

ABSTRACT
Accumulation of DNA damage in resting cells is an emerging cause of human disease. We identified a mechanism of DNA double-strand break (DSB) formation in non-replicating cells, which strictly depends on transcription. These transcriptional DSBs arise from the twinned processing of R-loops and topoisomerase I and may underlie neurological disorders and cancers.
KEYWORDS: DNA double-strand break, DNA repair, R-loop, RNA/DNA hybrid, Senataxin, TDP1, XPF, Neurodegenerative disease, Cancer, Topoisomerase I, Transcription
DNA double-strand breaks (DSBs) are infrequent but among the most harmful genomic lesions. Their defective repair can induce genomic rearrangements, mutations or cell death, and they have been implicated in the pathogenesis of several human diseases, including neurodegenerative syndromes and cancers. In dividing cells, DSBs occur primarily during DNA replication. Hence, their mechanisms of formation have been well documented, such as in the events of replication of a damaged DNA template and during conflicts between replication and transcription machineries. However, in human, most cells are non-replicating, and increasing evidence indicates that DNA damage and genomic instability in such cells can cause disease. Accumulation of DNA breaks in post-mitotic neurons is the underlying cause of multiple neurodegenerative syndromes.1 Many solid tumors are also characterized by a subpopulation of quiescent cells, which represents a fuel for cancer diversity and evolution.2 Currently, very little is known about how DSBs are generated in non-replicating cells, even though such knowledge is likely to provide information on the aetiology of several human diseases.
Our recent work has advanced this field by identifying a mechanism for DSB formation in non-replicating cells occurring under both physiological and pathological conditions.3 DSBs arise from two nearby single-strand breaks (SSBs) on the opposing DNA strands, both produced during transcription. One SSB results from the repair of a transcription-blocking topoisomerase I cleavage complex (TOP1cc), and the other from the cleavage of an R-loop structure (Figure 1). TOP1 removes DNA superhelical tensions generated during transcription by producing transient TOP1ccs.4 We found that stabilization (trapping) of TOP1ccs on chromatin primes the formation of DSBs by blocking transcription to promote R-loop formation. Notably, TOP1ccs are often trapped by DNA modifications, such as oxidative base damage,4 and they are associated with human disease (Figure 1). The repair of TOP1ccs generates SSB intermediates, and we found that DSBs are produced by concurrent SSBs on the opposing DNA strand of the R-loops. R-loops are RNA/DNA hybrids with displaced single-stranded DNA. They are widespread structures that can play physiological roles, but their unscheduled formation is a source of DNA breaks.5 Recent work reported the involvement of xeroderma pigmentosum complementation group F (XPF, also known as ERCC4) and group G (XPG, also known as ERCC5) flap nucleases in the production of R-loop-dependent DSBs in replicating cells.6 Our study in non-replicating cells sheds new light on R-loop processing by nucleases.3 It identified that flap structure-specific endonuclease 1 (FEN1), XPF and XPG induce SSBs in the R-loops by the cleavage of one strand and the DSB is created when another SSB is present on the opposing DNA strand. Our study further demonstrated that the single-stranded DNA in the R-loop must be cleaved at both 3ʹ- and 5ʹ-extremities to induce DSBs and that this dual incision is mediated by XPF/XPG or XPF/FEN1. However, it is still unclear what defines the choice between these specific combinations of nucleases.
Figure 1.
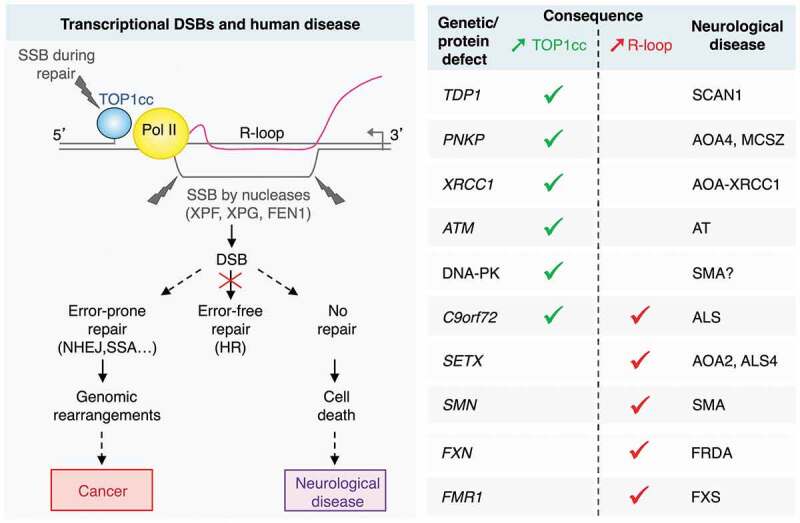
Outcomes of transcription-dependent DNA double-strand breaks (DSBs) in non-replicating cells. DSBs arise from two single-strand breaks (SSBs) on the opposing DNA strands: one SSB results from repair of a transcription-blocking topoisomerase I cleavage complex (TOP1cc), and the other from the cleavage of an R-loop by nucleases. In non-replicating cells, error-free homologous recombination (HR) is unavailable. Hence, DSBs could be repaired by error-prone pathways, such as non-homologous end joining (NHEJ) and single-strand annealing (SSA), which may promote genomic rearrangements and mutations. If left unrepaired, transcriptional DSBs may accumulate and induce cell death. The right-hand side table shows human neurological disorders, the genes (or protein expression) altered in these diseases and their association with increased TOP1ccs (green) or R-loops (red). AOA: ataxia with oculomotor apraxia; ALS: amyotrophic lateral sclerosis; AT: ataxia telangiectasia; ATM: ataxia telangiectasia mutated; C9orf72: chromosome 9 open reading frame 72; DNA-PK: DNA-dependent protein kinase; FEN1: flap structure-specific endonuclease 1; FRDA: Friedreich’s ataxia; FMR1: fragile X mental retardation 1; FXN: frataxin; FXS: fragile X syndrome; MCSZ: microcephaly, seizures, and developmental delay; PNKP: polynucleotide kinase 3ʹ-phosphatase; Pol II: RNA polymerase II, SCAN1: spinocerebellar ataxia with axonal neuropathy-1; SETX: senataxin; SMA: spinal muscular atrophy; SMN: survival motor neuron 1; TDP1: tyrosyl-DNA phosphodiesterase 1; XPF: xeroderma pigmentosum complementation group F; XPG: xeroderma pigmentosum complementation group G; XRCC1: X-ray repair cross-complementing 1.
These transcriptional DSBs have been detected in post-mitotic neurons7 and quiescent cells,3 and may underlie human disease. Indeed, we found that genetic defects in TOP1cc removal pathway [tyrosyl-DNA phosphodiesterase 1 (TDP1), polynucleotide kinase 3ʹ-phosphatase (PNKP), or X-ray repair cross-complementing 1 (XRCC1)] or in R-loop resolution [senataxin (SETX)] enhance transcriptional DSBs in non-replicating cells.3 Notably, all these genetic defects cause neurological disorders, primarily cerebellar ataxia and amyotrophic lateral sclerosis (Figure 1), suggesting that these transcriptional DSBs may be the underlying cause of several neurodegenerative syndromes. This is further supported by the observation that numerous other genetic (or protein) defects that act to increase TOP1ccs [ataxia telangiectasia mutated (ATM), DNA-dependent protein kinase (DNA-PK), or chromosome 9 open reading frame 72 (C9orf72) expansion repeat], and/or R-loops [C9orf72, frataxin (FXN) or fragile X mental retardation 1 (FMR1) expansion repeats, or survival motor neuron 1 (SMN)], are also associated with neurological disorders (Figure 1). A key question is why do these transcriptional DSBs primarily affect neuronal cells? First, neurons may be particularly prone to produce these breaks considering their high metabolic and transcriptional activity, which may promote reactive oxygen species (ROS)-dependent TOP1cc trapping4 and R-loop accumulation.3 Second, post-mitotic neurons are non-dividing cells, and as such have reduced repair capability compared to dividing cells. Homologous recombination (HR) and non-homologous end joining (NHEJ) are the main pathways for the repair of DSBs. HR is the preferred pathway to repair DSBs occurring in transcribed regions,8 however it is unavailable in non-dividing cells due to absence of sister chromatids for recombination (Figure 1). Our work further highlights that genetic defects in TOP1cc removal (TDP1) or R-loop resolution (SETX) prevent the repair of transcriptional DSBs in non-replicating cells.3 This raises the possibility that in neurons, transcriptional DSBs accumulate over time due to enhanced production and defective repair. Both will contribute to the neurodegenerative phenotype. Consistent with this, we found that such DSBs can kill non-replicating cells.3
It is conceivable that the transcriptional DSBs that depend on R-loops and TOP1ccs may contribute to other pathologies, including cancers. Indeed, many tumors contain a subpopulation of quiescent cells,2 and increasing evidence indicates that genetic alterations in cancer cells are linked to R-loop formation9 and TOP1 activity.4 Notably, many cancers have an altered cell metabolism resulting in increased ROS levels,10 which may further trap TOP1ccs,4 and an altered expression/mutation of many R-loop resolving factors [e.g. SETX, DExH-box helicase 9 (DHX9), breast cancer 1 and 2 (BRCA1 and BRCA2), fanconi anemia (FA) proteins],9 which may further stabilize R-loops. In quiescent cancer cells, transcriptional DSBs may promote tumor heterogeneity and progression because they occur in transcribed regions and they can’t use error-free HR repair. Their repair may rely on error-prone pathways, which may lead to mutations and genomic rearrangements (Figure 1).
Altogether, our work provides a new paradigm for the occurrence of DNA damage and genomic instability in resting cells in the context of human disease.
Acknowledgments
OS lab is supported by the Fondation pour la Recherche Médicale (FRM) [Equipe labellisée FRM (DEQ20170839117)] and the Ligue Nationale Contre le Cancer (LNCC) Comité Départemental 31. NG lab is supported by the Royal Society University Research fellowship. AC is supported by the EPA Research Fund (Sir William Dunn School of Pathology, University of Oxford) and Ataxia UK grant to NG. We thank Nick Proudfoot for critically reading the manuscript.
Disclosure of Potential Conflicts of Interest
The authors declare no conflict of interest
References
- 1.Rass U, Ahel I, West SC.. Defective DNA repair and neurodegenerative disease. Cell. 2007;130:1–3. doi: 10.1016/j.cell.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M.. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1740936. doi: 10.1155/2016/1740936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cristini A, Ricci G, Britton S, Salimbeni S, Huang SN, Marinello J, Calsou P, Pommier Y, Favre G, Capranico G, et al. Dual processing of R-loops and topoisomerase I induces transcription-dependent DNA double-strand breaks. Cell Rep. 2019;28:3167–3181 e3166. doi: 10.1016/j.celrep.2019.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y, Sun Y, Huang SN, Nitiss JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol. 2016;17:703–721. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia-Muse T, Aguilera A. R-loops: from physiological to pathological roles. Cell. 2019;179:604–618. doi: 10.1016/j.cell.2019.08.055. [DOI] [PubMed] [Google Scholar]
- 6.Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell. 2014;56:777–785. doi: 10.1016/j.molcel.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sordet O, Redon CE, Guirouilh-Barbat J, Smith S, Solier S, Douarre C, Conti C, Nakamura AJ, Das BB, Nicolas E, et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009;10:887–893. doi: 10.1038/embor.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP, et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat Struct Mol Biol. 2014;21:366–374. doi: 10.1038/nsmb.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulianne B, Feldhahn N. Transcribing malignancy: transcription-associated genomic instability in cancer. Oncogene. 2018;37:971–981. doi: 10.1038/onc.2017.402. [DOI] [PubMed] [Google Scholar]
- 10.Sullivan LB, Gui DY, Vander Heiden MG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016;16:680–693. doi: 10.1038/nrc.2016.85. [DOI] [PubMed] [Google Scholar]