, Kim Y, Lee SH, Kim S, Kang SW, Yang JY, Baek IJ, Sung YH, Park YY, Hwang SW, O E, Kim KS, Liu S, Kamada N, Gao N and Kweon MN. Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe. 2018;24:833–846 e6. [DOI] [PubMed] [Google Scholar]
- 59.D’Ippolito G, Schiller PC, Ricordi C, Roos BA and Howard GA. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res. 1999;14:1115–22. [DOI] [PubMed] [Google Scholar]
- 60.Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, Zhang H, Knudsen E, Zhang GF, Hilton MJ and Karner CM. Glutamine Metabolism Regulates Proliferation and Lineage Allocation in Skeletal Stem Cells. Cell Metab. 2019;29:966–978 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tran KA, Dillingham CM and Sridharan R. The role of alpha-ketoglutarate-dependent proteins in pluripotency acquisition and maintenance. J Biol Chem. 2019;294:5408–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Herr CQ and Hausinger RP. Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem Sci. 2018;43:517–532. [DOI] [PMC free article] [PubMed] [Google Scholar]