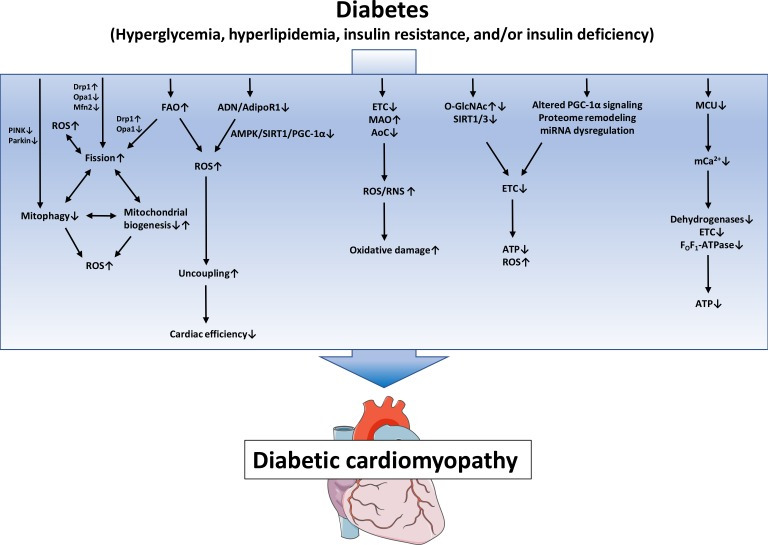
Fig. 1. Proposed mechanisms of mitochondrial dysfunction in diabetic cardiomyopathy. Defects in the electron transport chain (ETC), increased monoamine oxidases (MAO) activity and decreased antioxidative capacity lead to increased reactive oxygen species/reactive nitrogen species (ROS/RNS) generation and subsequent oxidative damage. Posttranslational mechanisms like altered protein O-linked beta-N-acetylglucosamine glycosylation (O-GlcNAcylation) and increased protein acylation due to impaired SIRT activity, as well as mitochondrial proteome remodeling, impaired peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) signaling and miRNA dysregulation contribute to impaired ETC activity, ultimately leading to energy depletion and oxidative stress. Increased fatty acid oxidation (FAO) and/or impaired adiponectin (ADN)/adiponectin receptor 1 (AdipoR1) signaling may contribute to mitochondrial uncoupling and decreased cardiac efficiency. Increased mitochondrial fission, decreased mitophagy and altered mitochondrial biogenesis contribute to mitochondrial ROS and energy depletion and are interrelated mechanisms that may modulate each other. Impaired mitochondrial calcium uniporter (MCU) activity decreases mitochondrial Ca2+ uptake and thereby impairs activity of Ca2+-dependent dehydrogenases and oxidative phosphorylation. PINK1, phosphatase and tensin homolog-induced putative kinase 1; Drp1, dynamin-related protein 1; Opa1, optic atrophy 1; Mfn2, mitofusion 2; AMPK, adenosine monophosphate-activated protein kinase; SIRT1, sirtuin 1; AoC, antioxidative capacity; ATP, adenosine triphosphate.