

Abstract
Hyperuricemia is characterized by elevated uric acid (UA) levels on blood, which can lead to gout, a common pathology. These high UA levels are associated with increased purine ingestion and metabolization and/or its decreased excretion. In this field, xanthine oxidase (XO), by converting hypoxanthine and xanthine to UA, plays an important role in hyperuricemia control. Based on limitations and adverse effects associated with the use of allopurinol and febuxostat, the most known approved drugs with XO inhibitory effect, the search for new molecules with XO activity is growing. However, despite the high number of studies, it was found that the majority of tested products with relevant XO inhibition were left out, and no further pharmacological evaluation was performed. Thus, in the present review, available information published in the past six years concerning isolated molecules with in vitro XO inhibition complemented with cytotoxicity evaluation as well as other relevant studies, including in vivo hypouricemic effect, and pharmacokinetic/pharmacodynamic profile was compiled. Interestingly, the analysis of data collected demonstrated that molecules from natural sources or their mimetics and semisynthetic derivatives constitute the majority of compounds being explored at the moment by means of in vitro and in vivo animal studies. Therefore, several of these molecules can be useful as lead compounds and some of them can even have the potential to be considered in the future clinical candidates for the treatment of hyperuricemia.
1. Introduction
Xanthine oxidase (XO) is a key enzyme in purine catabolism and, physiologically, catalyzes the oxidation of hypoxanthine to xanthine and then to uric acid (UA) with concomitant reactive oxygen species (ROS) production [1, 2]. Nonetheless, enzymes such as hypoxanthine-guanine phosphoribosyl transferase (HGPRT), phosphoribosyl pyrophosphate synthetase (PRPS), and phosphoribosyl pyrophosphate aminotransferase (PRPPAT) also participate in purine metabolic pathway [3]. The overproduction or low excretion of urate can lead to hyperuricemia and subsequently to gout [3, 4]. In the field of low excretion, anion-exchanging uptake transporters (OAT1, OCT2, URAT1, and GLUT9) have also been reported to play important roles in the regulation of serum UA [3].
The most relevant target conditioning serum UA levels is XO, and excessive activity of this enzyme can lead to a pathological UA overproduction [5]. Therefore, its inhibition is of high interest, mainly in the treatment of gout, being allopurinol, febuxostat, and topiroxostat (Figure 1), the most known XO inhibitors. In addition to these clinically used drugs, over the years, several other molecules with XO inhibitory activity have been described [6–9]. Despite the existence of some reviews in this topic [6, 7, 9–11], it is necessary to complement these works with a study focusing on the hit to lead evolution in the development of new XO inhibitors with improved potency and safety when compared with the clinically used drugs. Interestingly, as can be seen in this review, natural molecules and semisynthetic analogues and derivatives constitute a large group of compounds being explored at the moment in this context.
Figure 1.
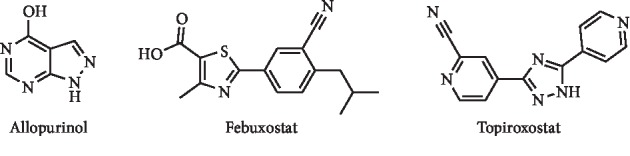
Structures of the clinically used XO inhibitors.
2. Materials and Methods
The present review covers the literature published in the last 6 years and develops the most relevant studies that focus not only in XO inhibition but also in the integration of these data with other relevant information for the hit to lead evolution in the development of new XO inhibitors. In this view, cytotoxicity studies, in vivo hypouricemic effect and/or pharmacokinetic/pharmacodynamic profile, are also discussed. For this, a research on scientific databases Web of Science, Pubmed, Scopus, and others was carried out using associations of the following key terms and synonyms: XO, cytotoxicity, hyperuricemia, hypouricemic, or pharmacokinetic profile.
The review is organized according to the origin of the compounds (synthetic and natural/semisynthetics/mimetics of natural structures) and then according to their chemical structures. The structures of the scaffolds and/or the specific compounds with the most promising results are presented in figures.
3. Results
3.1. Synthetic Derivatives
3.1.1. Purine Nucleus Analogues
Based on allopurinol structure, Rangappa and coworkers reported four 5H-thiadiazolopyrimidin-5-one analogues (Figure 2) as potent XO inhibitors with IC50 values in the range from 269 to 634 nM (XO from bovine milk) [12]. These compounds were later investigated on oxonate-treated rats, and it was observed that serum UA and creatinine levels significantly dropped at doses of 50 and 100 mg/kg. On the other hand, these pyrimidine analogues could significantly attenuate liver and kidney necrosis in oxonate-treated rats [13].
Figure 2.
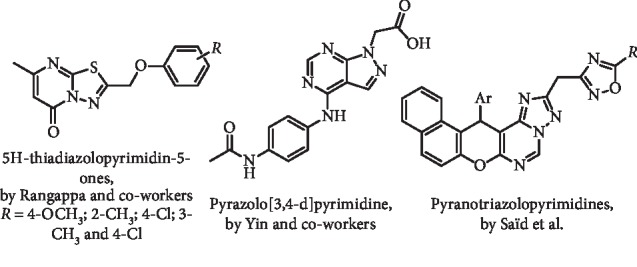
Structures of analogues of the purine nucleus.
A series of pyrazolo[4,3-d]pyrimidine analogues (Figure 2) was prepared and evaluated by Yin and coworkers as XO inhibitors and was determined an IC50 value of 33.77 μg/mL for the most potent inhibitor. Furthermore, most of these pyrimidines presented IC50 values higher than 64 μg/mL against two human hepatocellular carcinoma cells [14]. More recently, this research group described novel purine derivatives with antitumoral effects and attenuated in vitro XO inhibition [15].
Saïd et al. performed an in vitro screening of novel pyranotriazolopyrimidines (Figure 2) at 100 μM for the inhibition of XO activity and determined their cytotoxic effect against four cancer cell lines. According to the results, the XO% of inhibition at 100 μM ranged between 4.4 and 25.5%, and relevant antiproliferative effects were observed [16].
3.1.2. Febuxostat Analogues
Based on the success of the drug febuxostat, two series of 1-phenyl-pyrazole-4-carboxylic acid derivatives (Figure 3) have been designed and synthesized by Li et al. They reported a high in vitro XO inhibition, with IC50 values in the nanomolar range, acting by a mixed-type inhibition mechanism. The most potent inhibitors (IC50 of 4.2 and 5.7 nM) were further in vivo tested in mice with induced acute hyperuricemia, in comparison with the standard inhibitors febuxostat and Y-700 (Figure 3). Both studied compounds decreased the UA levels from approximately 1000 to 200 μM after 5 hours at the dose of 5 mg/kg. Interestingly, the compound with IC50 = 5.7 nM (Figure 3, R = 1-piperidin-1-yl) presented the most promising hypouricemic effect, which was similar to that observed with febuxostat. However, the pharmacokinetic studies demonstrated that further strategy to improve the plasma concentration of this compound is required because the Cmax and AUC0−∞ were remarkably lower than that determined for Y-700 [17].
Figure 3.
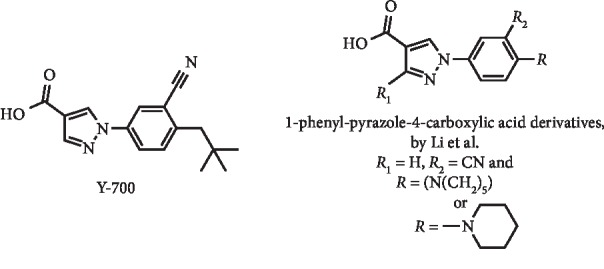
Structures of pyrazole febuxostat analogues.
In a study aiming to develop other febuxostat analogues with improved pharmacological properties, Xu et al. prepared a series of compounds structurally similar to this drug mainly exploring different 5-membered heterocyclic rings. Considering these data from in vitro XO inhibition, it was demonstrated that 2-phenylthiazole-4-carboxylic acid can be considered a new scaffold for this activity. Of the compounds studied, the best result was observed with 2-(4-isobutoxy-3-nitrophenyl)thiazole-4-carboxylic acid (IC50 = 48.6 μM; IC50 for febuxostat = 4.8 μM). Interestingly, a hypouricemic effect of this compound (Figure 4) in potassium oxonate hypoxanthine-induced hyperuricemic mice was observed, however, inferior to that observed with febuxostat [18].
Figure 4.
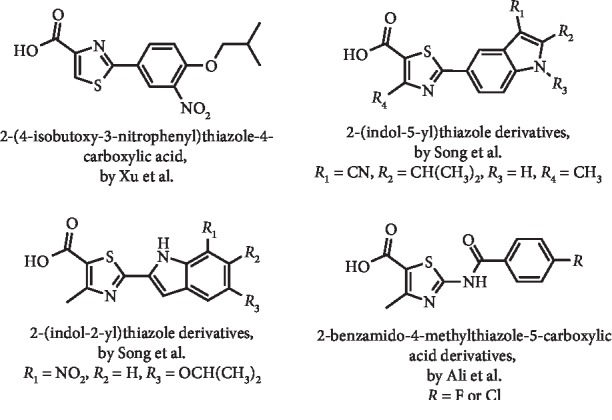
Structures of thiazole febuxostat analogues.
Also considering the febuxostat structure, Song et al. described a series of thiazole derivatives bearing the 2-(indol-5-yl) or 2-(indol-2-yl) moiety (Figure 4) [19, 20]. In the first study, they explored the structure-activity relationship for 2-(indol-5-yl)thiazoles, which presented XO inhibition levels similar to febuxostat, with IC50 values between 3.0 and 16 nM in most of the cases. However, not all of these compounds have this activity confirmed in an oxonic acid–induced high-uric acid in vivo model. In this context, 2-(3-cyano-2-isopropylindol-5-yl)-4-methylthiazole-5-carboxylic acid (Figure 4) exhibits the best compromise between in vitro XO inhibitory activity (IC50 of 3.5 nM) and plasma UA-lowering activity (60% at 1 h and 10 mg/kg). The pharmacokinetics profile study of this compound showed excellent oral bioavailability and long half-life. Therefore, according to the authors, these results evidenced the interest in performing clinical studies with this compound [19]. More recently, the same research group presented structure-activity relationship data for analogous 2-(indol-2-yl)thiazoles (Figure 4). From the observed results, although not being the most promising in vitro (IC50 of 5.1 nM) compound, 2-(7-nitro-5-isopropoxy-indol-2-yl)-4-methylthiazole-5-carboxylic acid (Figure 4) exhibited the most potent UA-lowering activity in the potassium oxonate–induced hyperuricemic rat model (43% at 1 h and 10 mg/kg). Similarly to which was observed with the previously referred 2-(indol-5-yl)thiazole derivative, this 2-(indol-2-yl)thiazole has excellent oral bioavailability in pharmacokinetic studies [20].
Ali et al. synthesized a different series of substituted 2-benzamido-4-methylthiazole-5-carboxylic acid derivatives (Figure 4) as potential XO inhibitors and free radical scavengers. In vitro studies revealed that the presence of a fluoro or chloro group at the para position of the benzamide led to an excellent XO inhibitory activity, with IC50 values of 0.57 (in a mixed-type inhibition) and 0.91 μM, respectively. These results were confirmed in a potassium oxonate–induced hyperuricemic in vivo rat model, with 62 and 53% of UA inhibition being observed after 1 h at a dose of 10 mg/kg [21].
Mao et al. reported a series of 2-phenyl-1,6-dihydropyrimidine-5-carboxylic acid derivatives (Figure 5) as excellent XO inhibitors with calculated IC50 values between 0.0181 and 0.5677 μM. These IC50 values were substantially better than that reported for allopurinol (IC50 = 7.5902 μM) and comparable to that of febuxostat (IC50 = 0.0236 μM). The authors chose 2-(3-cyano-4-isopentyloxyphenyl)-6-imino-1,6-dihydropyrimidine-5-carboxylic acid (Figure 5), with an IC50 value of 0.0240 μM, as a representative compound to continue the study. This derivative act as a mixed-type inhibitor, with a Ki value of 0.0042 μM. Also, this compound was able to significantly reduce the serum concentration of UA at a single oral dose of 5 mg/kg on potassium oxonate-induced hyperuricemic mice, with a hypouricemic potency similar to allopurinol (dose = 10 mg/kg). In an acute oral toxicity study, this derivative did not lead to behavioral abnormality 24 h after the administration of a single dose of 2000 mg/kg, which suggested that the LD50 value might be higher than 2000 mg/kg, being near 400 times over the effective dose (5 mg/kg) [22].
Figure 5.
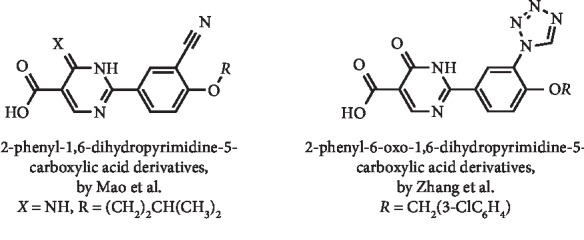
Structures of dihydropyrimidine febuxostat analogues.
Zhang et al. designed and prepared a group of 2-phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylic acid derivatives (Figure 5) bearing a tetrazol-1-yl and alkoxyl groups bounded to positions 3 and 4 of the phenyl ring, respectively, aiming to develop febuxostat analogues with higher selectivity for XO. Interestingly, all compounds had in vitro XO inhibitory properties (IC50 values ranging from 28.8 to 629 nM), and the most potent of these, 2-{4-[(3-chlorobenzyl)oxy]-3-(1H-tetrazol-1-yl)phenyl}-6-oxo-1,6-dihydropyrimidine-5-carboxylic acid (Figure 5), had an activity comparable to that observed with febuxostat (IC50 = 23.6 nM) and acted as a mixed-type inhibitor. The in vivo hypouricemic activity evaluation in the potassium oxonate–induced hyperuricemic rat model revealed that this compound (at an oral dose of 5 mg/kg) effectively reduced serum UA levels. However, its hypouricemic action was slightly lower than using febuxostat (5 mg/kg) and allopurinol (10 mg/kg). Furthermore, an acute oral toxicity study revealed that this dihydropyrimidine-5-carboxylic acid derivative was nontoxic to mice and could be tolerated at a dose up to 2000 mg/kg [23].
3.1.3. Dihydropyrimidinone Derivatives
Taking into account their structural resemblance with the pyrimidine ring of xanthine, a series of dihydropyrimidinones (Figure 6) were prepared and evaluated by Zafar et al. as potential XO inhibitors. Interestingly, from a set of 25 derivatives, 22 were found to be good to weak XO inhibitors (IC50 values ranging from 14.4 to 418 μM). These compounds were found to act by a competitive, noncompetitive, or mixed manner for XO inhibition. The best IC50 value was described for ethyl 4-ethyl-6-methyl-2-oxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (Figure 6), being a noncompetitive inhibitor, with a Ki = 20.5 μM. Additionally, no cytotoxic effects on mouse fibroblasts 3T3 at the concentration of 100 μM were observed for the most promising dihydropyrimidinones [24].
Figure 6.
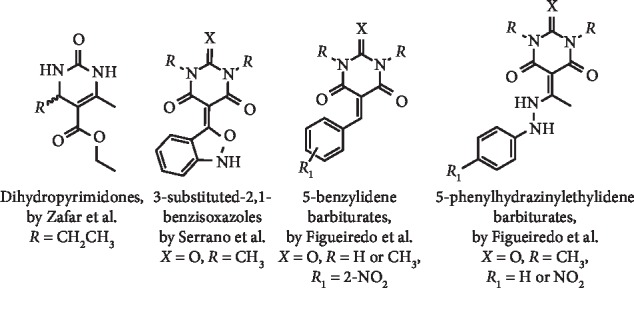
Structures of dihydropyrimidinones with XO inhibitory activity.
Also bearing the pyrimidine scaffold in their structure, several barbiturate and thiobarbiturate derivatives (Figure 6) were described as moderate XO inhibitors [25–27]. The XO inhibition of 3-substituted-2,1-benzisoxazoles (Figure 6) was demonstrated by Serrano et al. with an IC50 value of 22.10 μM being determined for the best compound. In addition, a marked reduction in proliferation at a 30 μM concentration induced by this barbiturate in the MCF-7 cell line was observed [25]. Moreover, 5-benzylidene [26] and phenylhydrazinylethylidene [26, 27] 1,3-disubstituted barbiturates (Figure 6) were described by Figueiredo et al. as moderate XO inhibitors, with IC50 values between 24.3 and 31.5 μM [26, 27]. The authors also demonstrated that these barbiturates have low cytotoxicity in normal human dermal fibroblasts. In fact, in a screening at 30 μM, a relative cellular proliferation ranging from 65 to 79% was observed, and an IC50 value of 82.02 μM was determined for the most cytotoxic barbiturate [26, 27].
3.1.4. Other Organic Synthetic Derivatives
3H-Quinazolin-4-one derivatives (Figure 7) were synthesized by El-Sayed et al., and in vitro studies for XO inhibition demonstrated that none of these compounds evidenced better activity than allopurinol. For the best compound (Figure 7), an IC50 of 3.0 μg/mL was calculated, which is a higher value than that observed for allopurinol (0.6 μg/mL). Cytotoxicity studies on colorectal cancer HT-29 and SW620 cell lines showed cellular viability nearly 40% at the concentration of 30 μg/mL [28].
Figure 7.
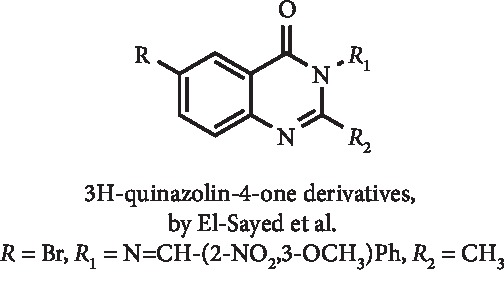
Structure of a 3H-quinazolin-4-one derivatives with XO inhibitory activity.
Benzylidene nicotino/isonicotinohydrazide derivatives (Figure 8) were reported by Zafar et al. as being good in vitro XO inhibitors, with IC50 values of 0.96, 10.0, and 12.4 μM being determined for the three most potent compounds. In addition, it was demonstrated that these hydrazides act by a competitive mode of inhibition. These compounds were also found to be noncytotoxic against a mouse fibroblast cell line and thus were selected for in vivo studies. Interestingly, these authors demonstrated that two isonicotinohydrazides (Figure 8) were able to in vivo inhibit XO by 28 and 44% (at 50 mg/kg) against 100% observed for allopurinol at the same concentration [29].
Figure 8.
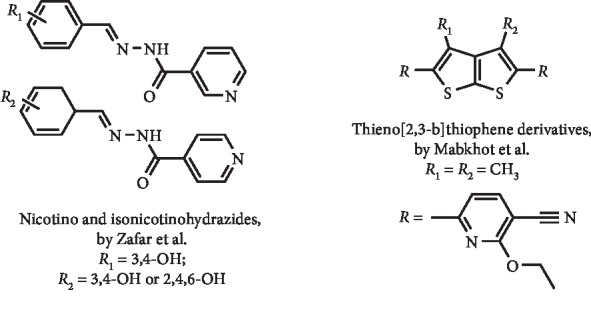
Structures of pyridine synthetic derivatives with XO inhibitory activity.
A series of compounds incorporating the thieno[2,3-b]thiophene moiety (Figure 8) were synthesized and tested against several enzymes by Mabkhot et al. Three of these compounds demonstrated to be selective XO inhibitors, being determined for the most potent one (Figure 8) an IC50 value of 14.4 μM. This compound was also found to be noncytotoxic in a human prostate cancer cell line [30].
Under the paradigm “old drug, new indication,” Niu et al. described olsalazine sodium (Figure 9), a commercial anti-ulcerative-colitis drug, as a serum UA levels reducer. For this, the author performed molecular docking to virtually screen potential XO inhibitors from a small approved drugs library. The in vitro studies showed olsalazine sodium as a promising compound in reducing XO activity, with an IC50 value of 3.4 mg/L (approximately 9.5 μM), acting by a hybrid-type inhibition mode. Finally, it was observed in in vivo studies that this drug decreased serum UA levels and serum/hepatic XO activities after intraperitoneal administration. Nevertheless, as the onset of hypouricemic action for olsalazine occurred 4 h after administration, the authors suggested that this drug might be particularly suitable for gout prevention and long-term treatment [31].
Figure 9.
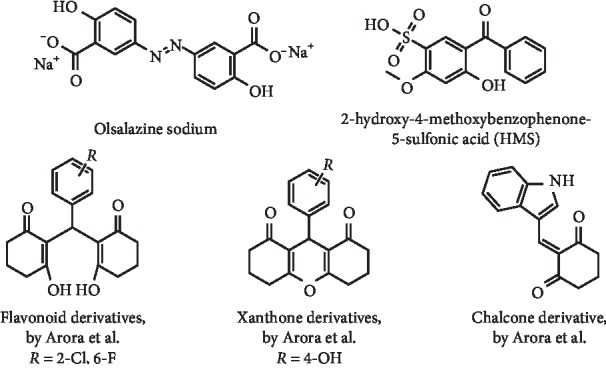
Structures of olsalazine sodium, 2-hydroxy-4-methoxybenzophenone-5-sulfonic acid, and Knoevenagel/tandem Knoevenagel and Michael adducts of cyclohexane-1,3-dione and aryl aldehydes.
2-Hydroxy-4-methoxybenzophenone-5-sulfonic acid (HMS) (Figure 9), a popular ultraviolet filter used in sunscreens, was studied as a potential hypouricemic agent by Zuo and coworkers. After an in silico molecular docking study, the XO inhibition by HMS was partially confirmed in vitro, and an IC50 of 36.1 μM was obtained, in comparison with 11.4 μM determined for allopurinol. In in vivo studies, it was observed that a 20 mg/kg dose of HMS administered to hyperuricemic mice leads to a significant reduction of serum UA levels. Possibly, this result was not only due to XO inhibition but also due to an upregulation of OAT1 and downregulation of GLUT9 mRNA and protein expression. Additionally, toxicity studies showed no negative impact in mice body weight growth and in kidney function [32].
Arora et al. designed and prepared Knoevenagel/tandem Knoevenagel and Michael adducts of cyclohexane-1,3-dione and aryl aldehydes (Figure 9) as XO inhibitors with a slight structural similarity to flavonoid (IC50 range from 4.93 to 15.28 μM), xanthone (IC50 range from 4.08 to 13.14 μM), and chalcone (IC50 = 3.66 μM) scaffolds. Most of the described compounds presented better IC50 values than allopurinol (IC50 = 7.08 μM). 2,2-((2-chloro-6-fluorophenyl)methylene)-bis(3-hydroxycyclohex-2-en-1-one) and 2-((1H-indol-3-yl)methylene)cyclohexane-1,3-dione (Figure 9) acted as noncompetitive and competitive-type inhibitors, respectively. Additionally, no significant cytotoxicity at 10 μM on normal cells (HEK293) was observed for these compounds [33].
3.1.5. Metal Complexes
A dinuclear cyclam complex (Figure 10) was described as a XO inhibitor (IC50 = 3.70 μM) by Zafar et al. This complex acted by a noncompetitive type of inhibition and was also found to be in vitro noncytotoxic and led to a marked inhibition of in vivo XO activity [34]. Özerkan et al. reported the synthesis and evaluation of novel palladium (II) complexes with tetradentate thiosemicarbazones (Figure 10). Two compounds are interesting XO inhibitors (uncompetitive inhibitory mode), with IC50 of 0.4 and 0.7 μg/mL, better values than the control allopurinol (IC50 = 1.1 μg/mL). In addition, in vitro cytotoxicity studies on healthy 3T3 fibroblast cells demonstrated IC50 values of 7.1 and 6.2 μg/mL for these two compounds, respectively [35].
Figure 10.
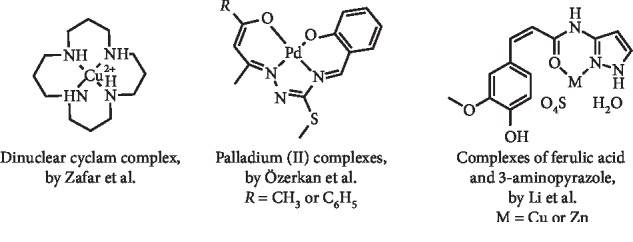
Structures of metal complexes with XO inhibitory activity and decreasing effect of serum UA levels.
Complexes resulting from the combination of ferulic acid, a known XO inhibitor, and 3-aminopyrazole, a molecule with anti-inflammatory effects, and copper or zinc (Figure 10) were prepared by Li et al. These two compounds exhibited inhibitory effects of XO activity in mouse liver homogenates, with better results being observed for the copper complex. In addition, studies in hyperuricemia model mice revealed that the elevated levels of blood UA could be decreased by both complexes (10 mg/kg, intraperitoneally injected) and that their effect is similar to the observed with febuxostat. Furthermore, it was evidenced that these complexes had no relevant effect on serum creatinine values, indicative of potential low renal side effects [36].
3.2. Natural and Semisynthetic Compounds and Mimetics of Natural Structures
3.2.1. Phenolic Compounds and Analogues
(1) Simple Phenolic Compounds. Aiming to develop a novel XO inhibitor with a potent activity and low toxicity, Lü et al. described a series of natural catechols and analogues. Of these, 3,4-dihydroxy-5-nitrobenzaldehyde (DHNB) (Figure 11), structurally similar to protocatechuic aldehyde, demonstrated to be the most potent XO inhibitor, with an IC50 value of 3 μM, acting by a mixed-type inhibition. In addition, DHNB effectively reduced serum UA levels after oral administration in allantoxanamide-induced hyperuricemic mice at a dose of 100 mg/kg. Nevertheless, in vivo toxicity studies demonstrated that a large oral dose of 500 mg/kg of DHNB did not lead to any evident side effects, in contrast with 42% of death in mice treated with the same dose of allopurinol [37]. The hypouricemic effect of 2,5-dihydroxyacetophenone (Figure 11), a compound computationally screened from Ganoderma applanatum was explored by Liang et al. For this, the in vitro and in vivo XO inhibitory activity was studied, evidence that this compound is an inhibitor of this enzyme (in vitro IC50 = 8.12 μM). In hyperuricemic mice, it was also demonstrated that after oral administration of this phenolic compound (20, 40, and 80 mg/kg) serum UA was markedly reduced and that blood nitrogen and creatinine levels were lower than that observed in hyperuricemic and allopurinol controls. By means of RT-PCR and Western blot, further studies on the mechanism of action were performed, and it was verified that RNA and protein expressions of OAT1 (organic anion transporter 1) were upregulated and that the expressions of GLUT9 (glucose transporter 9), URAT1 (uric acid transporter 1), and CNT2 (gastrointestinal concentrative nucleoside transporter 2) were downregulated [38].
Figure 11.
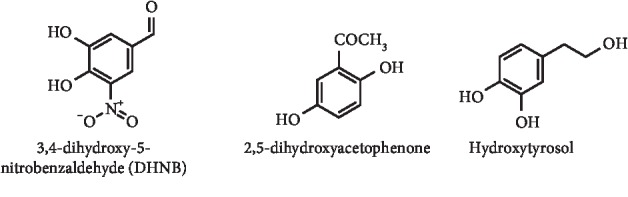
Structures of simple phenolic compounds with XO inhibitory activity and decreasing effect of serum UA levels.
Hydroxytyrosol (Figure 11) is another natural catecholic compound that was studied by Wan et al. against XO by in vivo animal model and in vitro inhibition assay. Interestingly, hydroxytyrosol has XO inhibitory activity, with a determined IC50 value of 8.75 mM. This molecule can also decrease serum UA levels and adjust the mRNA transcription levels of UA transporter genes. In fact, this compound reduced reabsorption transporter genes mRNA and increased secretion transporter genes mRNA to the normal [39].
(2) Phenolic Acids and Derivatives, Including Salvianolic Acids. Several phenolic acids and derivatives were studied as XO and cyclooxygenase-2 inhibitors by Nile et al. According to these authors, all compounds showed good inhibition of XO activity. The most interesting IC50 values were found for sinapic acid and propyl and stearyl gallate (Figure 12). In addition, generally, they did not show significant in vitro cytotoxicity at 10 μM except stearyl gallate; however, many of these ferulic acid derivatives revealed relevant toxicity at higher concentrations (50 μM). Furthermore, interesting anti-inflammatory action was also observed for some of these compounds [40]. Caffeic acid (Figure 12), a known antioxidant belonging to the family of cinnamic acids and being present in numerous plants, also has XO inhibitory properties. Recently, Wan et al. evaluated its reducing effects on plasmatic UA levels in hyperuricemia rats and explored potential mechanisms of action in this context. Interestingly, it was demonstrated that serum UA levels were reduced after intragastric administration of caffeic acid (100 mg/kg) to hyperuricemia rats. In addition, blood urea nitrogen and serum creatinine levels decreased as well as in vivo XO and adenosine deaminase activities. Moreover, in vitro assays confirmed the XO inhibitory properties of this phenolic acid (IC50 = 53.45 μM) and evidenced that it is a competitive inhibitor of this enzyme, which suffers structural changes (e.g. reduction of α-helix content) in the presence of this compound. Furthermore, it also acts by regulating the mRNA transcription of the renal uric acid transporters. In fact, the transcription levels of URAT1 and GLUT9 mRNA significantly increased, and the transcription levels of OAT1, UAT, and ABCG2 mRNA were significantly reduced than that observed in the blank group [41].
Figure 12.
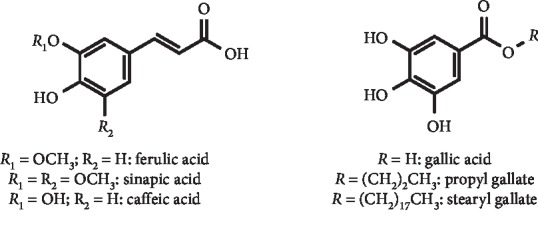
Structures of phenolic acids with XO inhibitory activity and decreasing effect of serum UA levels.
Verbascoside, a cinnamate ester glycoside (Figure 13), also has in vitro inhibitory effect on XO (IC50 = 81.11 mg/mL). Additionally, a dose of 54 mg/kg of verbascoside in a hyperuricemic in vivo model could reduce serum UA to normal levels [42]. 1,2,3,4,6-Penta-O-galloyl-β-D-glucopyranose, another glycoside derivative, demonstrated in vitro inhibition of XO activity (IC50 = 2.8 μM) in a noncompetitive manner (Ki = 3.1 μM) with a potency closely to the observed with allopurinol (IC50 = 2.3 μM). Serum UA levels on hyperuricemic mice at a dose of 40 mg/kg were also lowered [43].
Figure 13.
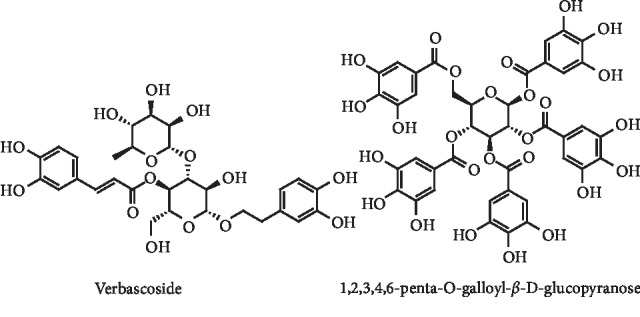
Structures of verbascoside and 1,2,3,4,6-penta-O-galloyl-β-D-glucopyranose.
A combination of in silico, in vitro, and in vivo studies aiming to explore the interaction between ten representative chemicals identified in Salvia miltiorrhiza and XO were applied by Tang et al. Of these compounds, salvianolic acid C (Figure 14) proved to be the most potent in vitro XO inhibitor (IC50 = 8.79 μM), acting by a mix-competitive manner. In vivo studies demonstrated a dose-dependent hypouricemic action of this compound in potassium oxonate–induced mice. Interestingly, it was observed that salvianolic acid C and allopurinol exhibit comparable in vitro XO inhibitory activity, but the hypouricemic effect of salvianolic acid C was lower [44]. Although with lower potency, salvianolic acid A (Figure 14) was also described as in vitro XO inhibitor, with an IC50 value of 73.17 μM, and also led to a decrease UA levels in acute myocardial infarction rats [45, 46].
Figure 14.
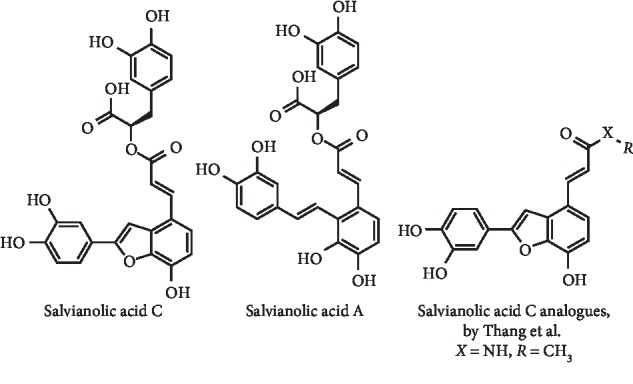
Structures of salvianolic acids C A and C analogues.
Thang et al. designed and synthesized a new series of 2-arylbenzo[b]furan derivatives (Figure 14), considering the structure and known bioactivity of salvianolic acid C. Most of these compounds exhibited potent in vitro XO inhibitory effect, particularly (E)-3-(2-(3,4-dihydroxyphenyl)-7-hydroxybenzofuran-4-yl)-N-methylacrylamide (IC50 = 4.45 μM; IC50 of allopurinol = 10.61 μM), which induced a mixed-type XO inhibition with a Ki of 3.5 μM. Additionally, this compound (Figure 14) exhibit a dose-dependent hypouricemic action in potassium oxonate–induced hyperuricemic mice [47].
(3) Hydroxylated Chalcones, 6-Shogaol, Curcumin, and Analogues. The chalcone scaffold, found in many naturally occurring compounds, have a wide range of bioactive properties and have also been prepared by synthesis [48, 49]. As an example, Li and co-workers [50–52] prepared several chalcones and evaluated their XO inhibitory activity. Of these, 3,5,2′,4′-tetrahydroxychalcone (Figure 15) displayed an interesting XO inhibition in a competitive manner, and IC50 and Ki values of 22.5 and 17.4 μM, respectively, were determined [52]. In addition, in vivo intragastric administration of this chalcone significantly reduced serum UA levels in hyperuricemic mice in a dose-dependent manner. The best effect occurred with doses between 2 and 4 mg/kg [50–52]. Studies performed by the authors suggested that this chalcone act by a dual mechanism [50]. In fact, this in vivo hypouricemic action can be associated not only with the inhibition of key enzymes (XO, PRPS, PRPPAT, and HGPRT) in the purine metabolism but also with an enhancement in UA excretion by inhibiting the expression of GLUT9 in the kidney [50, 51]. Finally, an acute toxicity study in mice showed that 3,5,2′,4′-tetrahydroxychalcone was safe at a dose up to 5 g/kg, making it suitable for future studies [52]. Other hydroxylated chalcones prepared by a Claisen–Schmidt condensation were described by Hofmann et al., and IC50 values between 1.2 and 93 μM as XO inhibitors were determined. Cytotoxicity tests revealed that the most active of these chalcones is noncytotoxic. However, further structure modifications on the chalcone scaffold are needed to optimize the XO inhibitory activity [53]. Later, Xie et al. described a synthetic series of hydroxychalcones (Figure 15) with moderate XO inhibition, and in vitro IC50 values of 47.3 and 56.8 μM were calculated for the most potent ones. These hydroxychalcones, which act by a mixed XO inhibition type, were tested in vivo in potassium oxonate–induced hyperuricemic mice (10 and 50 mg/kg, intragastrically administered). The results demonstrated that they led to a significant serum UA and XO activity reduction in both cases, with the highest significance at 50 mg/kg. Acute toxicity in vivo studies revealed no evident toxicity at doses up to 5 g/kg [54].
Figure 15.
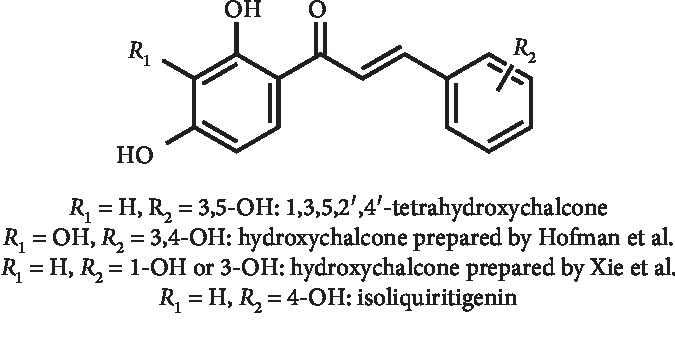
Structures of hydroxychalcones with XO inhibitory activity and decreasing effect of serum UA levels.
The XO inhibition in a mixed manner by isoliquiritigenin (Figure 15) was initially described by Cheng and coworkers (IC50 and Ki values of 55.8 and 17.4 μM, respectively) [55]. However, its poor absolute in vivo bioavailability was later reported [56]. In order to change that, Zang et al. developed an isoliquiritigenin-loaded self-microemulsifying drug delivery system. In fact, the in vivo oral bioavailability was enhanced in about 4.7 times, with an improvement in plasmatic concentrations. Additionally, the use of self-microemulsification proved to significantly improve the antihyperuricemic effect of isoliquiritigenin in rats. At the administered doses of 50–150 mg/kg, a reduction in serum UA levels from 25.2–51.9 to 41.7–78.8% for free drug and its drug delivery system was, respectively, demonstrated [57].
A study conducted by Peng et al. demonstrated that phenolic compounds isolated from ginger inhibit the XO activity at the concentration of 125 μM. 6-Shogaol (Figure 16), one of these phenols, also presented IC50 values of relative cell proliferation between 7.4 and 100.0 μM in the different tested cancer cell lines [58]. Later, Nile and Park reported again the XO inhibition by 6-shogaol, with an IC50 value of 15.2 μM versus 8.4 μM of allopurinol [59]. Due to the poor water solubility of 6-shogaol, Yu, Xu, and coworkers developed 6-shogaol-loaded solid lipid nanoparticles [60] and a 6-shogaol-loaded self-microemulsifying drug delivery system [61] to enhance its oral bioavailability. In fact, the nanoencapsulation led to a significant improvement of in vivo oral absorption, bioavailability, and acting time of 6-shogaol in healthy rats. 6-Shogaol–loaded solid lipid nanoparticles also could lower the serum UA levels in a higher extension than free drug in hyperuricemia/gouty arthritis rats at a 120 mg/kg dose. In addition, loaded solid lipid nanoparticles allowed an improvement in the organ protection effects of 6-shogaol [60]. The use of a 6-shogaol–loaded self-microemulsifying drug delivery system led to an increment of 571.18% in the oral bioavailability when compared with the administration of the free drug. In addition, an increment in the reduction of serum UA levels in hyperuricemic rats treated with 25, 50, and 100 mg/kg of dose was observed. As example, the highest dose (10 mg/Kg) led to a reduction in UA levels by 71.1% by the microemulsification and 60.0% by free drug. The 6-shogaol–loaded self-microemulsifying drug delivery system was also pointed as promising to reduce the kidney damage caused by hyperuricemia model [61].
Figure 16.
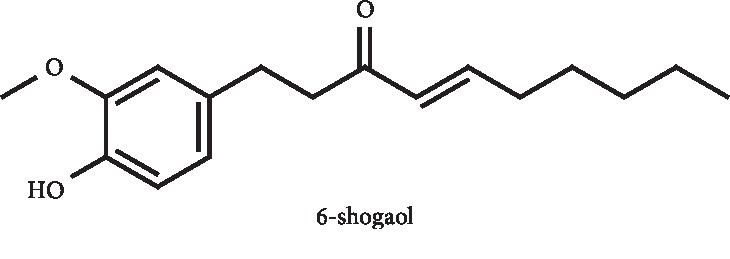
Structure of 6-shogaol.
Curcumin (Figure 17) was described as a XO inhibitor, with IC50 of 117.3 μM (IC50 of allopurinol = 28.9 μM) by Peng et al. [58]. Later, Chen et al. reported its in vivo activity on potassium oxonate–induced hyperuricemia mice. Oral administration in a dose of 20 or 40 mg/kg leads to a decrease of serum UA and an effective inhibition of serum and liver XO levels and to a reduction of kidney inflammation by NLRP3 inflammasome suppression [62]. α,β-Unsaturated cyclohexanone and cyclopentanone analogues of curcumin (Figure 17) were described as potential antihyperuricemia agents, acting by dual inhibition of XO and URAT1 by Ao et al. The most promising compound (Figure 17) with in vitro dual inhibition also demonstrated to reduce serum UA levels in a hyperuricemic in vivo model. Interestingly, at the doses of 10 and 20 mg/kg, this curcumin derivative increased the urinary UA excretion and decreased the serum and hepatic UA levels as well as XO activity and URAT1 protein levels. According to these authors, this compound is a first-in-class of dual inhibitors and may serve as a reference compound for further design of antihyperuricemic drugs [63].
Figure 17.
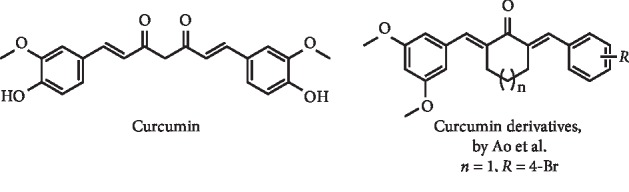
Structure of curcumin and analogues with XO inhibitory activity and decreasing effect of serum UA levels.
(4) Flavonoids and Analogues. Natural plant flavonoids have always been molecules of great interest [64]. Their hypouricemic action in mice was also evidenced [65] as well as of some semisynthesized derivatives [66]. In fact, the flavonoids luteolin, apigenin, diosmetin, chrysin, O3′,O7-dihexyl diosmetin, O4′,O7-dihexyl apigenin, and O7-hexyl chrysin (Figure 18) showed XO inhibition, with IC50 values between 4.5 and 8.1 μg/mL. In addition, anti-inflammatory action and a relatively low cytotoxicity were found for these flavonoids [66]. Other series of apigenin flavonoid derivatives have been prepared by Su et al. by a coupling of the carboxyl alkyl group to 4′-, 5- or 7-hydroxyl groups of apigenin. The 4′-modified derivatives (Figure 18) were demonstrated to be more potent than apigenin in XO inhibition, with IC50 values between 0.098 and 0.82 μM, versus 3.2 μM. The insertion of a carboxyl n-hexyl group in this position (Figure 18) led to the best in vitro results. This molecule was evaluated for its hypouricemic effects on the potassium oxonate–induced hyperuricemic mouse model. Interestingly, the serum UA levels were significantly decreased by an intraperitoneal dose of 10 mg/kg, with no significant effects being observed with apigenin. Nevertheless, the in vitro results presented higher XO inhibition for apigenin derivatives than allopurinol, but in vivo tests did not confirm these results. The authors pointed to the poor bioavailability or short half-life of the compound as the cause of this reduced activity [67].
Figure 18.

Structures of flavonoids with XO inhibitory activity and decreasing effect of serum UA levels.
Four flavones with XO inhibitory activity were described by Metoui et al. from the Tunisian Artemisia campestris leaves. 2′,4′,5,7-Tetrahydroxy-5′,6-dimethoxyflavone, eupatilin, dimethoxycentaureidin, and cirsiliol (Figure 18) exhibited higher potency for XO inhibition than allopurinol (IC50 = 8.2 μM), with IC50 values of 5.5, 3.3, 6.8, and 5.5 μM, respectively. In addition, these four flavones were in vitro tested against four cancer cell lines at a concentration of 15 μM to verify their cytotoxic activity. A proliferation value below or close to 50% was found for all tested flavones at this single concentration [68].
Luteolin (Figure 18) has been reported to have a significant XO inhibitory activity in comparison with allopurinol, with an IC50 value of 4.8 μM [69]. An inhibition effect assay performed by Dong et al. evidenced that luteolin and a luteolin–manganese(II) complex (Figure 18) reversibly inhibited XO in a competitive manner. Interestingly, the complex had a more remarkable hypouricemic effect than luteolin and both compounds proved to be noncytotoxic to a human liver cancer cell line [70]. A glycosylated derivative, luteolin-4′-glucoside (Figure 18), also inhibited XO, with an in vitro IC50 value of 0.26 μg/mL [71]. In addition, this molecule had potent uricosuric effects in hyperuricemic mice with renal mURAT1 and decreasing XO activity, as well as anti-inflammatory effects. Furthermore, it was noted that, at the same concentration of luteolin and luteolin-4′-O-glucoside, a better therapeutic effect was observed for luteolin [72]. 6-Hydroxyluteolin (Figure 18) was also described as an inhibitor of XO activity, with an IC50 value of 7.52 μM [73]. In vivo studies evidenced that this flavonol led to a significant dose-dependent reduction on the serum UA level of hyperuricemic rats at doses of 0.05, 0.1, and 0.3 mmol/kg. It was also demonstrated significant inhibition of rat liver XO (about 80%) in the highest dose [74].
de Araújo et al. described the improvement of in vitro XO inhibitory activity after enzymatic deglycosylation of rutin (Figure 18). However, this hydrolysis also leads to an increase in the cytotoxicity of this flavonoid against several tested cell lines. The total deglycosylated derivative, quercetin (Figure 18), was demonstrated to be the most potent XO inhibitor and had intermediary cytotoxicity [75]. In this context, it was previously demonstrated that quercetin has high XO inhibitory effects, with an IC50 value of 1.9 μM [76]. Glycosylated anthocyanins from purple sweet potato [77–80] as well as other glycosylated flavonoids and coumaroylspermidines from rape bee pollen [81] were described by Zhang research group as good XO inhibitors. In fact, these authors demonstrated that these compounds, particularly highly acylated anthocyanins, can reduce the kidney inflammation in hyperuricemic mice in addition to their hypouricemic effect [77, 80].
The flavonoids hesperetin (Figure 18), hesperidin, and G-hesperidin were also tested as XO inhibitors by De Souza et al. These authors demonstrated that hesperetin has more potent in vitro XO inhibitory activity than the glycosylated derivatives (hesperidin and G-hesperidin). This compound acted by a competitive inhibition mode, and IC50 and Ki values of 53 and 17 μM, respectively, were determined. In addition, in vivo studies demonstrated that monoglucuronides of hesperetin were the major forms present in plasma after the ingestion of this flavonoid [82].
(−)-Epigallocatechin-3-gallate (EGCG) (Figure 18), important catechin of green tea, was evaluated as XO inhibitor, with a determined IC50 of 12.5 μM by Lin et al. These authors described that EGCG may act as “suicide substrate,” similarly to allopurinol [83]. More recently, Zhu et al. demonstrated that EGCG displayed inhibitory effects on hepatic XO at the dose of 50 mg/kg. In addition, this compound had a significant dose-dependent effect on lowering serum UA levels and in the regulation of GLUT9 and URAT1 mRNA expression levels in hyperuricemic mice, when compared with the model group. Furthermore, this molecule had effective renal protective effects in the prevention of glomeruli and kidney tubules damages, as evidenced by the analysis of histopathologic sections of hyperuricemic mice [84].
Two series of benzoflavone derivatives (Figure 19) were designed, synthesized, and evaluated for their XO inhibitory potential by Singh et al. Some of these derivatives had significant in vitro XO inhibition, with IC50 values lower than 10 μM. Among the series, the most potent compounds on in vitro studies were 7,8- and 5,6-benzoflavones (Figure 19), which demonstrated IC50 values of 0.6 and 5.2 μM, respectively. These derivatives act as mixed-type inhibitors and has a higher potency than allopurinol (IC50 = 8.7 μM). Of these two compounds, in vivo studies demonstrated that only the 5,6-benzoflavone derivative presented the ability to reduce the serum UA to its normal level at a dose of 10 mg/kg. Interestingly, the authors explored six other derivatives on in vivo studies that evidenced the potential of at least three compounds as clinical candidates for the treatment of hyperuricemia. These compounds, which presented in vitro IC50 values of 4.9, 8.9, and 7.7 μM, showed to be able to reduce in vivo serum UA to values at a level comparable to that observed with febuxostat (dose of 5 mg/kg). Additionally, two of these benzoflavones did not led to behavioral abnormality on in vivo acute toxicity study [85].
Figure 19.
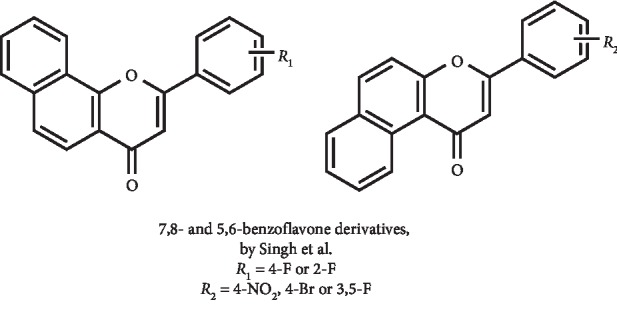
Benzoflavone derivatives with XO inhibitory activity and decreasing effect of serum UA levels.
(E)-2-(4-Bromophenyl)-1-(2,4-dihydroxyphenyl)ethanone oxime (BDEO) (Figure 20), a novel compound with a chemical structure similar to flavonoids, was described as an inhibitor of XO activity, with an IC50 value of 3.33 μM. Its UA uptake via URAT1 was evaluated in vitro, and the results demonstrated that this absorption was effectively inhibited. According to this study, BDEO effectively decreased hepatic XO activity and downregulated renal URAT1 protein expression in hyperuricemic mice, especially at doses of 10 and 20 mg/kg. Nevertheless, those indicators were not changed by BDEO in normal mice when compared with allopurinol and benzbromarone, reflecting its marked advantages in treating hyperuricemia and predicting its safety. Therefore, the authors suggested that BDEO may serve as a dual XO and URAT1 inhibitor for the treatment of hyperuricemia [86]. Considering the structure of flavonoids and synthetic intermediates for their preparation with antihyperuricemia effects, XO inhibition and also immune-regulating actions, several benzoxazole analogues of these compounds (Figure 20) were developed aiming to improve these activities. Specifically, two series of benzoxazole deoxybenzoin oxime derivatives were prepared as dual inhibitors of innate immune sensors and XO. In vitro studies evidenced that the majority of compounds suppressed XO activity, and the best result was observed with compound (E)-1-(6-methoxybenzo[d]oxazol-2-yl)-2-(4-methoxyphenyl)ethanone oxime (IC50 = 3.7 μM; IC50 for allopurinol = 2.9 μM). Enzyme kinetics studies demonstrated that this compound (Figure 20) acted as a competitive-type XO inhibitor. Using mice with oxonate-induced hyperuricemia, this compound reduced serum UA levels in a dose-dependent manner, consistent with in vitro studies. In addition, it was demonstrated that this benzoxazole has higher safety than allopurinol in normal mice and that it has antiacute gouty arthritis effect in vivo [87].
Figure 20.
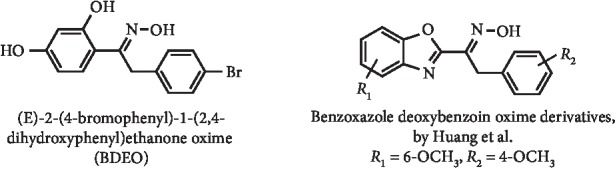
Structures of oxime analogues of flavonoids with XO inhibitory activity and decreasing effect of serum UA levels.
From a set of compounds with a chemical structure similar to flavonoids, 4-(2-(4-chlorophenyl)-1-((4-chlorophenyl)amino)ethyl)benzene-1,3-diol (CBED) (Figure 21) was found to be a dual XO and NLRP3 inhibitor after molecular docking studies by Liu and coworkers. In vitro results for XO inhibitory activity demonstrated an IC50 value of 3.9 μM, close to that obtained for allopurinol (IC50 = 2.1 μM). On the other hand, in vivo results demonstrated a reduction in serum UA levels at the dose of 20 mg/kg. Additionally, a remarkably suppression of NLRP3 inflammasome activation and regulation on hepatic XO activity were observed. Thus and according to the authors, CBED can lead not only to a hypouricemic effect but also to a reduction in kidney inflammation caused by high UA levels [88].
Figure 21.
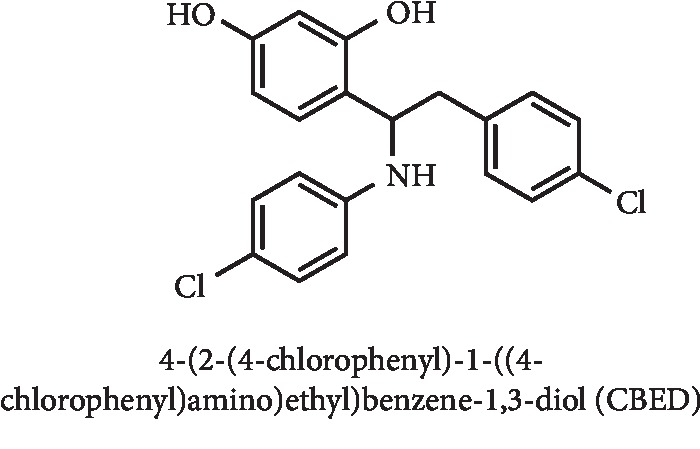
Structure of 4-(2-(4-chlorophenyl)-1-((4-chlorophenyl)amino)ethyl)benzene-1,3-diol (CBED).
(5) Phenolic Coumarin and Xanthone Derivatives. To understand structure-activity relationships on a series of coumarin derivatives, Fais et al. prepared a series of twenty 3-arylcoumarins and eight 3-heteroarylcoumarins. Of these, 5,7-dihydroxy-3-(3′-hydroxyphenyl)coumarin (Figure 22) was proved to be the best XO inhibitor with a determined IC50 value 7-fold better than that observed for allopurinol (IC50 of 2.1 and 14.7 μM, respectively). In vitro kinetics studies revealed an uncompetitive inhibition mode and a Ki value of 0.4 μM. Finally, studies on 3T3 normal fibroblasts revealed no cytotoxic effect caused by this compound [89].
Figure 22.
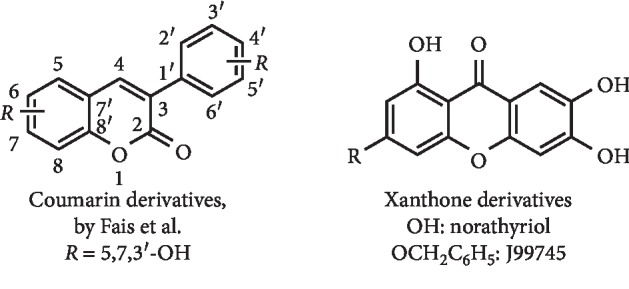
Structure of phenolic coumarin and of xanthone derivatives with XO inhibitory activity.
Norathyriol (Figure 22), a xanthone analogue, was described by Lin et al. as an uncompetitive concentration-dependent XO inhibitor, with IC50 of 7.8 μM, close to that observed for allopurinol (IC50 = 76.3 μM). The uncompetitive inhibition was appointed by the authors as a beneficial point in comparison with the competitive or mixed-type inhibition of allopurinol and febuxostat, respectively. In addition, an intragastric dose of 2.0 mg/kg of norathyriol was enough to reduce the serum UA levels in hyperuricemic mice to the normal values of healthy mice. In vivo results demonstrated also that norathyriol acted not only by XO inhibition but also by OAT1 activation [90]. Qin et al. demonstrated for the first time that the norathyriol 3-benzyloxy derivative J99745 (Figure 22) presented an in vitro XO inhibition, with a determined IC50 value of 3.297 μM. Experiments involving hyperuricemia mice showed that J99745 at doses of 10 and 30 mg/kg significantly reduced serum urate levels, enhanced UA excretion, and provided higher nephroprotective effects than allopurinol. These authors suggested that this molecule exerts a urate-lowering effect by the inhibition of XO activity and a decrease in URAT1 expression [91].
3.2.2. Terpenes and Dioscin
Lin et al. isolated a triterpenoid and an alkamide from Ganoderma tsuga and prepared four terpenoid derivatives by semisynthesis from 3-oxo-5α-lanosta-8,24-dien-21-oic acid (Figure 23). In addition, these compounds were evaluated as XO inhibitors, antioxidants, and antiproliferative agents against human prostatic cancer cells. Of these, two compounds with interesting XO inhibition (IC50 values of 313.3 and 43.9 μM) in a concentration-dependent manner were identified. The compound with the best activity (3-oxo-5α-lanosta-8-en-21-oic acid) (Figure 23), prepared by semisynthesis, was as a mixed inhibitor of XO, with a Ki value of 3.2 μM. The authors also reported the cytotoxicity of this compound against prostatic cancer cells, with an IC50 value of 23.9 μM being determined [92]. In previous works of this research group, the lanostanoid used as starting material had already been isolated from Ganoderma tsugae [92, 93]. In addition, five another lanostanoids were isolated and tested as XO inhibitors and as DPPH scavengers, and their cytotoxicity was evaluated against prostatic cancer cells and keratinocytes. Of these, three compounds inhibited the XO activity in a concentration-dependent manner with IC50 values of 90.2, 116.1, and 181.8 μM. The compound with the highest XO inhibition (3α-acetoxy-22-oxo-5α-lanosta-8,24-dien-21-oic acid) (Figure 23) is a mixed inhibitor with Ki of 0.6 μM. However, this compound also presented the most interesting result in human prostatic cancer cells, also enhancing the cytotoxicity induced by cisplatin [93]. Since a long time, the glycoside dioscin (Figure 23) is known as a weak XO inhibitor with an IC50 value of 115 μM [94]. Recently, it was evidenced that it has pronounced antihyperuricemic effects in mice by reducing serum UA levels over 4 h at 25 and 50 mg/kg of oral dose [95, 96]. This activity can be explained by the regulation of levels of mOAT1, mURAT1, mOCT2, and GLUT-9 expressions in the kidney [95, 96].
Figure 23.

Terpenes and dioscin with XO inhibitory activity and decreasing effect of serum UA levels.
Ursolic acid (Figure 23), a bioactive pentacyclic triterpene extracted from the Tribulus arabicus hexane fraction, was found to be a potent XO inhibitor. The authors described an in vitro IC50 value of 10.3 μg/mL for the inhibition of XO activity (IC50 = 6.5 μg/mL for allopurinol). In addition, this in vitro activity was confirmed in vivo on mice with potassium oxonate–induced hyperuricemia. Results demonstrated an effective hypouricemic effect at a dose of 5 and 10 mg/kg with the reduction of UA levels in 56.1 and 79.9%, respectively [97]. Given its potential as a hypouricemic agent, ursolic acid is already patented in China for gout treatment [98]. In a recent study on triterpenic acids from apple pomace by Kai and coworkers, an in vitro cytotoxicity with IC50 values between 5.6 and 20.8 μg/mL in four cancer cell lines was described for ursolic acid. From this natural source, other compounds were isolated and tested against XO. In fact, beyond the most active betulinic acid (IC50 = 12.6 μg/mL), oleanolic acid (IC50 = 17.8 μg/mL), maslinic acid (IC50 = 21.5 μg/mL), erythrodiol (IC50 = 25.8 μg/mL), and uvaol (IC50 = 30.2 μg/mL) were also described (Figure 23). Nevertheless, all the described triterpenic acids did not show higher activity than allopurinol (IC50 = 9.6 μg/mL). By means of kinetic studies, a noncompetitive type of inhibition for betulinic acid was revealed. Additionally, in vitro cytotoxic effects on four cancer cell lines and IC50 values from 6.2 to 40.1 μg/mL were obtained [99].
3.2.3. Other Natural and Semisynthetic Compounds
α-Lipoic acid (Figure 24) was found by Hameed and Ramadhan to be a potent XO inhibitor, with an IC50 value of 2.9 μg/mL when compared with 1.7 μg/mL of allopurinol. In vivo results on potassium oxonate–induced hyperuricemic mice demonstrated that a 10 mg/kg dose of α-lipoic acid and allopurinol led to a serum UA reduction from 4.4 mg/dL to 2.3 and 1.7 mg/dL, respectively [100].
Figure 24.
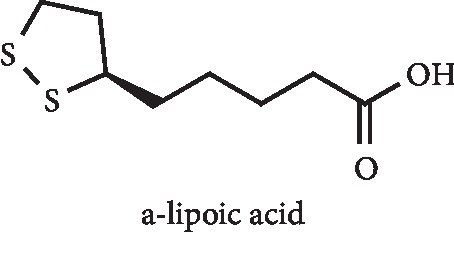
Structure of α-lipoic acid.
The alkaloids from Alphonsea elliptica barks atherospermidine (IC50 = 46.3 μg/mL), liriodenine (IC50 = 7.7 μg/mL), N-methylouregidione (IC50 = 42.1 μg/mL), and kinabaline (IC50 = 50.7 μg/mL) (Figure 25) were described as possessing moderate inhibition of XO activity by Aldulaimi et al. In addition, the cytotoxicity on MCF-7 human breast cancer cells was evaluated, with determined IC50 values greater than 62.0 μg/mL [101].
Figure 25.

Structures of alkaloids with XO inhibitory activity.
The effect of lobetyolin (Figure 26) on XO inhibition was presented by Yoon and Cho. Even though a weak in vitro mixed XO inhibition (IC50 = 2985 μM), an in vivo significant hepatic XO activity reduction at a dose of 50 mg/kg was observed [102].
Figure 26.
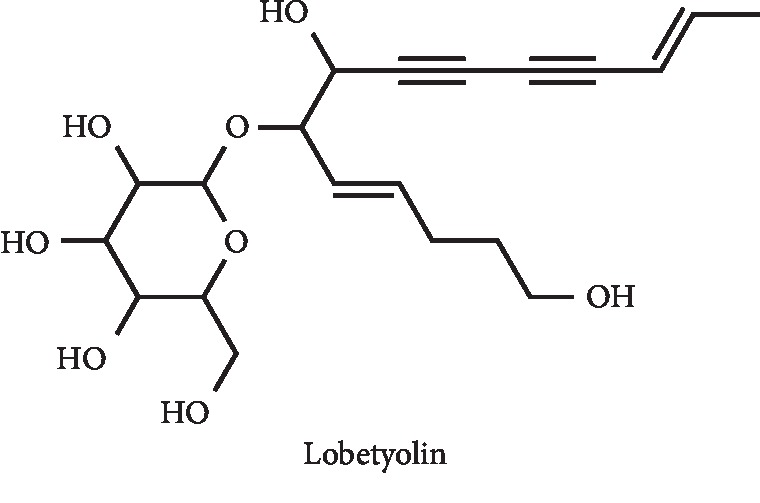
Structure of lobetyolin.
In the field of small natural molecules, Feng et al. demonstrated that the administration of 1% taurine (Figure 27), a sulfur-containing semiessential amino acid, in drinking water could efficiently ameliorate kidney injury and decrease uric acid levels by regulating uric acid formation and excretion in hyperuricemic rats [103]. The significant inhibition of XO activity by taurine was already been evidenced [104].
Figure 27.

Structures of the amino acid taurine and of the peptide RDP-1.
A new peptide, RDP-1 (Figure 27), was isolated from the extract of shelled fruits of Oryza sativa and structurally characterized. In vivo biological evaluation evidenced that after intragastric administration, this compound (10 μg/Kg) reduced hyperuricemia induced by potassium oxonate in rats. In addition, a reduction of creatinine levels and an ameliorating effect on hyperuricemic nephropathy was observed. It was also demonstrated that this peptide inhibited XO in vitro and in vivo, probably by occupying the binding site to xanthine (docking studies). Furthermore, no acute toxicity in rats was observed, and authors also evidenced that RDP-1 was stable in several temperatures [105].
4. Conclusions
Given the relevance of XO as a validated target to reduce increased serum UA levels and also due to the fact that the commercialized drugs with XO inhibitory effects can have serious side effects, in recent years a large number of new alternative XO inhibitors were described. Despite the fact that the majority of tested products with relevant in vitro XO inhibition do not advance for further pharmacological evaluation, it is clear in this review that a large number of compounds are being explored not only in in vitro studies but also in in vivo biological evaluation, and very promising molecules can be recognized and used in future studies. It is also possible to conclude that the majority of compounds being explored in more advanced biological evaluation studies are natural structures or their mimetics and semisynthetic derivatives. In addition, it is clear that frequently the observed in vitro XO inhibition results do not have a clear correspondence with the in vivo hypouricemic effects. This fact evidences the relevance of performing in vivo biological evaluation of the efficacy of the molecules in addition to the in vitro XO inhibition studies and access, as far as possible, other possible mechanisms of action, as well as the pharmacokinetics and toxicity profile of the molecules under study.
Acknowledgments
This work was supported by FEDER funds through the POCI—COMPETE 2020—Operational Programme Competitiveness and Internationalization in Axis I—Strengthening research, technological development and innovation (Project no. 007491) and National Funds by FCT—Foundation for Science and Technology (Project UID/Multi/00709). J. L. Serrano and J. Figueiredo acknowledge a fellowship from Santander-Totta/UBI (BID/ICI-UID FC/Santander Universidades-UBI/2017).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Chen C., Lü J.-M., Yao Q. Hyperuricemia-related diseases and xanthine oxidoreductase (XOR) inhibitors: an overview. Medical Science Monitor. 2016;22:2501–2512. doi: 10.12659/msm.899852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison R. Physiological roles of xanthine oxidoreductase. Drug Metabolism Reviews. 2004;36(2):363–375. doi: 10.1081/DMR-120037569. [DOI] [PubMed] [Google Scholar]
- 3.Maiuolo J., Oppedisano F., Gratteri S., Muscoli C., Mollace V. Regulation of uric acid metabolism and excretion. International Journal of Cardiology. 2016;213:8–14. doi: 10.1016/j.ijcard.2015.08.109. [DOI] [PubMed] [Google Scholar]
- 4.Wortmann R. L. Gout and hyperuricemia. Current Opinion in Rheumatology. 2002;14(3):281–286. doi: 10.1097/00002281-200205000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Jin M., Yang F., Yang I., et al. Uric acid, hyperuricemia and vascular diseases. Frontiers in Bioscience. 2012;17(1):656–669. doi: 10.2741/3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Šmelcerović A., Tomović K., Šmelcerović Ž., et al. Xanthine oxidase inhibitors beyond allopurinol and febuxostat; an overview and selection of potential leads based on in silico calculated physico-chemical properties, predicted pharmacokinetics and toxicity. European Journal of Medicinal Chemistry. 2017;135:491–516. doi: 10.1016/j.ejmech.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 7.Kumar R., Darpan, Sharma S., Singh R. Xanthine oxidase inhibitors: a patent survey. Expert Opinion on Therapeutic Patents. 2011;21(7):1071–1108. doi: 10.1517/13543776.2011.577417. [DOI] [PubMed] [Google Scholar]
- 8.Ojha R., Singh J., Ojha A., Singh H., Sharma S., Nepali K. An updated patent review: xanthine oxidase inhibitors for the treatment of hyperuricemia and gout (2011–2015) Expert Opinion on Therapeutic Patents. 2017;27(3):311–345. doi: 10.1080/13543776.2017.1261111. [DOI] [PubMed] [Google Scholar]
- 9.Borges F., Fernandes E., Roleira F. Progress towards the discovery of xanthine oxidase inhibitors. Current Medicinal Chemistry. 2002;9(2):195–217. doi: 10.2174/0929867023371229. [DOI] [PubMed] [Google Scholar]
- 10.Luna G., Dolzhenko A. V., Mancera R. L. Inhibitors of xanthine oxidase: scaffold diversity and structure-based drug design. ChemMedChem. 2019;14(7):714–743. doi: 10.1002/cmdc.201900034. [DOI] [PubMed] [Google Scholar]
- 11.Mehmood A., Ishaq M., Zhao L., et al. Natural compounds with xanthine oxidase inhibitory activity: a review. Chemical Biology & Drug Design. 2019;93(4):387–418. doi: 10.1111/cbdd.13437. [DOI] [PubMed] [Google Scholar]
- 12.Sathisha K. R., Khanum S. A., Chandra J. N. N. S., et al. Synthesis and xanthine oxidase inhibitory activity of 7-methyl-2-(phenoxymethyl)-5H-[1,3,4]thiadiazolo[3,2-a]pyrimidin-5-one derivatives. Bioorganic & Medicinal Chemistry. 2011;19(1):211–220. doi: 10.1016/j.bmc.2010.11.034. [DOI] [PubMed] [Google Scholar]
- 13.Sathisha K. R., Gopal S., Rangappa K. S. Antihyperuricemic effects of thiadiazolopyrimidin-5-one analogues in oxonate treated rats. European Journal of Pharmacology. 2016;776:99–105. doi: 10.1016/j.ejphar.2016.02.039. [DOI] [PubMed] [Google Scholar]
- 14.Yuan L., Song C., Li C., Li Y., Dong L., Yin S. Synthesis and biological evaluation of pyrazolo[4,3-d]pyrimidine analogues. European Journal of Medicinal Chemistry. 2013;67:152–157. doi: 10.1016/j.ejmech.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 15.Li Y., Cao T.-T., Guo S., et al. Discovery of novel allopurinol derivatives with anticancer activity and attenuated xanthine oxidase inhibition. Molecules. 2016;21(6):p. 771. doi: 10.3390/molecules21060771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saïd A. B., Romdhane A., Elie N., Touboul D., Jannet H. B., Bouajila J. Design, synthesis of novel pyranotriazolopyrimidines and evaluation of their anti-soybean lipoxygenase, anti-xanthine oxidase, and cytotoxic activities. Journal of Enzyme Inhibition and Medicinal Chemistry. 2016;31(6):1277–1285. doi: 10.3109/14756366.2015.1118684. [DOI] [PubMed] [Google Scholar]
- 17.Li J., Wu F., Liu X., et al. Synthesis and bioevaluation of 1-phenyl-pyrazole-4-carboxylic acid derivatives as potent xanthine oxidoreductase inhibitors. European Journal of Medicinal Chemistry. 2017;140:20–30. doi: 10.1016/j.ejmech.2017.08.047. [DOI] [PubMed] [Google Scholar]
- 18.Xu X., Deng L., Nie L., et al. Discovery of 2-phenylthiazole-4-carboxylic acid, a novel and potent scaffold as xanthine oxidase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2019;29(4):525–528. doi: 10.1016/j.bmcl.2019.01.005. [DOI] [PubMed] [Google Scholar]
- 19.Song J. U., Choi S. P., Kim T. H., et al. Design and synthesis of novel 2-(indol-5-yl)thiazole derivatives as xanthine oxidase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2015;25(6):1254–1258. doi: 10.1016/j.bmcl.2015.01.055. [DOI] [PubMed] [Google Scholar]
- 20.Song J. U., Jang J. W., Kim T. H., et al. Structure-based design and biological evaluation of novel 2-(indol-2-yl) thiazole derivatives as xanthine oxidase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2016;26(3):950–954. doi: 10.1016/j.bmcl.2015.12.055. [DOI] [PubMed] [Google Scholar]
- 21.Ali M. R., Kumar S., Afzal O., et al. 2-Benzamido-4-methylthiazole-5-carboxylic acid derivatives as potential xanthine oxidase inhibitors and free radical scavengers. Archiv der Pharmazie. 2017;350(2) doi: 10.1002/ardp.201600313.1600313 [DOI] [PubMed] [Google Scholar]
- 22.Mao Q., Dai X., Xu G., et al. Design, synthesis and biological evaluation of 2-(4-alkoxy-3-cyano)phenyl-6-oxo-1,6-dihydropyrimidine-5-carboxylic acid derivatives as novel xanthine oxidase inhibitors. European Journal of Medicinal Chemistry. 2019;181 doi: 10.1016/j.ejmech.2019.07.061.111558 [DOI] [PubMed] [Google Scholar]
- 23.Zhang B., Dai X., Bao Z., et al. Targeting the subpocket in xanthine oxidase: design, synthesis, and biological evaluation of 2-[4-alkoxy-3-(1H-tetrazol-1-yl) phenyl]-6-oxo-1,6-dihydropyrimidine-5-carboxylic acid derivatives. European Journal of Medicinal Chemistry. 2019;181 doi: 10.1016/j.ejmech.2019.07.062.111559 [DOI] [PubMed] [Google Scholar]
- 24.Zafar H., Iqbal S., Javaid S., Khan K. M., Choudhary M. I. Xanthine oxidase inhibitory and molecular docking studies on pyrimidones. Medicinal Chemistry. 2018;14(5):524–535. doi: 10.2174/1573406413666171129224919. [DOI] [PubMed] [Google Scholar]
- 25.Serrano J. L., Cavalheiro E., Barroso S., Romão M. J., Silvestre S., Almeida P. A synthetic route to novel 3-substituted-2,1-benzisoxazoles from 5-(2-nitrobenzylidene)(thio)barbiturates. Comptes Rendus Chimie. 2017;20(11-12):990–995. doi: 10.1016/j.crci.2017.10.002. [DOI] [Google Scholar]
- 26.Figueiredo J., Serrano J. L., Cavalheiro E., et al. Trisubstituted barbiturates and thiobarbiturates: synthesis and biological evaluation as xanthine oxidase inhibitors, antioxidants, antibacterial and anti-proliferative agents. European Journal of Medicinal Chemistry. 2018;143:829–842. doi: 10.1016/j.ejmech.2017.11.070. [DOI] [PubMed] [Google Scholar]
- 27.Figueiredo J., Serrano J. L., Soares M., et al. 5-Hydrazinylethylidenepyrimidines effective against multidrug-resistant Acinetobacter baumannii: synthesis and in vitro biological evaluation of antibacterial, radical scavenging and cytotoxic activities. European Journal of Pharmaceutical Sciences. 2019;137 doi: 10.1016/j.ejps.2019.104964.104964 [DOI] [PubMed] [Google Scholar]
- 28.El-Sayed N. N. E., Almaneai N. M., Ben Bacha A., et al. Synthesis and evaluation of anticancer, antiphospholipases, antiproteases, and antimetabolic syndrome activities of some 3H-quinazolin-4-one derivatives. Journal of Enzyme Inhibition and Medicinal Chemistry. 2019;34(1):672–683. doi: 10.1080/14756366.2019.1574780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zafar H., Hayat M., Saied S., et al. Xanthine oxidase inhibitory activity of nicotino/isonicotinohydrazides: a systematic approach from in vitro, in silico to in vivo studies. Bioorganic & Medicinal Chemistry. 2017;25(8):2351–2371. doi: 10.1016/j.bmc.2017.02.044. [DOI] [PubMed] [Google Scholar]
- 30.Mabkhot Y. N., Barakat A., Yousuf S., et al. Substituted thieno[2,3-b]thiophenes and related congeners: synthesis, β-glucuronidase inhibition activity, crystal structure, and POM analyses. Bioorganic & Medicinal Chemistry. 2014;22(23):6715–6725. doi: 10.1016/j.bmc.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 31.Niu Y., Li H., Gao L., et al. Old drug, new indication: olsalazine sodium reduced serum uric acid levels in mice via inhibiting xanthine oxidoreductase activity. Journal of Pharmacological Sciences. 2017;135(3):114–120. doi: 10.1016/j.jphs.2017.10.007. [DOI] [PubMed] [Google Scholar]
- 32.Yong T., Li D., Li M., et al. Anti-hyperuricemic effect of 2-hydroxy-4-methoxy-benzophenone-5-sulfonic acid in hyperuricemic mice through XOD. Molecules. 2018;23(10):p. 2671. doi: 10.3390/molecules23102671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arora S., Joshi G., Kalra S., et al. Knoevenagel/tandem knoevenagel and michael adducts of cyclohexane-1,3-dione and aryl aldehydes: synthesis, DFT studies, xanthine oxidase inhibitory potential, and molecular modeling. ACS Omega. 2019;4(3):4604–4614. doi: 10.1021/acsomega.8b03060. [DOI] [Google Scholar]
- 34.Zafar H., Hamdi N., Lachkar M., Bali B. E., Khan A., Choudhary M. I. Dinuclear cyclam complex as a non-cytotoxic, anti-hyperurecemic lead: in vitro to in vivo studies. Medicinal Chemistry. 2017;13(6):585–591. doi: 10.2174/1573406413666170309095104. [DOI] [PubMed] [Google Scholar]
- 35.Özerkan D., Ertik O., Kaya B., Kuruca S. E., Yanardag R., Ülküseven B. Novel palladium (II) complexes with tetradentate thiosemicarbazones. Synthesis, characterization, in vitro cytotoxicity and xanthine oxidase inhibition. Investigational New Drugs. 2019;37(6):1187–1197. doi: 10.1007/s10637-019-00751-1. [DOI] [PubMed] [Google Scholar]
- 36.Li L.-Z., Zhou G.-X., Li J., Jiang W., Liu B.-L., Zhou W. Compounds containing trace element copper or zinc exhibit as potent hyperuricemia inhibitors via xanthine oxidase inactivation. Journal of Trace Elements in Medicine and Biology. 2018;49:72–78. doi: 10.1016/j.jtemb.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 37.Lü J.-M., Yao Q., Chen C. 3,4-dihydroxy-5-nitrobenzaldehyde (DHNB) is a potent inhibitor of xanthine oxidase: a potential therapeutic agent for treatment of hyperuricemia and gout. Biochemical Pharmacology. 2013;86(9):1328–1337. doi: 10.1016/j.bcp.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liang D., Yong T., Chen S., et al. Hypouricemic effect of 2,5-dihydroxyacetophenone, a computational screened bioactive compound from Ganoderma applanatum, on hyperuricemic mice. International Journal of Molecular Sciences. 2018;19(5):p. 1394. doi: 10.3390/ijms19051394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan Y., Liang Y.-X., Zou B., Fu G.-M., Xie M.-Y. The possible mechanism of hydroxytyrosol on reducing uric acid levels. Journal of Functional Foods. 2018;42:319–326. doi: 10.1016/j.jff.2018.01.009. [DOI] [Google Scholar]
- 40.Nile S. H., Ko E. Y., Kim D. H., Keum Y.-S. Screening of ferulic acid related compounds as inhibitors of xanthine oxidase and cyclooxygenase-2 with anti-inflammatory activity. Revista Brasileira de Farmacognosia. 2016;26(1):50–55. doi: 10.1016/j.bjp.2015.08.013. [DOI] [Google Scholar]
- 41.Wan Y., Wang F., Zou B., et al. Molecular mechanism underlying the ability of caffeic acid to decrease uric acid levels in hyperuricemia rats. Journal of Functional Foods. 2019;57:150–156. doi: 10.1016/j.jff.2019.03.038. [DOI] [Google Scholar]
- 42.Wan Y., Zou B., Zeng H., Zhang L., Chen M., Fu G. Inhibitory effect of verbascoside on xanthine oxidase activity. International Journal of Biological Macromolecules. 2016;93:609–614. doi: 10.1016/j.ijbiomac.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 43.Yuk H., Lee Y.-S., Ryu H., Kim S.-H., Kim D.-S. Effects of Toona sinensis leaf extract and its chemical constituents on xanthine oxidase activity and serum uric acid levels in potassium oxonate-induced hyperuricemic rats. Molecules. 2018;23(12):p. 3254. doi: 10.3390/molecules23123254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang H., Yang L., Li W., Li J., Chen J. Exploring the interaction between Salvia miltiorrhiza and xanthine oxidase: insights from computational analysis and experimental studies combined with enzyme channel blocking. RSC Advances. 2016;6(114):113527–113537. doi: 10.1039/c6ra24396g. [DOI] [Google Scholar]
- 45.Fu Y., Mo H.-Y., Gao W., et al. Affinity selection-based two-dimensional chromatography coupled with high-performance liquid chromatography-mass spectrometry for discovering xanthine oxidase inhibitors from Radix Salviae miltiorrhizae. Analytical and Bioanalytical Chemistry. 2014;406(20):4987–4995. doi: 10.1007/s00216-014-7902-9. [DOI] [PubMed] [Google Scholar]
- 46.Wang H., Li X., Zhang W., et al. Mechanism-based pharmacokinetic–pharmacodynamic modeling of salvianolic acid A effects on plasma xanthine oxidase activity and uric acid levels in acute myocardial infarction rats. Xenobiotica. 2017;47(3):208–216. doi: 10.1080/00498254.2016.1180440. [DOI] [PubMed] [Google Scholar]
- 47.Tang H.-J., Li W., Zhou M., et al. Design, synthesis and biological evaluation of novel xanthine oxidase inhibitors bearing a 2-arylbenzo[b]furan scaffold. European Journal of Medicinal Chemistry. 2018;151:849–860. doi: 10.1016/j.ejmech.2018.01.096. [DOI] [PubMed] [Google Scholar]
- 48.Kar Mahapatra D., Asati V., Bharti S. K. An updated patent review of therapeutic applications of chalcone derivatives (2014–present) Expert Opinion on Therapeutic Patents. 2019;29(5):385–406. doi: 10.1080/13543776.2019.1613374. [DOI] [PubMed] [Google Scholar]
- 49.Zhuang C., Zhang W., Sheng C., Zhang W., Xing C., Miao Z. Chalcone: a privileged structure in medicinal chemistry. Chemical Reviews. 2017;117(12):7762–7810. doi: 10.1021/acs.chemrev.7b00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Niu Y., Zhou Y., Lin H., et al. Inhibition of 3,5,2′,4′-tetrahydroxychalcone on production of uric acid in hypoxanthine-induced hyperuricemic mice. Biological & Pharmaceutical Bulletin. 2018;41(1):99–105. doi: 10.1248/bpb.b17-00655. [DOI] [PubMed] [Google Scholar]
- 51.Niu Y. F., Liu K., Gao L. H., et al. Effects of 3,5,2′,4′-tetrahydroxychalcone on serum uric acid levels and the content of hepatic XOD/XDH in mice. Chinese Pharmaceutical Journal. 2015;50:34–38. [Google Scholar]
- 52.Niu Y., Zhu H., Liu J., et al. 3,5,2′,4′-tetrahydroxychalcone, a new non-purine xanthine oxidase inhibitor. Chemico-Biological Interactions. 2011;189(3):161–166. doi: 10.1016/j.cbi.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 53.Hofmann E., Webster J., Do T., et al. Hydroxylated chalcones with dual properties: xanthine oxidase inhibitors and radical scavengers. Bioorganic & Medicinal Chemistry. 2016;24(4):578–587. doi: 10.1016/j.bmc.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie Z., Luo X., Zou Z., et al. Synthesis and evaluation of hydroxychalcones as multifunctional non-purine xanthine oxidase inhibitors for the treatment of hyperuricemia. Bioorganic & Medicinal Chemistry Letters. 2017;27(15):3602–3606. doi: 10.1016/j.bmcl.2017.01.053. [DOI] [PubMed] [Google Scholar]
- 55.Kong L. D., Zhang Y., Pan X., Tan R. X., Cheng C. H. K. Inhibition of xanthine oxidase by liquiritigenin and isoliquiritigenin isolated from Sinofranchetia chinensis. Cellular and Molecular Life Sciences. 2000;57(3):500–505. doi: 10.1007/pl00000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qiao H., Zhang X., Wang T., Liang L., Chang W., Xia H. Pharmacokinetics, biodistribution and bioavailability of isoliquiritigenin after intravenous and oral administration. Pharmaceutical Biology. 2014;52(2):228–236. doi: 10.3109/13880209.2013.832334. [DOI] [PubMed] [Google Scholar]
- 57.Zhang K., Wang Q., Yang Q., et al. Enhancement of oral bioavailability and anti-hyperuricemic activity of isoliquiritigenin via self-microemulsifying drug delivery system. AAPS PharmSciTech. 2019;20(5):1265–1276. doi: 10.1208/s12249-019-1421-0. [DOI] [PubMed] [Google Scholar]
- 58.Peng F., Tao Q., Wu X., et al. Cytotoxic, cytoprotective and antioxidant effects of isolated phenolic compounds from fresh ginger. Fitoterapia. 2012;83(3):568–585. doi: 10.1016/j.fitote.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 59.Nile S. H., Park S. W. Chromatographic analysis, antioxidant, anti-inflammatory, and xanthine oxidase inhibitory activities of ginger extracts and its reference compounds. Industrial Crops and Products. 2015;70:238–244. doi: 10.1016/j.indcrop.2015.03.033. [DOI] [Google Scholar]
- 60.Wang Q., Yang Q., Cao X., et al. Enhanced oral bioavailability and anti-gout activity of [6]-shogaol-loaded solid lipid nanoparticles. International Journal of Pharmaceutics. 2018;550(1-2):24–34. doi: 10.1016/j.ijpharm.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 61.Yang Q., Wang Q., Feng Y., et al. Anti-hyperuricemic property of 6-shogaol via self-micro emulsifying drug delivery system in model rats: formulation design, in vitro and in vivo evaluation. Drug Development and Industrial Pharmacy. 2019;45(8):1265–1276. doi: 10.1080/03639045.2019.1594885. [DOI] [PubMed] [Google Scholar]
- 62.Chen Y., Li C., Duan S., Yuan X., Liang J., Hou S. Curcumin attenuates potassium oxonate-induced hyperuricemia and kidney inflammation in mice. Biomedicine & Pharmacotherapy. 2019;118 doi: 10.1016/j.biopha.2019.109195.109195 [DOI] [PubMed] [Google Scholar]
- 63.Ao G.-Z., Zhou M.-Z., Li Y.-Y., et al. Discovery of novel curcumin derivatives targeting xanthine oxidase and urate transporter 1 as anti-hyperuricemic agents. Bioorganic & Medicinal Chemistry. 2017;25(1):166–174. doi: 10.1016/j.bmc.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 64.Karak P. Biological activities of flavonoids: an overview. International Journal of Pharmaceutical Sciences and Research. 2019;10:1567–1574. doi: 10.13040/ijpsr.0975-8232. [DOI] [Google Scholar]
- 65.Mo S.-F., Zhou F., Lv Y.-Z., Hu Q.-H., Zhang D.-M., Kong L.-D. Hypouricemic action of selected flavonoids in mice: structure-activity relationships. Biological & Pharmaceutical Bulletin. 2007;30(8):1551–1556. doi: 10.1248/bpb.30.1551. [DOI] [PubMed] [Google Scholar]
- 66.Nile S. H., Keum Y. S., Nile A. S., Jalde S. S., Patel R. V. Antioxidant, anti-inflammatory, and enzyme inhibitory activity of natural plant flavonoids and their synthesized derivatives. Journal of Biochemical and Molecular Toxicology. 2018;32(1) doi: 10.1002/jbt.22002.e22002 [DOI] [PubMed] [Google Scholar]
- 67.Su Z.-R., Fan S.-Y., Shi W.-G., Zhong B.-H. Discovery of xanthine oxidase inhibitors and/or α-glucosidase inhibitors by carboxyalkyl derivatization based on the flavonoid of apigenin. Bioorganic & Medicinal Chemistry Letters. 2015;25(14):2778–2781. doi: 10.1016/j.bmcl.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 68.Metoui R., Bouajila J., Znati M., Cazaux S., Neffati M., Akrout A. Bioactive flavones isolated from Tunisian Artemisia campestris L. Leaves. Cellular and Molecular Biology. 2017;63(11):86–91. doi: 10.14715/cmb/2017.63.11.15. [DOI] [PubMed] [Google Scholar]
- 69.Yan J., Zhang G., Hu Y., Ma Y. Effect of luteolin on xanthine oxidase: inhibition kinetics and interaction mechanism merging with docking simulation. Food Chemistry. 2013;141(4):3766–3773. doi: 10.1016/j.foodchem.2013.06.092. [DOI] [PubMed] [Google Scholar]
- 70.Dong H., Yang X., He J., Cai S., Xiao K., Zhu L. Enhanced antioxidant activity, antibacterial activity and hypoglycemic effect of luteolin by complexation with manganese(II) and its inhibition kinetics on xanthine oxidase. RSC Advances. 2017;7(84):53385–53395. doi: 10.1039/c7ra11036g. [DOI] [Google Scholar]
- 71.Zhang H.-J., Hu Y.-J., Xu P., et al. Screening of potential xanthine oxidase inhibitors in Gnaphalium hypoleucum DC. by immobilized metal affinity chromatography and ultrafiltration-ultra performance liquid chromatography-mass spectrometry. Molecules. 2016;21(9):p. 1242. doi: 10.3390/molecules21091242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin Y., Liu P.-G., Liang W.-Q., et al. Luteolin-4′-O-glucoside and its aglycone, two major flavones of Gnaphalium affine D. Don, resist hyperuricemia and acute gouty arthritis activity in animal models. Phytomedicine. 2018;41:54–61. doi: 10.1016/j.phymed.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 73.Cheng L. C., Murugaiyah V., Chan K. L. In vitro xanthine oxidase inhibitory studies of Lippia nodiflora and isolated flavonoids and phenylethanoid glycosides as potential uric acid-lowering agents. Natural Product Communications. 2015;10(6):945–948. doi: 10.1177/1934578x1501000638. [DOI] [PubMed] [Google Scholar]
- 74.Cheng L.-C., Murugaiyah V., Chan K.-L. Flavonoids and phenylethanoid glycosides from Lippia nodiflora as promising antihyperuricemic agents and elucidation of their mechanism of action. Journal of Ethnopharmacology. 2015;176:485–493. doi: 10.1016/j.jep.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 75.de Araújo M. E. M. B., Moreira Franco Y. E., Alberto T. G., et al. Enzymatic de-glycosylation of rutin improves its antioxidant and antiproliferative activities. Food Chemistry. 2013;141(1):266–273. doi: 10.1016/j.foodchem.2013.02.127. [DOI] [PubMed] [Google Scholar]
- 76.Nguyen M. T. T., Nguyen N. T. A new lupane triterpene from Tetracera scandens L., xanthine oxidase inhibitor. Natural Product Research. 2013;27(1):61–67. doi: 10.1080/14786419.2011.652960. [DOI] [PubMed] [Google Scholar]
- 77.Zhang Z.-C., Su G.-H., Luo C.-L., et al. Effects of anthocyanins from purple sweet potato (Ipomoea batatas L. cultivar Eshu no. 8) on the serum uric acid level and xanthine oxidase activity in hyperuricemic mice. Food & Function. 2015;6(9):3045–3055. doi: 10.1039/C5FO00499C. [DOI] [PubMed] [Google Scholar]
- 78.Zhang Z.-C., Wang H.-B., Zhou Q., Hu B., Wen J.-H., Zhang J.-L. Screening of effective xanthine oxidase inhibitors in dietary anthocyanins from purple sweet potato (Ipomoea batatas L. cultivar Eshu no. 8) and deciphering of the underlying mechanisms in vitro. Journal of Functional Foods. 2017;36:102–111. doi: 10.1016/j.jff.2017.06.048. [DOI] [Google Scholar]
- 79.Zhang J.-L., Luo C.-L., Zhou Q., Zhang Z.-C. Isolation and identification of two major acylated anthocyanins from purple sweet potato (Ipomoea batatas L. cultivar Eshu no. 8) by UPLC-QTOF-MS/MS and NMR. International Journal of Food Science & Technology. 2018;53(8):1932–1941. doi: 10.1111/ijfs.13780. [DOI] [Google Scholar]
- 80.Zhang Z.-c., Zhou Q., Yang Y., Wang Y., Zhang J.-l. Highly acylated anthocyanins from purple sweet potato (Ipomoea batatas L.) alleviate hyperuricemia and kidney inflammation in hyperuricemic mice: possible attenuation effects on allopurinol. Journal of Agricultural and Food Chemistry. 2019;67(22):6202–6211. doi: 10.1021/acs.jafc.9b01810. [DOI] [PubMed] [Google Scholar]
- 81.Wang R.-d., Su G.-h., Wang L., et al. Identification and mechanism of effective components from rape (Brassica napus L.) bee pollen on serum uric acid level and xanthine oxidase activity. Journal of Functional Foods. 2018;47:241–251. doi: 10.1016/j.jff.2018.05.064. [DOI] [Google Scholar]
- 82.de Souza V. T., de Franco É. P. D., de Araújo M. E. M. B., et al. Characterization of the antioxidant activity of aglycone and glycosylated derivatives of hesperetin: an in vitro and in vivo study. Journal of Molecular Recognition. 2016;29(2):80–87. doi: 10.1002/jmr.2509. [DOI] [PubMed] [Google Scholar]
- 83.Lin J.-K., Chen P.-C., Ho C.-T., Lin-Shiau S.-Y. Inhibition of xanthine oxidase and suppression of intracellular reactive oxygen species in HL-60 cells by theaflavin-3,3′-digallate, (−)- epigallocatechin-3-gallate, and propyl gallate. Journal of Agricultural and Food Chemistry. 2000;48(7):2736–2743. doi: 10.1021/jf000066d. [DOI] [PubMed] [Google Scholar]
- 84.Zhu C., Xu Y., Liu Z.-H., Wan X.-C., Li D.-X., Tai L.-L. The anti-hyperuricemic effect of epigallocatechin-3-gallate (EGCG) on hyperuricemic mice. Biomedicine & Pharmacotherapy. 2018;97:168–173. doi: 10.1016/j.biopha.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 85.Singh J. V., Mal G., Kaur G., et al. Benzoflavone derivatives as potent antihyperuricemic agents. MedChemComm. 2019;10(1):128–147. doi: 10.1039/c8md00512e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hu Q., Zhou M., Zhu H., et al. (E)-2-(4-Bromophenyl)-1-(2,4-dihydroxyphenyl)ethanone oxime is a potential therapeutic agent for treatment of hyperuricemia through its dual inhibitory effects on XOD and URAT1. Biomedicine & Pharmacotherapy. 2017;86:88–94. doi: 10.1016/j.biopha.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Huang J., Zhou Z., Zhou M., Miao M., Li H., Hu Q. Development of benzoxazole deoxybenzoin oxime and acyloxylamine derivatives targeting innate immune sensors and xanthine oxidase for treatment of gout. Bioorganic & Medicinal Chemistry. 2018;26(8):1653–1664. doi: 10.1016/j.bmc.2018.02.013. [DOI] [PubMed] [Google Scholar]
- 88.Zhou M., Li S., Song L., Hu Q., Liu W. 4-(2-(4-chlorophenyl)-1-((4-chlorophenyl)amino)ethyl)benzene-1,3-diol is a potential agent for gout therapy as a dual inhibitor of XOD and NLRP3. Phytomedicine. 2018;42:9–17. doi: 10.1016/j.phymed.2018.03.007. [DOI] [PubMed] [Google Scholar]
- 89.Fais A., Era B., Asthana S., et al. Coumarin derivatives as promising xanthine oxidase inhibitors. International Journal of Biological Macromolecules. 2018;120:1286–1293. doi: 10.1016/j.ijbiomac.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 90.Lin H., Tu C., Niu Y., et al. Dual actions of norathyriol as a new candidate hypouricaemic agent: uricosuric effects and xanthine oxidase inhibition. European Journal of Pharmacology. 2019;853:371–380. doi: 10.1016/j.ejphar.2019.04.034. [DOI] [PubMed] [Google Scholar]
- 91.Qin Z., Wang S., Lin Y., et al. Antihyperuricemic effect of mangiferin aglycon derivative J99745 by inhibiting xanthine oxidase activity and urate transporter 1 expression in mice. Acta Pharmaceutica Sinica B. 2018;8(2):306–315. doi: 10.1016/j.apsb.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lin K.-W., Maitraie D., Huang A.-M., Wang J.-P., Lin C.-N. Triterpenoids and an alkamide from Ganoderma tsugae. Fitoterapia. 2016;108:73–80. doi: 10.1016/j.fitote.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 93.Lin K.-W., Chen Y.-T., Yang S.-C., Wei B.-L., Hung C.-F., Lin C.-N. Xanthine oxidase inhibitory lanostanoids from Ganoderma tsugae. Fitoterapia. 2013;89:231–238. doi: 10.1016/j.fitote.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 94.Chiang H.-C., Chen Y.-Y. Xanthine oxidase inhibitors from the roots of eggplant (Solanum melongena L.) Journal of Enzyme Inhibition. 1993;7(3):225–235. doi: 10.3109/14756369309040765. [DOI] [Google Scholar]
- 95.Su J., Wei Y., Liu M., et al. Anti-hyperuricemic and nephroprotective effects of rhizoma Dioscoreae septemlobae extracts and its main component dioscin via regulation of mOAT1, mURAT1 and mOCT2 in hypertensive mice. Archives of Pharmacal Research. 2014;37(10):1336–1344. doi: 10.1007/s12272-014-0413-6. [DOI] [PubMed] [Google Scholar]
- 96.Zhang Y., Jin L., Liu J., et al. Effect and mechanism of dioscin from Dioscorea spongiosa on uric acid excretion in animal model of hyperuricemia. Journal of Ethnopharmacology. 2018;214:29–36. doi: 10.1016/j.jep.2017.12.004. [DOI] [PubMed] [Google Scholar]
- 97.Abu-Gharbieh E., Shehab N. G., Almasri I. M., Bustanji Y. Antihyperuricemic and xanthine oxidase inhibitory activities of Tribulus arabicus and its isolated compound, ursolic acid: in vitro and in vivo investigation and docking simulations. PLoS One. 2018;13(8) doi: 10.1371/journal.pone.0202572.e0202572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guo E., Jiang A., Chen W. Application of ursolic acid to preparation of medicine for treating gout. 2016. Patent No. CN106727602A.
- 99.Nile S. H., Nile A., Liu J., Kim D. H., Kai G. Exploitation of apple pomace towards extraction of triterpenic acids, antioxidant potential, cytotoxic effects, and inhibition of clinically important enzymes. Food and Chemical Toxicology. 2019;131 doi: 10.1016/j.fct.2019.110563.110563 [DOI] [PubMed] [Google Scholar]
- 100.Hameed B. J., Ramadhan U. H. Xanthine oxidase inhibitory, antihyperuricemic, anti-inflammatory, antinociceptive activity of α-lipoic acid in gouty arthritis model. Asian Journal of Pharmaceutical and Clinical Research. 2018;11(12):483–487. doi: 10.22159/ajpcr.2018.v11i12.29160. [DOI] [Google Scholar]
- 101.Aldulaimi A. K. O., Azziz S. S. S. A., Bakri Y. M., et al. Alkaloids from Alphonsea elliptica barks and their biological activities. Journal of Global Pharma Technology. 2018;10(8):270–275. [Google Scholar]
- 102.Yoon I.-S., Cho S.-S. Effects of lobetyolin on xanthine oxidase activity in vitro and in vivo: weak and mixed inhibition. Natural Product Research. 2019:1–4. doi: 10.1080/14786419.2019.1622108. , In press. [DOI] [PubMed] [Google Scholar]
- 103.Feng Y., Sun F., Gao Y., et al. Taurine decreased uric acid levels in hyperuricemic rats and alleviated kidney injury. Biochemical and Biophysical Research Communications. 2017;489(3):312–318. doi: 10.1016/j.bbrc.2017.05.139. [DOI] [PubMed] [Google Scholar]
- 104.Das J., Sil P. C. Taurine ameliorates alloxan-induced diabetic renal injury, oxidative stress-related signaling pathways and apoptosis in rats. Amino Acids. 2012;43(4):1509–1523. doi: 10.1007/s00726-012-1225-y. [DOI] [PubMed] [Google Scholar]
- 105.Liu N., Wang Y., Yang M., et al. New rice-derived short peptide potently alleviated hyperuricemia induced by potassium oxonate in rats. Journal of Agricultural and Food Chemistry. 2019;67(1):220–228. doi: 10.1021/acs.jafc.8b05879. [DOI] [PubMed] [Google Scholar]