
Fig. (2).
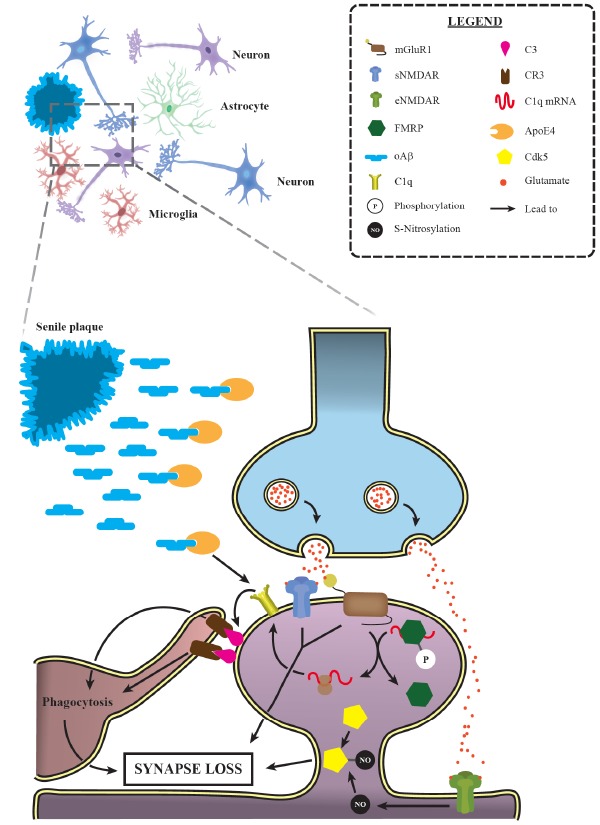
Activation of synaptic pruning mechanisms is relevant for AD progression. Progressive synaptic loss is one of the main morphological features of Alzheimer’s disease, observed even earlier than senile plaque deposition. Several studies have demonstrated the prominent role of the classical complement cascade in such event. Indeed, it has been shown that ApoE4, a major AD risk factor, leads to C1q accumulation in animal models. Additionally, mGluR1 activation allows a greater C1q mRNA translation by promoting the dephosphorylation of FMRP, an RNA-binding protein that represses translation. These data provide a suitable explanation for the increased levels of C1q and its downstream complement factor C3 in AD, which, in turn, trigger targeted synapses phagocytosis by microglial cells. Moreover, NMDAR and group I mGluRs activation have also been involved in the excessive synaptic loss observed during AD. Interestingly, the activation of extrasynaptic NMDAR (eNMDAR) induces the production of the S-nitrosylation of Cdk5, another factor implicated in AD excessive synaptic elimination. mGluR1: metabotropic glutamate receptor 1; sNMDAR: synaptic N-methyl-D-aspartate receptor; eNMDAR: extrasynaptic N-methyl-D-aspartate receptor; FMRP: fragile X mental retardation protein; oAβ: oligomeric amyloid β; CR3: complement receptor 3; ApoE4: Apolipoprotein E ε4; Cdk5: cyclin-dependent kinase 5. (A higher resolution / colour version of this figure is available in the electronic copy of the article).