

Abstract
Fluc family fluoride channels are assembled as primitive antiparallel homodimers. Crystallographic studies revealed a cation bound at the center of the protein, where it is coordinated at the dimer interface by mainchain carbonyl oxygens from the mid-membrane breaks in two corresponding transmembrane helices. Here, we show that this cation is a stably bound sodium ion, and, although it is not a transported substrate, its presence is required for the channel to adopt an open, fluoride conducting conformation. The interfacial site is selective for sodium over other cations, except for Li+, which competes with Na+ for binding, but does not support channel activity. The strictly structural role fulfilled by this sodium provides new context to understand the structures, mechanisms, and evolutionary origins of widespread Na+-coupled transporters.
Keywords: Membrane protein architecture, transporter, inverted repeat, dual topology
Graphical Abstract
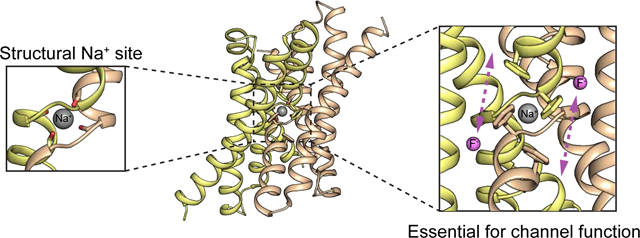
Introduction:
Fluc proteins are electrodiffusive fluoride channels that protect microbes against the cytoplasmic accumulation of toxic environmental fluoride ion[1–3]. Among membrane proteins of known structure, they possess a unique fold, forming an antiparallel dimer (Figure 1A)[4, 5]. Although this so-called “dual topology” architecture is highly unusual among membrane proteins, it is reminiscent of a very common architecture for transporters, the inverted repeat, in which two structurally homologous domains in a single polypeptide are arranged in antiparallel orientation[6]. It is likely that transporters with inverted repeat architectures evolved via a gene duplication and fusion, and that their evolutionary antecedents were dual topology homodimers like the Flucs [7, 8].
Figure 1.
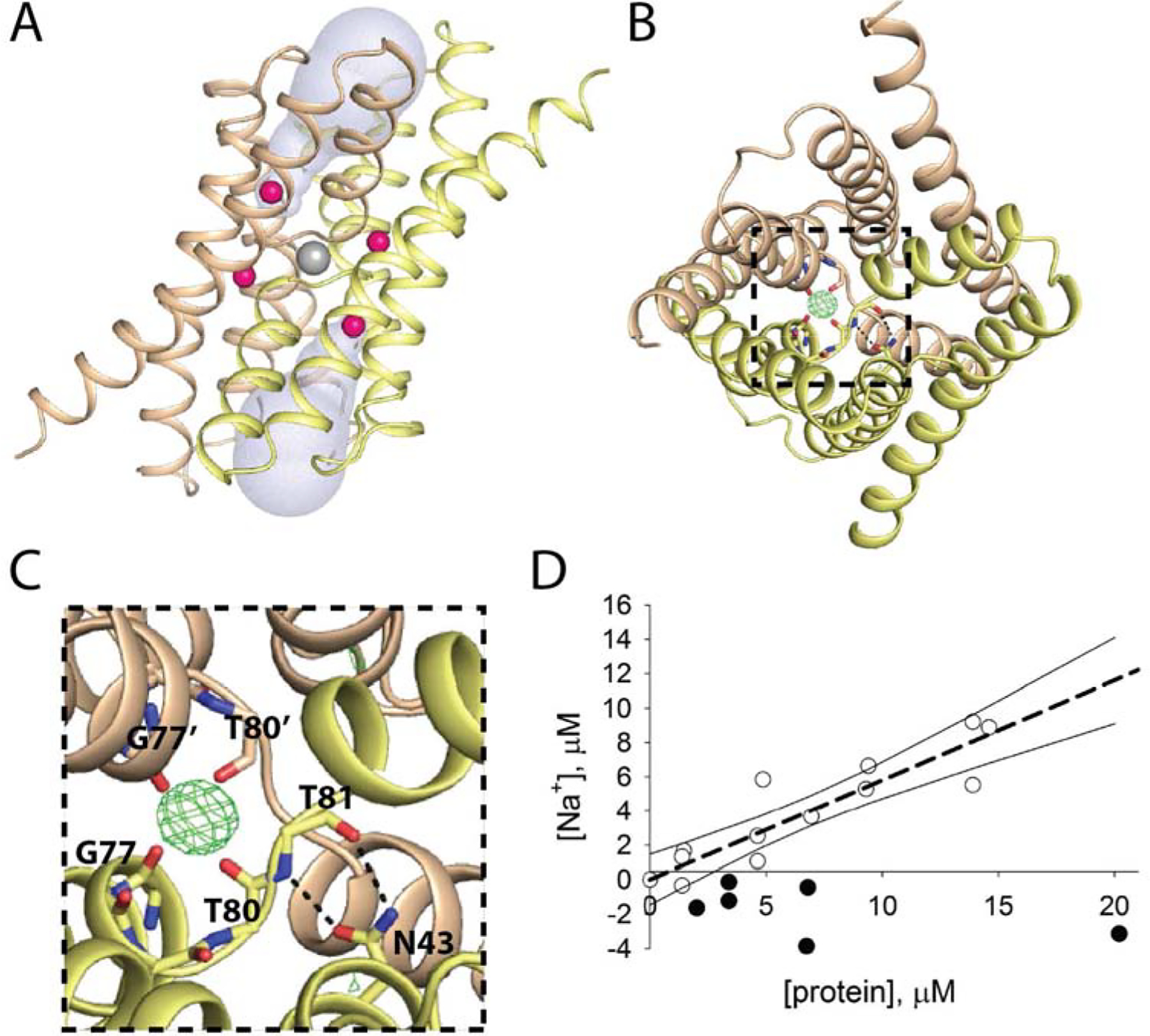
Central cation in Fluc-Bpe (PDB: 5NKQ) A. Fluc viewed through the plane of the membrane. Subunits colored wheat and yellow, fluoride ions in pink, central cation in gray, and aqueous vestibules in blue surface representation. B. Top-down view of Fluc with mFo-DFc map contoured at 4σ around the central cation. N43 sidechain and main chain atoms from G77-T81 are shown as sticks. C. Zoomed in view of panel B. Residues within coordination distance of the central cation or within H-bonding distance of N43 are labeled. D. ICP-MS measurements of Na+ in samples of Fluc (open circles) and an unrelated fluoride transporter from the CLC family, CLCF-Eca (closed circles). Raw ICP measurements were converted into molarity by comparison to a standard curve, which was linear over this concentration range. Background Na+ from the final purification buffer (typically ~2–6 μM) was measured for each protein sample and subtracted from the total to yield the final Na+ concentration. The dashed line indicates a linear regression with a slope of 0.6, and the solid lines show the 95% confidence intervals for the fit.
As a result of its dual topology architecture, the Fluc channels possess two-fold symmetry along an axis parallel to the plane of the membrane, as well as a pair of pores, both of which form functional fluoride-conducting pathways[9]. Four fluoride ion binding sites have been proposed based on structural and functional data [5, 10]. Fluoride ions access these pores via the protein’s deep electropositive vestibules (Figure 1A). Electrophysiological evidence shows that the channels are exceptionally selective for fluoride over other anions as well as small cations like sodium [2].
In addition to the presumed fluoride densities, maps from multiple different structures of two different Fluc homologues also revealed an additional fifth electron density (Figure 1B, C)[5, 11, 12] well removed from the fluoride permeation pathway. This ligand is plainly cationic, as it is tetrahedrally coordinated by four carbonyl oxygens. Its coordination sphere is constituted of backbone atoms from an equivalent pair of amino acids, G77 and T80 from each subunit, at the crossover of the TM3 helical breaks (Figure 1C). Thus, the density resides along the protein’s two-fold symmetry axis, where it is coordinated by both subunits, as if it is buttoning the dimer interface together. Based on the charge and chemical considerations of the coordination sphere, along with the fact that the purification buffers and crystallization solutions all contained Na+, the density was assigned as a sodium ion[5]. However, crystallographic identification of this cation is uncertain, and no additional experimental evidence has been put forth to positively identify the ion. Moreover, it is unknown whether the ion plays any kind of functional role in the protein. Wildtype channels continue to conduct fluoride ion after sodium-containing solutions are exchanged with solutions containing a bulky organic cation, N-methyl glucamine (NMG+) [5]. But it is unclear whether this negative result occurs because Na+ does not impact channel function, or because the ion, which is not accessible to aqueous solution in the crystal structures, cannot be exchanged on the ~tens of minutes timescale typical for electrophysiology experiments.
In this study, we provide functional evidence that the central density is, as hypothesized, a sodium ion, and we construct mutants in which this central Na+ can be exchanged rapidly. We show that the central Na+ binding site is specific for Na+ over most other cations including K+ and Mg2+, and that, although the Na+ is not transported, its presence is essential for fluoride channel function. Thus, for this simplest of membrane transport proteins, a bound Na+ plays an essential structural role. Similarities between the sodium binding site in Fluc and sodium binding sites in other membrane transport proteins provoke the question of whether these other proteins employ Na+ in a similar structural capacity – either today, or at some point in the fold’s evolutionary history.
Results:
Spectroscopic identification of central sodium ion
In order to detect and quantify sodium ion stably bound to fluoride channel proteins, we employed a spectroscopic technique, inductively-coupled plasma mass spectrometry (ICP-MS). Proteins were purified in stringently Na+-free conditions (≤ 6 μM measured with ICP-MS), denatured with nitric acid, and the Na+ content of the samples was determined based on comparison to a standard curve (Figure 1D). Whereas a structurally characterized fluoride transporter from the CLCF family [13] showed no enrichment of Na+ relative to buffer, Fluc samples had elevated Na+ concentrations that were linearly related to protein concentration. However, the Fluc dimer: Na+ ratio was sub-stoichiometric, with Na+ concentrations ~60% of expected for a stable 1 dimer: 1 Na+ complex. We speculate that the ratio is sub-stoichiometric due to Na+ dissociation during the ~2 hour purification process; however, the difficulty of performing Na+-free ICP-MS measurements precluded extensive investigation into the dissociation kinetics of the bound Na+. Nonetheless, these experiments suggested that Na+ is probably bound to the wildtype dimer with a long residence time of at least several hours in detergent (and perhaps longer in lipid bilayers).
Influence of central sodium ion on fluoride channel function
The slow dissociation kinetics make it impractical to assess the functional role of Na+ in the wildtype protein using electrophysiology or liposome efflux measurements. Therefore, in an effort to increase the rate of Na+ ion dissociation, we endeavored to identify a mutant that destabilized the Na+ binding site. Since backbone carbonyl oxygens comprise the entire coordination sphere, we tested adjacent sidechains for destabilization of the Na+ binding site. In the crystal structure, the N43 sidechain is within hydrogen bonding distance of both the backbone carbonyl and backbone amide of T81, the amino acid immediately adjacent to T80, whose carbonyl contributes to the Na+ binding site (Figure 1B, C). We investigated two mutations at this position, N43S and N43D. The N43 sidechain also plays a role in fluoride permeation, and introduction of serine or aspartate at this position slows fluoride throughput so that it falls within a range that can be monitored using liposome efflux experiments[5].
N43S and N43D both required Na+ for full activity. In the presence of 30 mM Na+, these proteins transport F− ion with a rate of ~2500 s−1. But after 30 minutes dialysis in a solution in which the Na+ is replaced with NMG+, F− efflux from liposomes was substantially reduced (Figure 2A, Figure S1A). Both mutants displayed saturable, Na+-dependent activity that fits a single-site binding isotherm (Figure 2B, Figure S1B), with Kd values of 1.5 mM for Bpe-N43D and 3.6 mM for Bpe-N43S. Na+ exchange is rapid, occurring within about 10 seconds for these mutants (Figure S2). Otherwise, the biochemical behavior of these proteins resembled WT: crosslinking experiments showed that, like WT, N43S remains a dimer in sodium-free detergent solutions (Figure 2C). Planar lipid bilayer electrophysiology confirmed that Na+ is impermeant. N43S retains WT-like selectivity for F− over Na+, with a reversal potential of 51.8 ± 1 mV in an 8-fold NaF activity gradient (Figure 2D).
Figure 2.
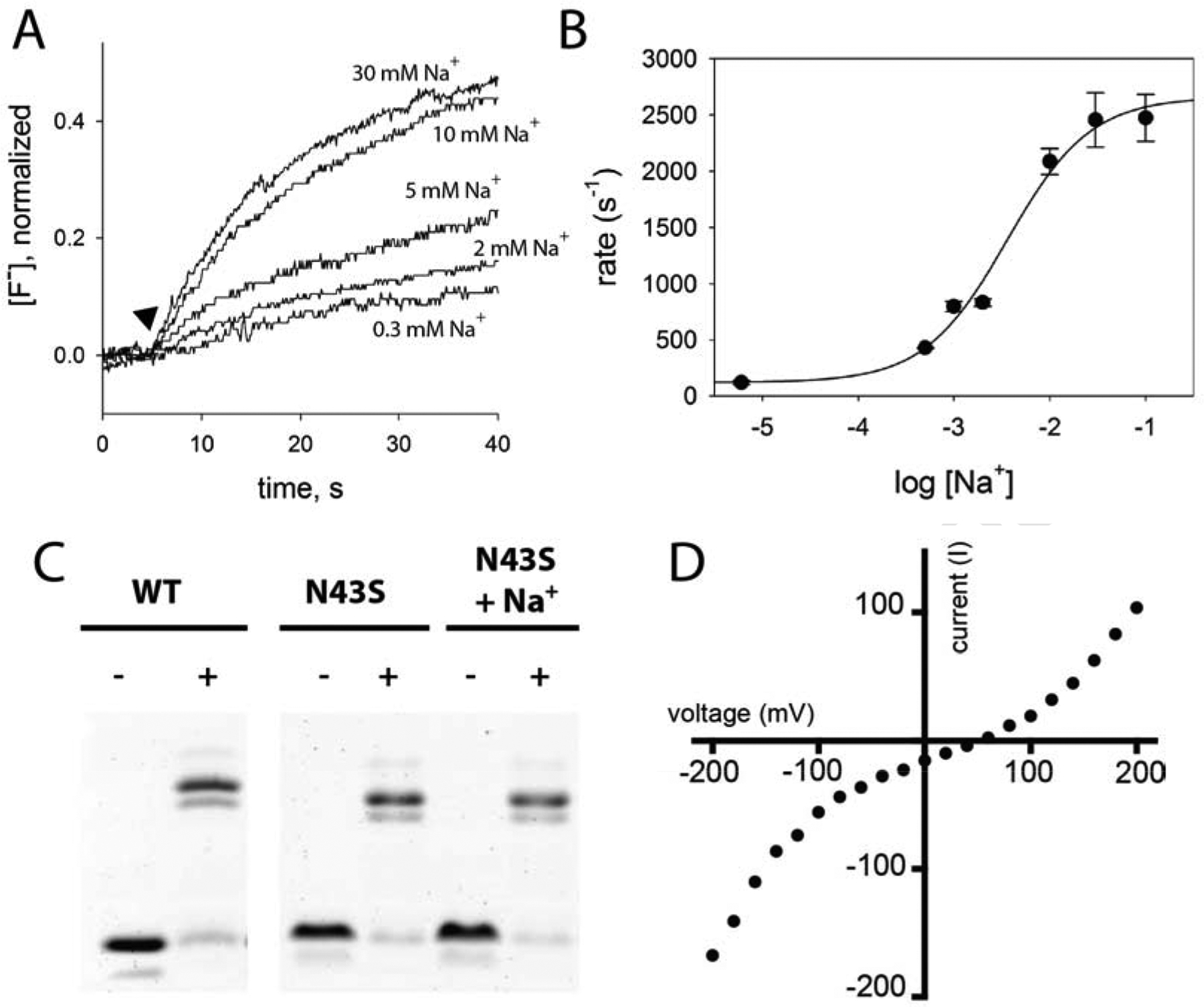
Na+ dependence of fluoride transport by Fluc-N43S. A. Fluoride efflux measured with a fluoride-specific electrode in the presence of varying Na+-isethionate, as indicated. Transport is initiated by addition of valinomycin (closed triangle). Traces are normalized with respect to total encapsulated fluoride. B. Rate of fluoride transport as a function of Na+ concentration. Solid line shows fit to a single-site binding model with a Kd of 3.6 mM, and limiting rates of 120 s−1 and 2660 s−1 for Na+-free and saturating Na+ conditions, respectively. Error bars represent the standard error of measurement (n=3). C. WT and N43S proteins purified in the absence of Na+ were crosslinked with glutaraldehyde where indicated, and run on an SDS-PAGE gel. Bands correspond to monomer and dimer positions. Where indicated, protein was incubated with 10 mM NaCl for one hour prior to crosslinking experiment. D. Representative macroscopic current-voltage relationship under an 8-fold NaF activity gradient (300 mM NaF/30 mM NaF). Reversal potential (51.8 ± 1 mV) is the mean and SEM of measurements from 5 independent bilayers.
In order to assess the specificity of the central site, we performed fluoride efflux experiments with the Na+ replaced by a series of different cations. For proteoliposomes incubated with the following cations, fluoride transport rates were indistinguishable from those incubated with NMG+: Rb+, Cs+, Sr+, Mg2+, Zn2+, NH4+, hydrazine, and hydroxylamine. In addition, since fluoride efflux assays are performed in the presence of 300 mM K+, we assumed that K+, too, is incapable of substituting for Na+. To validate this assumption, we monitored macroscopic fluoride currents for N43S using planar lipid bilayer electrophysiology. On a background of 10 mM Na+, we did not observe any change in fluoride currents upon addition of 10 mM K+ isethionate (Figure S3A).
Of cations tested, only Li+ exerted any influence on F− transport kinetics, but in contrast to Na+, it acted to inhibit F− efflux from liposomes (Figure S3B). To further assess channel inhibition by Li+, we monitored fluoride efflux from N43D and N43S proteoliposomes, varying Li+ on a background of near-saturating Na+ (Figure 3A, Figure S4A). In these experiments, increasing Li+ depressed F− transport, even in the presence of high Na+ (Figure 3B, Figure S4B). It seems probable that Na+ and Li+ compete for the same site. From the apparent Kd values of 5.8 mM (in the presence of 20 mM Na+) for N43D, and 960 μM (in the presence of 10 mM Na+) for N43S, we estimated Ki values of 670 μM and 250 μM, respectively. We also sought to exchange Na+ for Li+ in the WT protein, but consistent with our hypothesis that the Na+ exchanges slowly, we did not observe any Li+-dependent changes in F− currents using planar lipid bilayer electrophysiology (Figure S5).
Figure 3.
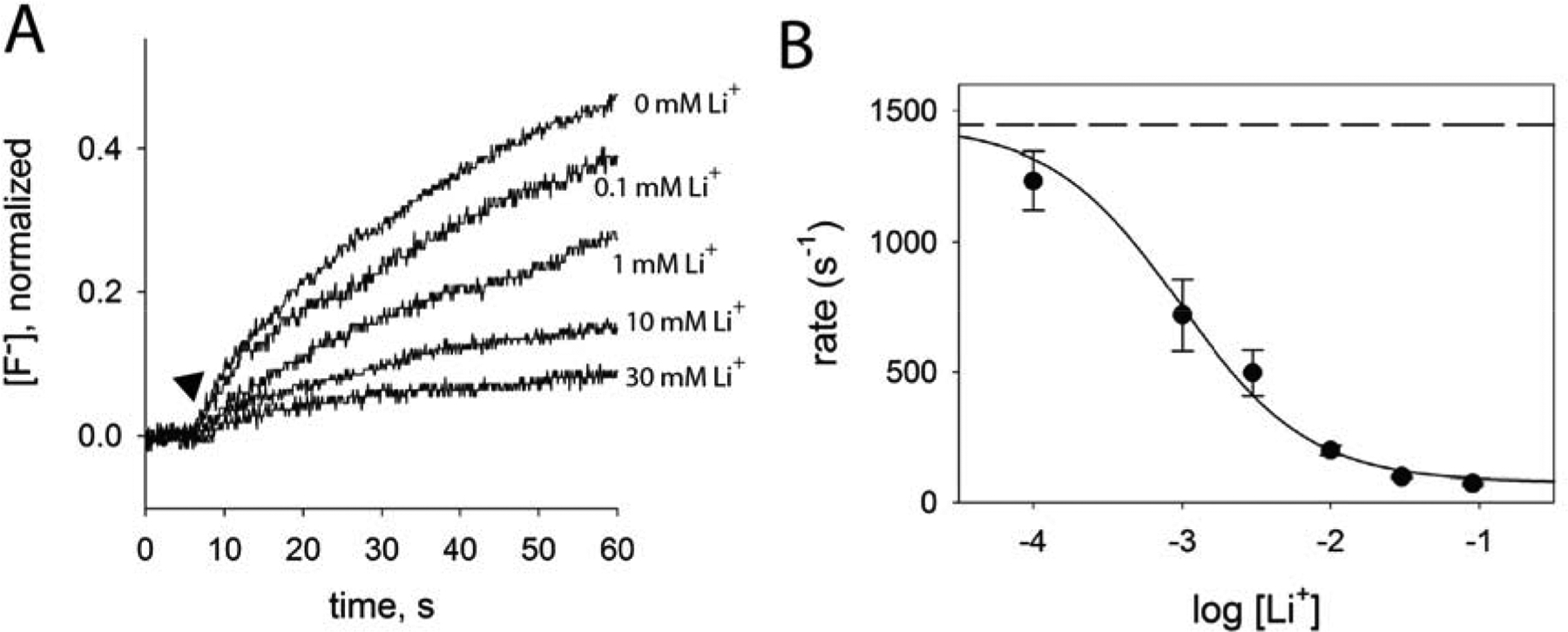
Li+ dependence of fluoride transport by Fluc-N43S. A. Fluoride efflux measured with a fluoride-specific electrode in the presence of 10 mM Na+-isethionate and indicated concentrations of Li+ isethionate. Transport is initiated by addition of valinomycin (closed triangle). Traces are normalized with respect to total encapsulated fluoride. B. Rate of fluoride transport as a function of Li+ concentration. Solid line shows fit to a single-site binding model, with an apparent Kd of 960 μM and a limiting rate in saturating Li+ of 80 s−1. The maximum rate (dashed line) was defined according to the rate of fluoride efflux with 10 mM Na+ and 0 mM Li+ (1450 s−1). Error bars represent the standard error of measurement (n=3).
In addition to N43, we also identified a second position, R22, for which mutation (to lysine) introduced Na+ dependency to fluoride transport. We hypothesize that this highly conserved Arg acts as an electrostatic barrier to Na+ dissociation. It is positioned in the aqueous vestibule, about 5 Å above the Na+, and tethered in place by H-bond interactions involving both subunits of the dimer (Figure 4A). Like N43, R22 might also help to stabilize the helical break near the Na+ binding site, since two of its H-bond partners are immediately adjacent to Na+-coordinating residues from the helical break. R22K liposomes required long, overnight dialysis times in Na+-free buffer to observe cation-dependent changes in fluoride transport. However, qualitatively similar behavior was observed for this mutant: fluoride efflux rates were reduced in the absence of Na+, and transport was further inhibited in the presence of Li+ (Figure 4B).
Figure 4.
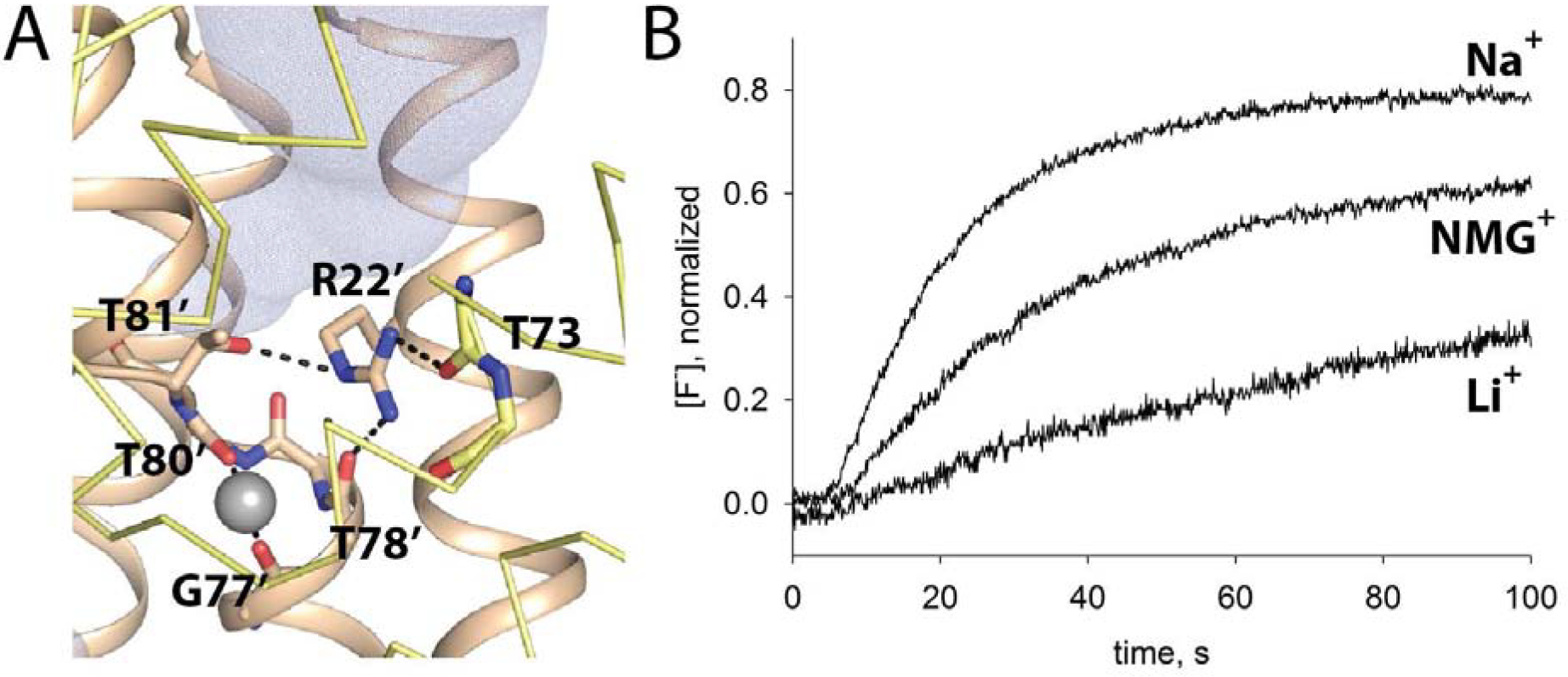
Cation dependence of fluoride transport by Fluc-R22K. A. R22 coordination. Coloring as in Figure 1. Yellow subunit is shown in ribbon format for clarity, except for T73 main chain atoms, which are shown as sticks. Residues within coordination distance of the central cation or within H-bonding distance of R22 are labeled. R22 and T81 sidechain, and G77-T80 main chain atoms are shown as sticks. B. Fluoride efflux from Bpe-R22K proteoliposomes incubated for 12 hours with 10 mM Na+, NMG+, or Li+ isethionate. Efflux monitored with a fluoride-specific electrode.
This set of mutant proteins shows that Na+ promotes F− efflux function, Li+ binding inhibits F− efflux function, and that the kinetics of cation exchange vary depending on the construct. If the Na+ remains stably bound because of a kinetic barrier to dissociation, then it is possible that elevated temperature might increase the rate of ion exchange so that the cation dependence of wildtype proteins can be evaluated. To test this, we dialyzed wildtype Bpe proteoliposomes at 38 °C for 16 hours in the presence of 10 mM Na+ with increasing concentrations of Li+. (Protein dialyzed at elevated temperature without either Na+ or Li+ tended to precipitate). We then tested fluoride efflux under the usual room temperature conditions. Whereas functional differences between wildtype proteins dialyzed with Na+ or Li+ at room temperature were minor (Figure 5A), protein dialyzed at elevated temperature recapitulated the cation dependence of fluoride transport described for the N43 and R22 mutants (Figure 5B). WT proteins dialyzed with Na+ alone exhibit familiar step-like fluoride efflux, reflecting a F− transport rate that exceeds the response time of the F− electrode[2]. As Li+ was increased on this Na+ background, the fraction of liposomes exhibiting protein-dependent fluoride efflux activity decreased, and the fluoride released during the second step, detergent disruption of the liposomes, was correspondingly higher (Figure 5B). We also tested whether a return to the Na+-only condition could rescue function of proteins dialyzed in 10 mM Na+ with 30 mM Li+. Indeed, after a second overnight dialysis step at elevated temperatures, in the presence of 10 mM Na+ only, fluoride transport was partially restored to ~50% of the original level. This partial loss of function may reflect loss of activity during the long dialysis at elevated temperatures, or incomplete exchange of Li+ by Na+. Nonetheless, it is qualitatively clear that Na+ and Li+ can be exchanged for one another in WT proteins, and that WT Bpe, and the N43 and R22 mutants, experience similar effects on fractional activity at similar concentrations of Na+ and Li+.
Figure 5.
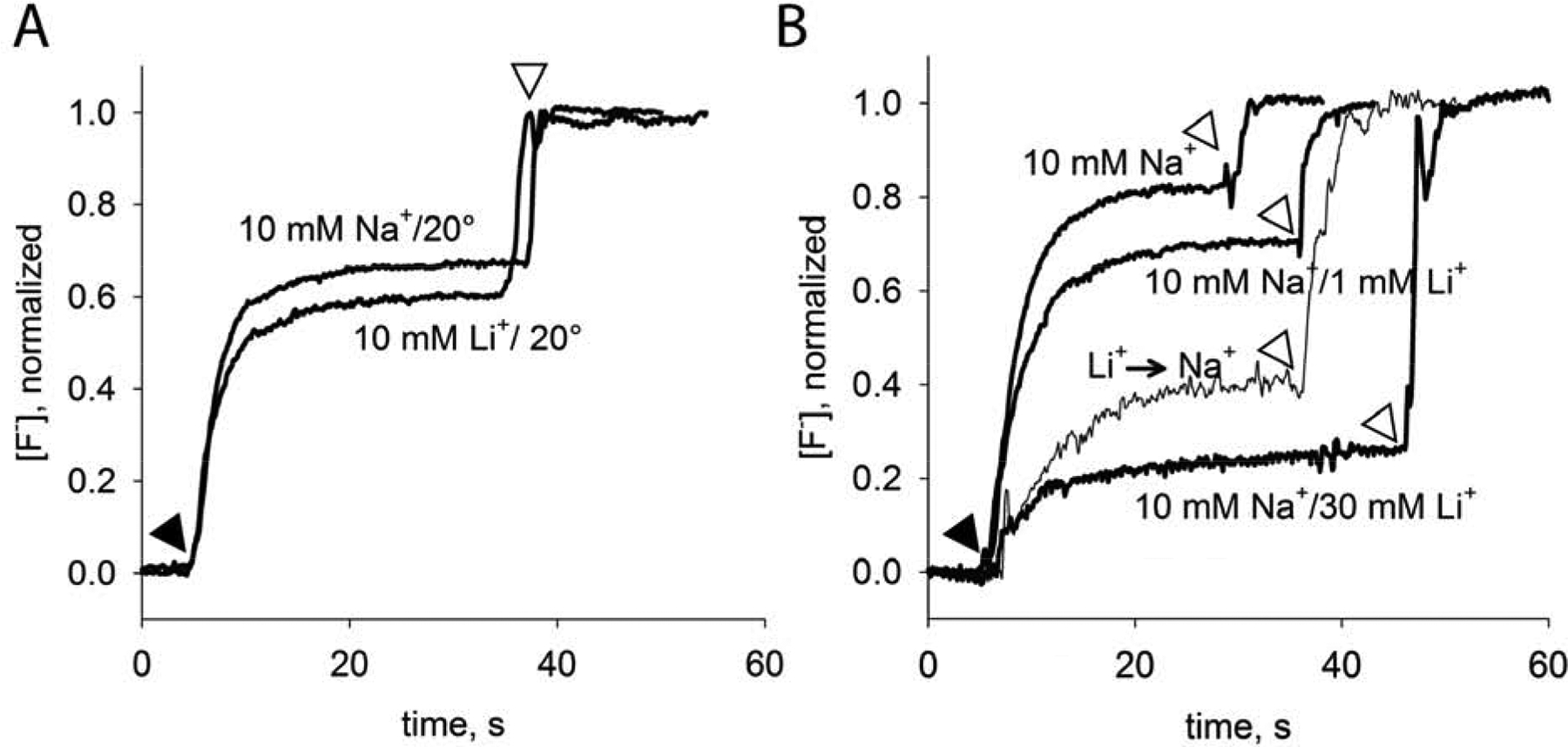
Cation dependence of fluoride transport by WT protein dialyzed at elevated temperatures. Fluoride efflux monitored with a F− selective electrode at room temperature. Valinomycin addition indicated by black triangle, and detergent addition indicated by open triangle. A. Fluoride efflux from WT Bpe proteoliposomes dialyzed for 16 hours at room temperature (21 °C) with 10 mM NaCl LiCl. B. Fluoride efflux from WT Bpe proteoliposomes dialyzed for 16 hours at 38 °C with 10 mM NaCl and the indicated concentration of LiCl. Liposomes dialyzed in 10 mM Na+/30 mM Li+ were returned to dialysis buffer containing 10 mM Na+ for overnight dialysis at 38 °C, indicated by the label “Li+ → Na+.”
Identification of structurally similar sodium binding sites in other membrane proteins
The experiments presented so far show that, in Fluc proteins, a Na+ ion crosslinks symmetry-related helical breaks at the dimer interface, and that it is required for the channels to adopt an open, conducting conformation. We wondered whether similar interfacial Na+ binding sites might exist in other membrane proteins. The presence of such sites could indicate a more general role for cation binding in stabilizing this common structural feature – either now, or at some point in the fold’s evolutionary history.
We analyzed Na+ binding sites in Na+-coupled transporters with a straightforward, two-domain inverted repeat architecture. Since the two domains are able to drift independently after duplication, a highly symmetric site like that observed in Fluc is unlikely. But such sites should nonetheless feature Na+ ions stabilized, in part, by carbonyl oxygens at breaks in the transmembrane helices, and would be close to the pseudosymmetry axis of the inverted repeat, simultaneously stabilizing discontinuous helices from two different domains. While many transporters do not possess sites meeting these criteria, we did identify examples in Na+-coupled transporters representing three different transporter folds: the vcINDY fold, the NhaA fold, and the LeuT fold (at the so-called Na1 site [14, 15]). Figure 6 shows three examples: citrate transporter CitS (vcINDY fold) [16], the Neisseria meningitidis homologue of the apical sodium-dependent bile acid transporter, ASBT (NhaA fold) [17], and the Drosophila dopamine transporter, DAT (LeuT fold) [17]. The inverted repeat’s pseudosymmetry axis is indicated in each view[18]. Although the perfect symmetry has degraded, and, in most cases, amino acids sidechains have encroached on the coordination sphere, these sites persist at the interface between the two inverted domains, where they are coordinated by carbonyl oxygens from pseudosymmetry-related helical discontinuities.
Figure 6.
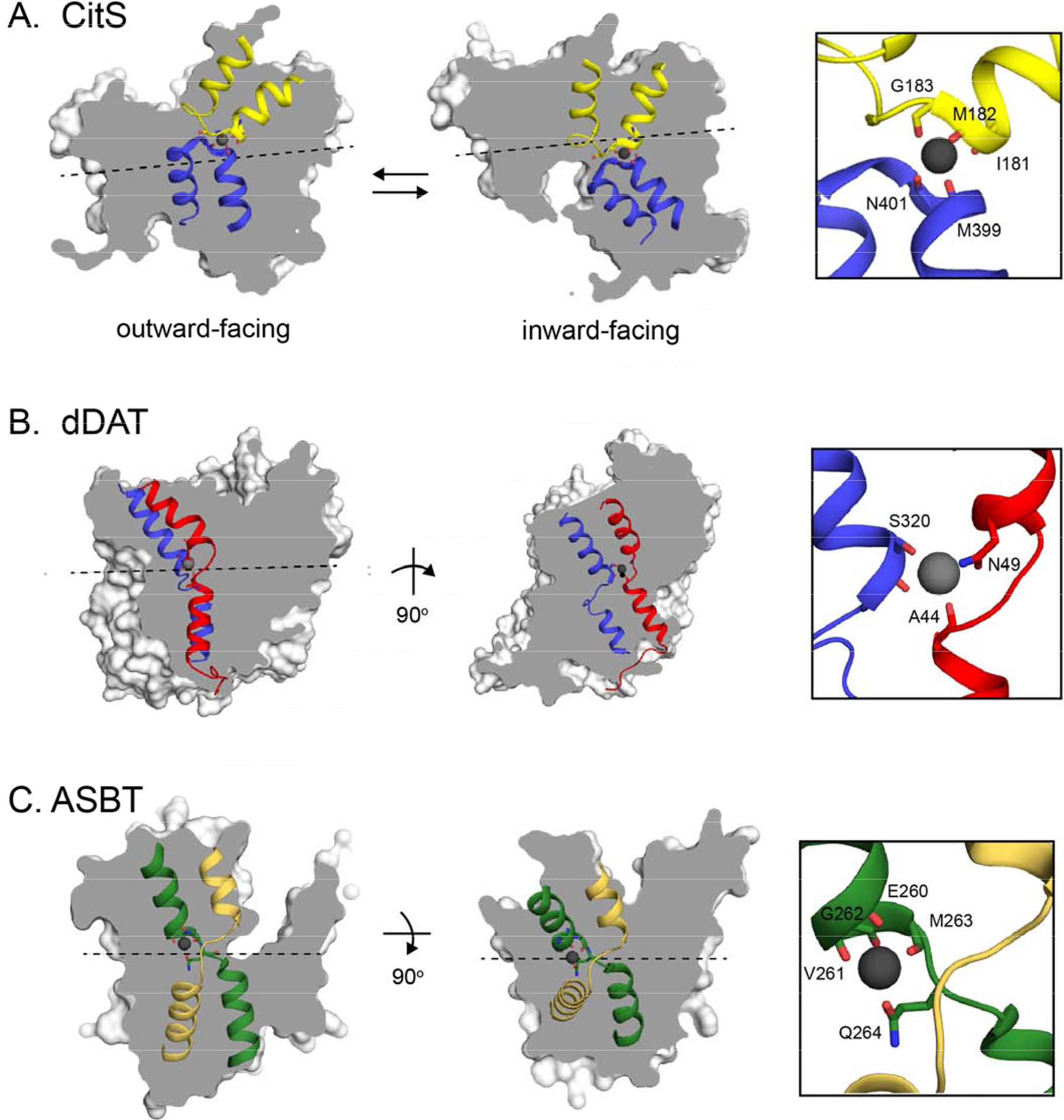
Sodium binding sites in sodium-coupled transporters with inverted repeat assembly. Twofold pseudosymmetry axes relating the inverted repeat domains were detected using encoMPASS[18] and are indicated. The overall structures are shown as surfaces, and the broken/hairpin helices related by twofold pseudosymmetry are shown as cartoons. Na+ ions are show as gray spheres, and coordinating ligands are shown as sticks. A. The Salmonella enterica citrate transporter CitS (pdb: 5A1S [16]). Hairpin helices 6 and 12, which are related by pseudosymmetry, are shown in blue and yellow. Two views shown are from two different protomers in the structure, representing two different steps in the transport cycle. The Na+ nexus is thought to move vertically with respect to the membrane during the transport cycle, with an average position on the two-fold pseudosymmetry axis at the interface between the two antiparallel domains. Main chain atoms of I181-G184 (yellow) and M399-R402 (blue) are shown as sticks, with the Na+ coordinated by backbone carbonyl oxygens from I181 and G183 from hairpin 6, and M399 and N401 from hairpin 12. B. The Na1 site in the Drosophila melanogaster dopamine transporter DAT (pdb: 4M48[14]. Helices 1 and 6, which are related by inverted symmetry, are shown in red and blue. Two views of the sodium binding site are shown: left, through the plane of the membrane and perpendicular to the pseudosymmetry axis, and right, rotated 90° so that the view is through the plane of the membrane and along the pseudosymmetry axis (the axis is represented as a black dot and located just below the sodium). Main chain atoms of A44-V45 (red) and S320 (blue), along with sidechains N49 (red) and S320 (blue) are shown as sticks. The Na+ is coordinated by backbone oxygens from A44 and S320, along with the S320 and N49 sidechains. For both breaks, the backbone atom that coordinates the Na+ is also the atom that initiates the discontinuity in the substrate-binding helix. C. The Neisseria meningitidis homologue of the apical sodium-dependent bile acid Transporter, ASBT (pdb: 3ZUX [17]). Broken helices 4 and 9, which are related by pseudosymmetry, are shown in green and yellow. A view parallel to the plane of the membrane (left) and a top-down view (right) are shown. Main chain atoms of E260-Q264, and the Q264 sidechain (green subunit) are shown as sticks. The Na+ is coordinated at the crossover of the breaks by the backbone oxygens from E260, V261, and M263, along with the Q264 sidechain.
Discussion:
We provide evidence here that a Na+ ion, buried at the dimer interface, is an essential structural component of the Fluc fluoride channels. Although not itself transported, its presence is required for Fluc to adopt a conducting conformation. This finding has implications both for understanding the mechanism of fluoride conduction by this idiosyncratic family of fluoride channels, and more broadly, for understanding the structures, mechanisms, and evolutionary origins of Na+ binding sites in widespread membrane transport proteins.
Sequestered from aqueous solution, we imagine that the sodium ion is inserted during protein assembly and locked in place electrostatically by a conserved arginine in the vestibule, which provides a kinetic barrier to cation dissociation that can be overcome in wildtype proteins by incubation at elevated temperature. Although the kinetics of exchange for WT proteins are much slower than for the N43 mutants, precluding quantitative analysis of cation binding to WT proteins, the cations exert similar effects on the fractional activity of the WT and mutant proteins at similar concentrations. We therefore expect that the equilibrium constant for cation binding is likely similar to that measured for the N43 mutants (in the mM range), which is in harmony with biological Na+ concentrations.
The Na+ contributes to the highly electropositive central region of the channel, and as such, likely contributes to electrostatic stabilization of permeant fluoride ions. However, positive charge alone is not sufficient to support fluoride transport since Li+ -bound channels do not conduct fluoride. Na+ and Li+ differ in coordination distance by about 0.2 Å, and subtle alterations in geometry to account for this difference might have meaningful allosteric effects on F− throughput. Li+ is a common competitor for Na+ in protein binding sites[19], and for this site in particular, the tetrahedral geometry appears optimal for Li+. Na+, in contrast, prefers five ligands in its coordination sphere. However, coordination by four ligands is not prohibited, and is observed in about 15% of sodium-containing proteins in the protein databank[20, 21]. These geometric preferences might explain why Li+ binds ~5–10-fold tighter than Na+. And while Li+ is probably not a biologically relevant ligand – it is not prevalent in biological systems – it should be noted that in 2 of the 5 unique structures of Fluc channels [5, 11, 12], Li+ is present together with Na+ in the crystallization condition, making crystallographic assignment of the bound cation uncertain. Reexamination of these structures did not reveal any obvious differences between structures obtained with and without Li+, and several lines of evidence suggest that these crystal structures probably represent the conducting conformation of the channel [11, 22]. Unfortunately, we were unable to obtain N43S or R22K crystals that diffracted to high resolution.
In addition to its role in supporting F− conduction, it remains to be explored whether the Na+ ion might also contribute to fluoride channel assembly and/or biogenesis. At the subunit concentrations and timescales of these experiments, Fluc remains dimeric with or without Na+. However, at biological dilutions of protein, destabilization of the dimer interface by removing the central cation might have much larger implications for proper channel oligomerization[23]. A role for the Na+ could also be imagined during protein folding, stabilizing helical breaks in the nascent protein as the overall topology and fold are achieved. Indeed, one trick that has been employed in the de novo design of protein-protein interfaces is the inclusion of a metal coordination site, whose strict geometric requirements lends specificity to the assembly of the oligomer[24–26].
Finally, it is interesting to consider how the Fluc Na+ site compares to Na+ binding sites in other membrane proteins. Coordination by carbonyl oxygens is a common feature of biological Na+ binding sites[27, 28], and for a slew of Na+-coupled transporters, substrate Na+ ions are coordinated by several carbonyl oxygens made available by breaks in the transmembrane helices. At this position, the cations help stabilize both the helix dipole and the unsatisfied H-bonding potential of the carbonyls within the discontinuous helix. In turn, the helical breaks provide substrate binding capacity, flexibility for conformational changes, and help define aqueous pockets and vestibules [29]. Indeed, we identified examples of Na+ binding sites similar to Fluc’s in proteins representing three major structural classes of sodium-coupled transporters, vcINDY, NhaA, and LeuT.
Thus, what is remarkable about the Fluc Na+ site is not its chemical details, but that it is, to our knowledge, the first example of a sodium ion that has been identified as a strictly structural element, and not a transported substrate. Dual topology proteins like Fluc have been called “evolutionary fossils” due to their rudimentary assembly as antiparallel homodimers[30, 31]. This topology is an evolutionary forebear to the inverted repeat architecture that defines a plurality of membrane transporters[6]. The Fluc structural sodium raises the possibility that, for ancient membrane proteins, cation binding stabilized and preserved functionally useful discontinuities in transmembrane helices – and that some binding sites for Na+-coupled transport may have evolved from structural Na+ sites resembling that of Fluc.
Despite the structural similarities described above, it should be noted that a key difference between the sodium binding sites of Fluc and the secondary active transporters stands out as well: in the examples in Figure 6, the sodium ions are coordinated by 5 ligands, and in many other sodium-coupled transporters, the cation is coordinated by six. Why does Fluc only have four? We speculate that the number of ligands is a result of two factors: 1) the unusual two-fold symmetry that results from dual topology architecture, which demands an even number of ligands, and 2) the functional role of this protein in transporting anions. For sodium ion coordination spheres with 6 or more ligands, typically one of the ligands is negatively charged (a sidechain carboxylate, for example)[21]. However, introduction of a negatively charged sidechain near the center of Fluc might interfere electrostatically with fluoride binding and transport. Some eukaryotic members of the Fluc family possess a more conventional two-domain inverted repeat architecture[32]; it would be interesting to know whether these proteins maintain a tetrahedral Na+ binding site, or if, freed from the constraints of 2-fold homodimeric symmetry, they gain a 5th ligand.
Finally, our results also emphasize the importance of functional transport stoichiometry measurements to corroborate structural observations for Na+-coupled transporters. Another illustration of this was recently provided for the amine transporters possessing the LeuT fold (including DAT). Differences between the measured transport stoichiometry and the number of structurally observed Na+ ions provoked the unifying hypothesis that Na1 (the Na+ bound at the helical breaks along the pseudosymmetry axis in Figure 5C) is not released to the inner side of the membrane during the transport cycle, but is instead shuttled back across the membrane ahead of the next transport cycle [33–36]. Considered together with our experiments, this implies that Na+ ions might play structural roles in more membrane proteins than has been appreciated previously.
Materials and Methods:
Chemicals:
Most reagents were obtained from Sigma-Aldrich. High purity KCl (99.997% metals basis) was from Alfa-Aesar and ultrapure trace elemental analysis grade water and nitric acid were from Fisher and Fluka, respectively. Isethionate salts were prepared from isethionic acid (Wako Chemicals, Richmond, VA) and potassium hydroxide, sodium hydroxide, lithium hydroxide, or N-methyl-glucamine (NMG+) base.
Protein Purification and Crosslinking:
All experiments described were performed using Fluc channels from Bordetella pertussis, Fluc-Bpe, bearing two functionally neutral mutations, R29K and E94S, that increase protein yield[5]. Additional mutations (N43D, N43S, or R22K) were introduced to this background and verified using standard molecular biology techniques. Protein purification was carried out as described previously[2]. Briefly, His-tagged proteins were expressed in E. coli (C43) and purified via Co2+ affinity and size exclusion using a Superdex 200 column. Typically SEC buffer contained 100 mM KCl, 10 mM (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) -KOH pH 7, 10 mM NaF, 5 mM decyl β -D-maltopyranoside (DM) unless otherwise indicated. Crosslinking experiments were performed with proteins bearing an alanine-alanine insertion at the N-terminus, between the methionine start codon and the coding sequence. For crosslinking, ~0.1 mg/mL freshly purified protein in FPLC buffer was incubated with 0.125% glutaraldehyde (~500-fold molar excess) for 30 minutes. The reaction was quenched by addition of Tris-HCl, pH 7.5, and samples were run on a 15% SDS-PAGE gel.
Sodium-free protein purification and ICP-MS:
In order to purify protein without contaminating sodium ion, Na+ was replaced by K+ in purification buffers beginning at the cobalt column wash step. Since glassware is a common source of Na+ contamination, plasticware was used for all steps of the purification. Plasticware, chromatography tubing, and the size exclusion column were washed with 0.1 M HCl and trace elemental analysis grade water prior to use. FPLC buffer contained 100 mM KCl, 5 mM HEPES-KOH, 5 mM DM. Potassium salts of the highest purity (99.997% metals basis) were used. For ICP-MS experiments, fractions were collected from the main protein elution peak off the size exclusion column. These were combined with 0.1 M HNO3 (ICP-MS grade) and boiled for 10 minutes to denature and hydrolyze the protein. Sodium content was analyzed using a Perkin-Elmer Nexion 2000 ICP-MS instrument with Na+ filters. Raw ICP measurements were converted into molarity by comparison to a standard curve, which was linear over this concentration range. Background Na+ from the SEC purification buffer (typically ~2–6 μM) was measured for each protein sample and subtracted from the total to yield the final Na+ concentration. Samples were derived from 3 (CLC) or 5 (Fluc-Bpe) independent purifications. Errors in technical replicates were < 1%.
Fluoride efflux from proteoliposomes:
Fluoride efflux experiments were carried out as described previously [2, 37]. Protein was reconstituted in liposomes (10 mg/mL E. coli polar lipids, Avanti) at a protein to lipid ratio of 0.2 μg protein/mg lipid. Liposomes were formed by dialysis over 1.5 days against ~6 L of buffer containing 300 mM KF, 30 mM Na+ isethionate, 10 mM HEPES-KOH, pH 7. To exchange the cation, a fourth dialysis step was performed, with the 30 mM Na+ isethionate replaced by the test cation (0.1–30 mM as an isethionate or chloride salt, with the ionic strength maintained at 30 mM with NMG+ isethionate). The final dialysis was 30 minutes for N43 mutants, overnight for R22K, and overnight in a 38 °C oven for WT. Samples were freeze/thawed three times before extrusion 21 times through a 400 nm filter. External solution was exchanged by passing liposomes over a Sephadex G50 column equilibrated in experiment buffer (300 mM K+ isethionate, 10 mM HEPES-KOH pH 7, with test cation/NMG+ isethionate added to match the intraliposomal concentration). For the flux assay, liposomes were diluted 20-fold in matching experiment buffer. Fluoride efflux was initiated by addition of 1 μM valinomycin and monitored using a fluoride-selective electrode. After fluoride efflux reached steady-state, liposomes were disrupted by addition of 50 mM n-octyl-β- D-glucoside to release remaining encapsulated fluoride. Fluoride efflux was normalized against total encapsulated fluoride.
Planar bilayer electrophysiology:
For electrophysiological recordings in planar lipid bilayers, purified Fluc proteins were reconstituted into liposomes at 5 μg protein/mg lipid, as described[4, 22]. Cis- and trans-chambers buffers were prepared with ionic conditions described in the text, and contained 10 mM 3-(N-morpholino)propanesulfonic (MOPS)-KOH or –NMG-OH as appropriate, pH 7. Recordings were acquired at −200 mM holding voltage, electronically filtered at 1 kHz during acquisition, and digitally filtered to 500 Hz for analysis. Traces shown are representative of recordings from at least three independent bilayers.
Supplementary Material
Figure S1. Na+ dependence of fluoride transport by Bpe-N43D. A. Fluoride efflux measured with a fluoride-specific electrode in the presence of varying Na+-isethionate, as indicated. Transport is initiated by addition of valinomycin (closed triangle). Traces are normalized with respect to total encapsulated fluoride. B. Rate of fluoride transport as a function of Na+ concentration. Solid line shows fit to a single-site binding model with a Kd of 1.5 mM, and limiting rates of 210 s−1 and 2400 s−1 for Na+-free and saturating Na+ conditions, respectively. Error bars represent the standard error of measurement (n=3–5).
Figure S2. Fast kinetics of Na+ exchange for Bpe-N43S. Fluoride efflux measured with a fluoride-specific electrode. Proteoliposomes were incubated with 20 mM Na+-isethionate for the indicated time before transport is initiated by addition of valinomycin (closed triangle). After steady state is reached, remaining fluoride is released from the liposomes by addition of detergent (e.g., white triangle). Traces are normalized with respect to total encapsulated fluoride.
Figure S3. Fluoride transport by N43 mutants in the presence of different cations. A. Fluoride currents mediated by Fluc-N43S in response to addition of 10 mM K+ isethionate at the indicated time. Recording solution contains 10 mM Na+ and 290 mM NMG+ to maintain ionic strength. Zero current level indicated by the dashed line. B. Fluoride efflux from Fluc-N43D proteoliposomes with 10 mM Na+, NMG+, or Li+ isethionate, as indicated. Efflux monitored with a fluoride-specific electrode. 300 mM K+ is present in all experiments.
Figure S4. Li+ dependence of fluoride transport by Bpe-N43D. A. Fluoride efflux measured with a fluoride-specific electrode in the presence of 20 mM Na+-isethionate and indicated concentrations of Li+ isethionate. Transport is initiated by addition of valinomycin (closed triangle), and after steady state is reached, remaining fluoride is released from the liposomes by addition of detergent (e.g., white triangle). Traces are normalized with respect to total encapsulated fluoride. B. Rate of fluoride transport as a function of Li+ concentration. Solid line shows fit to a single-site binding model, with an apparent Kd of 5.8 mM. The maximum rate (dashed line) was defined according to the rate of fluoride efflux with 0 mM Li+ (2440 s−1). Error bars represent the standard error of measurement (n=3).
Figure S5. Electrophysiological recording of WT Fluc macroscopic currents. Currents recorded in 300 mM NMG+F− 10 mM Li+ isethionate added at the indicated time. Dashed line indicates zero current level. Recording is representative of experiments performed with three independent bilayers
Highlights:
Fluc family fluoride channels are assembled as primitive antiparallel homodimers.
A sodium ion is stably bound at the dimer interface.
This bound Na+ is essential for channels to adopt an open, conducting conformation.
Examples of similar Na+ binding sites exist in other transporters.
Acknowledgements:
We thank Melanie Ernst for comments on the manuscript. This work was supported by National Institutes of Health grant R35-GM128768 and Alfred P. Sloan Research Fellowship to R.B. Stockbridge. E.H. was supported by an Interdisciplinary Opportunities in Biophysics REU at the University of Michigan (PHY-1262920).
Abbreviations:
- TM
transmembrane
- ICP-MS
inductively-coupled plasma-mass spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests:
The authors declare no competing interests.
References:
- 1.Baker JL, Sudarsan N, Weinberg Z, Roth A, Stockbridge RB, Breaker RR. Widespread genetic switches and toxicity resistance proteins for fluoride. Science. 2012;335:233–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stockbridge RB, Robertson JL, Kolmakova-Partensky L, Miller C. A family of fluoride-specific ion channels with dual-topology architecture. Elife. 2013;2:e01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji C, Stockbridge RB, Miller C. Bacterial fluoride resistance, Fluc channels, and the weak acid accumulation effect. The Journal of General Physiology. 2014;144:257–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stockbridge RB, Koide A, Miller C, Koide S. Proof of dual-topology architecture of Fluc F- channels with monobody blockers. Nat Commun. 2014;5:5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stockbridge RB, Kolmakova-Partensky L, Shane T, Koide A, Koide S, Miller C, et al. Crystal structures of a double-barrelled fluoride ion channel. Nature. 2015;525:548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Forrest LR. Structural Symmetry in Membrane Proteins. Annu Rev Biophys. 2015;44:311–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasie I, Steiner-Mordoch S, Gold A, Schuldiner S. Topologically random insertion of EmrE supports a pathway for evolution of inverted repeats in ion-coupled transporters. J Biol Chem. 2010;285:15234–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller R, Ziegler C, Schneider D. When two turn into one: evolution of membrane transporters from half modules. Biol Chem. 2014;395:1379–88. [DOI] [PubMed] [Google Scholar]
- 9.Last NB, Kolmakova-Partensky L, Shane T, Miller C. Mechanistic signs of double-barreled structure in a fluoride ion channel. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Last NB, Sun S, Pham MC, Miller C. Molecular determinants of permeation in a fluoride-specific ion channel. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McIlwain BC, Newstead S, Stockbridge RB. Cork-in-Bottle Occlusion of Fluoride Ion Channels by Crystallization Chaperones. Structure. 2018;26:635–9 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turman DL, Cheloff AZ, Corrado AD, Nathanson JT, Miller C. Molecular Interactions between a Fluoride Ion Channel and Synthetic Protein Blockers. Biochemistry. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Last NB, Stockbridge RB, Wilson AE, Shane T, Kolmakova-Partensky L, Koide A, et al. A CLC-type F(−)/H(+) antiporter in ion-swapped conformations. Nat Struct Mol Biol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl--dependent neurotransmitter transporters. Nature. 2005;437:215–23. [DOI] [PubMed] [Google Scholar]
- 16.Wohlert D, Grotzinger MJ, Kuhlbrandt W, Yildiz O. Mechanism of Na(+)-dependent citrate transport from the structure of an asymmetrical CitS dimer. Elife. 2015;4:e09375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu NJ, Iwata S, Cameron AD, Drew D. Crystal structure of a bacterial homologue of the bile acid sodium symporter ASBT. Nature. 2011;478:408–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarti E, Aleksandrova AA, Ganta SK, Yavatkar AS, Forrest LR. EncoMPASS: an online database for analyzing structure and symmetry in membrane proteins. Nucleic Acids Res. 2019;47:D315–D21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudev T, Mazmanian K, Lim C. Competition between Li(+) and Na(+) in sodium transporters and receptors: Which Na(+)-Binding sites are “therapeutic” Li(+) targets? Chem Sci. 2018;9:4093–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lev B, Roux B, Noskov SY. Relative Free Energies for Hydration of Monovalent Ions from QM and QM/MM Simulations. J Chem Theory Comput 2013;9:4165–75. [DOI] [PubMed] [Google Scholar]
- 21.Lev B, Noskov SY. In: Kretsinger RH, Uversky VN, Permyakov EA, editors. Encyclopedia of Metalloproteins. New York, New York: Springer; 2013. p. 2112–8. [Google Scholar]
- 22.Turman DL, Stockbridge RB. Mechanism of single- and double-sided inhibition of dual topology fluoride channels by synthetic monobodies. J Gen Physiol. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chadda R, Krishnamani V, Mersch K, Wong J, Brimberry M, Chadda A, et al. The dimerization equilibrium of a ClC Cl(−)/H(+) antiporter in lipid bilayers. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salgado EN, Ambroggio XI, Brodin JD, Lewis RA, Kuhlman B, Tezcan FA. Metal templated design of protein interfaces. Proc Natl Acad Sci U S A. 2010;107:1827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Der BS, Edwards DR, Kuhlman B. Catalysis by a de novo zinc-mediated protein interface: implications for natural enzyme evolution and rational enzyme engineering. Biochemistry. 2012;51:3933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Der BS, Machius M, Miley MJ, Mills JL, Szyperski T, Kuhlman B. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J Am Chem Soc. 2012;134:375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torrance GM, Leader DP, Gilbert DR, Milner-White EJ. A novel main chain motif in proteins bridged by cationic groups: the niche. J Mol Biol. 2009;385:1076–86. [DOI] [PubMed] [Google Scholar]
- 28.Dudev T, Mazmanian K, Lim C. Factors controlling the selectivity for Na(+) over Mg(2+) in sodium transporters and enzymes. Phys Chem Chem Phys. 2016;18:16986–97. [DOI] [PubMed] [Google Scholar]
- 29.Screpanti E, Hunte C. Discontinuous membrane helices in transport proteins and their correlation with function. J Struct Biol. 2007;159:261–7. [DOI] [PubMed] [Google Scholar]
- 30.Schuldiner S What Can a Living Fossil Tell Us About Evolution and Mechanism of Ion-Coupled Transporters: The Story of Small Multidrug Transporters In: Kramer R, Ziegler C, editors. Membrane Transport Mechanism. Berlin: Springer; 2014. [Google Scholar]
- 31.Macdonald CB, Stockbridge RB. A topologically diverse family of fluoride channels. Curr Opin Struct Biol. 2017;45:142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li S, Smith KD, Davis JH, Gordon PB, Breaker RR, Strobel SA. Eukaryotic resistance to fluoride toxicity mediated by a widespread family of fluoride export proteins. Proceedings of the National Academy of Sciences. 2013;110:19018–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang YW, Tavoulari S, Sinning S, Aleksandrova AA, Forrest LR, Rudnick G. Structural elements required for coupling ion and substrate transport in the neurotransmitter transporter homolog LeuT. Proc Natl Acad Sci U S A. 2018;115:E8854–E62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coleman JA, Yang D, Zhao Z, Wen PC, Yoshioka C, Tajkhorshid E, et al. Serotonin transporter-ibogaine complexes illuminate mechanisms of inhibition and transport. Nature. 2019;569:141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudnick G, Sandtner W. Serotonin transport in the 21st century. J Gen Physiol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudnick G, Talvenheimo J, Fishkes H, Nelson PJ. Sodium ion requirements for serotonin transport and imipramine binding. Psychopharmacol Bull. 1983;19:545–9. [PubMed] [Google Scholar]
- 37.Brammer AE, Stockbridge RB, Miller C. F-/Cl- selectivity in CLCF-type F-/H+ antiporters. J Gen Physiol. 2014;144:129–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Na+ dependence of fluoride transport by Bpe-N43D. A. Fluoride efflux measured with a fluoride-specific electrode in the presence of varying Na+-isethionate, as indicated. Transport is initiated by addition of valinomycin (closed triangle). Traces are normalized with respect to total encapsulated fluoride. B. Rate of fluoride transport as a function of Na+ concentration. Solid line shows fit to a single-site binding model with a Kd of 1.5 mM, and limiting rates of 210 s−1 and 2400 s−1 for Na+-free and saturating Na+ conditions, respectively. Error bars represent the standard error of measurement (n=3–5).
Figure S2. Fast kinetics of Na+ exchange for Bpe-N43S. Fluoride efflux measured with a fluoride-specific electrode. Proteoliposomes were incubated with 20 mM Na+-isethionate for the indicated time before transport is initiated by addition of valinomycin (closed triangle). After steady state is reached, remaining fluoride is released from the liposomes by addition of detergent (e.g., white triangle). Traces are normalized with respect to total encapsulated fluoride.
Figure S3. Fluoride transport by N43 mutants in the presence of different cations. A. Fluoride currents mediated by Fluc-N43S in response to addition of 10 mM K+ isethionate at the indicated time. Recording solution contains 10 mM Na+ and 290 mM NMG+ to maintain ionic strength. Zero current level indicated by the dashed line. B. Fluoride efflux from Fluc-N43D proteoliposomes with 10 mM Na+, NMG+, or Li+ isethionate, as indicated. Efflux monitored with a fluoride-specific electrode. 300 mM K+ is present in all experiments.
Figure S4. Li+ dependence of fluoride transport by Bpe-N43D. A. Fluoride efflux measured with a fluoride-specific electrode in the presence of 20 mM Na+-isethionate and indicated concentrations of Li+ isethionate. Transport is initiated by addition of valinomycin (closed triangle), and after steady state is reached, remaining fluoride is released from the liposomes by addition of detergent (e.g., white triangle). Traces are normalized with respect to total encapsulated fluoride. B. Rate of fluoride transport as a function of Li+ concentration. Solid line shows fit to a single-site binding model, with an apparent Kd of 5.8 mM. The maximum rate (dashed line) was defined according to the rate of fluoride efflux with 0 mM Li+ (2440 s−1). Error bars represent the standard error of measurement (n=3).
Figure S5. Electrophysiological recording of WT Fluc macroscopic currents. Currents recorded in 300 mM NMG+F− 10 mM Li+ isethionate added at the indicated time. Dashed line indicates zero current level. Recording is representative of experiments performed with three independent bilayers