

Abstract
Introduction: Circulating tumor DNA (ctDNA) for monitoring the effects of chemotherapy and predicting prognosis in advanced gastric cancer have not been thoroughly investigated. Methods: We performed next-generation sequencing (NGS) of ctDNA from 23 gastric cancer patients. Then the genetic information and clinical information were statistically analyzed. Results: In this study, the frequency of TP53 was significantly different between the effective and ineffective groups (P = 0.040), and the number of TP53 mutations was more frequent in the ineffective group. Missense mutation was a significant difference between the treatment effect groups (P = 0.026). The number of gene mutations and the change in copy number levels were related to therapeutic effect. Among the ineffective group, there was a significant difference in the number of gene mutations (P = 0.0006). We further divided the number of gene mutations into an increase group and a decrease group, and found that there was a significant difference between the effective and ineffective groups (P = 0.038). Finally, it was found that patients with high mutation abundance of gastric cancer had a shorter overall survival than patients with low mutation abundance (P<0.05). Conclusion: ctDNA can be used as an effective tool to monitor the efficacy of chemotherapy and predict prognosis in advanced gastric cancer.
Keywords: Gastric cancer, ctDNA, next-generation sequencing, treatment effect
Introduction
Gastric cancer is the fifth most common malignant tumor in the world [1]. In China, most patients with gastric cancer have been diagnosed at the advanced stage. Chemotherapy is an important method of treatment of advanced gastric cancer. Sometimes we encounter situations in which tumor size temporarily increases despite the high anti-tumor activity. This pseudo-progression makes clinicians confused regarding the evaluation of treatment outcome [2-6]. Therefore, there is an urgent need for a detection method that can effectively monitor the effects of chemotherapy in real time.
Circulating free DNA (cfDNA) refers to partially degraded endogenous DNA that is free of extracellular cells and is found in circulating blood. Stroun [7] has confirmed the tumor-derived nature of the circulating DNA. Circulating tumor DNA (ctDNA) can be detected using highly sensitive detection techniques such as next-generation sequencing (NGS). Monitoring ctDNA levels can be used to understand tumor burden. At present, no report has been found on the correlation between ctDNA detection and the outcome of therapy for gastric cancer. In this study, we tested ctDNA of 23 gastric cancer patients by NGS and found that changes in tumor mutation have a specific relationship with the change of the disease and overall survival.
Materials and methods
Patients and sample collection
Patients with gastric adenocarcinoma diagnosed at Fujian Medical University Union Hospital from November 2005 to July 2018 were enrolled. They were prepared to receive chemotherapy-based comprehensive treatment. The survival time was expected to be longer than 3 months before and after chemotherapy. When preparing the imaging evaluation effect every 2-3 cycles (using the solid tumor efficacy evaluation standard RECIST version 1.1, in which stable disease and partial response were set to be effective in this study, and progressive disease was set to be ineffective), 10 ml of peripheral blood was collected for ctDNA detection. Each time the detection of ctDNA corresponded to a baseline time point, a first evaluation time point and a second evaluation time point, respectively. The clinical information was as follows: gender, age, (carcinoembryonic antigen) CEA, metastasis, surgery, postoperative pathology, the variant allele frequency (VAF), chemotherapy effect, and so on. The study was approved by the ethics committee of the study and written informed consent signed by all patients.
This retrospective study was approved by the Ethical Committees of Fujian Medical University Union Hospital. Informed consent was signed for all enrolled patients.
Sample processing, DNA extraction and quality control
Peripheral blood was collected in EDTA Vacutainer tubes (BD Diagnostics, Franklin Lakes, NJ, USA) and processed within 4 h. Plasma was separated by two centrifugations to remove remaining cell debris. Peripheral blood lymphocytes (PBLs) from the first centrifugation were used for the extraction of germline genomic DNA. Circulating tumor DNA (ctDNA) was isolated using QIAsymphony Circulating DNA Kit (Qiagen, Hilden, Germany), and peripheral blood lymphocytes (PBLs) DNA was extracted using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s protocol. DNA concentration was measured using the Qubit 3.0 fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Thermo Fisher Scientific Inc., Carlsbad, CA, USA). Every ctDNA sample was analyzed on the Agilent 2100 BioAnalyzer using the Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA, USA), and ctDNA fragments distributed with a dominant peak at approximately 170 bp.
Target capture and next-generation sequencing
Sequencing libraries of both cfDNA and PBL DNA were constructed using the KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA). Libraries were measured using an Agilent 2100 Bioanalyzer and an Applied Biosystems 7500 real-time PCR system (Thermo Fisher Scientific Inc., Carlsbad, CA, USA). Libraries were hybridized to the custom-designed biotinylated oligonucleotide probes (Roche NimbleGen, Madison, WI, USA) covering about 1.1 Mbp of sequence of 1021 genes. DNA sequencing was performed on the HiSeq3000 Sequencing System (Illumina, San Diego, CA, USA) with 2×75 bp paired-end reads.
Sequencing data analysis
From raw sequencing data, terminal adaptor sequences and low-quality reads were removed. The reads were aligned to the human genome build GRCh37 using BWA (a Burrows-Wheeler aligner) [11]. To mark PCR duplicates, Picard tools (http://broadinstitute.github.io/picard/) were used. Single nucleotide variants (SNVs) and small insertions and deletions (Indels) were called using MuTect (version 1.1.4) [8]. PBL DNA sequencing results were used to identify somatic mutations. All candidate somatic mutations identified by the bioinformatics pipeline were manually reviewed in the Integrative Genomics Viewer (IGV) [9] through assessing the quality of base calls, the mapping quality of the reads and the overall read depth at each mutation site. A mutation was identified as somatic mutation when (1) with VAF≥0.1%, and (2) at least 5 high-quality reads (Phred score ≥30, mapping quality ≥30, and without paired-end reads bias). For a given variant in plasma ctDNA, VAF = sequencing read count of alternate alleles/(sequencing read count of reference alleles + sequencing read count of alternate alleles) ×100%. Mutations were annotated to genes by ANNOVAR [10] software to identify the mutated protein-coding position and filtered intronic and silent changes. CONTRA [11] was used to detect copy number variants (CNVs). BreakDancer [12] was used to detect cancer-associated structure variants (SVs). The mutation abundance is represented by VAF.
Overall survival (OS) refers to the time from the start of randomization to the death of any cause. For subjects who have been lost to follow-up before death, the last follow-up time is usually counted as the time of death.
Statistical analysis
Comparative analysis of two component ratios used chi-square test. Inferred analysis of the two population distribution patterns used the Wilcoxon test. The overall survival analysis was performed using the Kaplan-Meier method. IBM SPSS software (23.0), GraphPad Prism (6.01) and R software (3.4.3) were used in statistical analysis. All tests were two-sided and considered significant at P<0.05.
Results
Clinicopathological characteristics of the 23 patients and cluster analysis
Peripheral blood samples were obtained from the 23 patients at three different time points corresponding to the baseline time point, the first evaluation time point and the second evaluation time point. There were a total of 46 assessments. The patients were divided into an effective and an ineffective treatment group according to the results of imaging evaluation. In the first assessment, there were 13 patients in the effective group and 10 patients in the ineffective group. In the overall assessment, there were 8 patients in the effective group and 15 patients in the ineffective group.
The relevant clinicopathological characteristics of the patients were summarized in Table 1. It was shown that the degree of tumor differentiation and the baseline level of CEA were related to therapeutic outcome, but other clinicopathological characteristics were not associated with therapeutic outcome. Unsupervised hierarchical clustered and analyzed was performed on each mutant gene and copy number change gene, and its mutation type and copy number. However, no specific mutations and clustering of mutation types were found in the effective and ineffective groups (Figure 1).
Table 1.
Clinicopathologic characteristics of patients
| Characteristics | Ineffective N = 7 | Effective N = 29 | P |
|---|---|---|---|
|
|
|
||
| N (15) | N (8) | ||
| Median age (range), years | 63 (37-81) | 59 (26-74) | 0.771 |
| Gender | 0.621 | ||
| male | 12 | 5 | |
| female | 3 | 3 | |
| Tumor location | 0.127 | ||
| proximal | 8 | 1 | |
| distal | 6 | 5 | |
| both | 1 | 2 | |
| Differentiation | 0.001 | ||
| Middle or high | 2 | 7 | |
| poor | 13 | 1 | |
| Operative treatment | 0.400 | ||
| yes | 8 | 6 | |
| no | 7 | 2 | |
| Baseline CEA | <0.001 | ||
| >5 | 12 | 0 | |
| ≤5 | 3 | 8 | |
| Metastatic lesion (baseline) | 1.000 | ||
| yes | 11 | 6 | |
| no | 4 | 2 | |
| Hematogenous metastasis | 0.089 | ||
| yes | 10 | 2 | |
| no | 5 | 6 | |
| Peritoneal metastasis | 0.657 | ||
| yes | 10 | 4 | |
| no | 5 | 4 | |
| Baseline VAF (%) | 0.657 | ||
| ≤10 | 9 | 6 | |
| >10 | 6 | 2 | |
| Total VAF (%) | 0.345 | ||
| ≤30 | 9 | 7 | |
| >30 | 6 | 1 |
CEA = carcinoembryonic antigen; VAF = Variant allele frequency.
Figure 1.
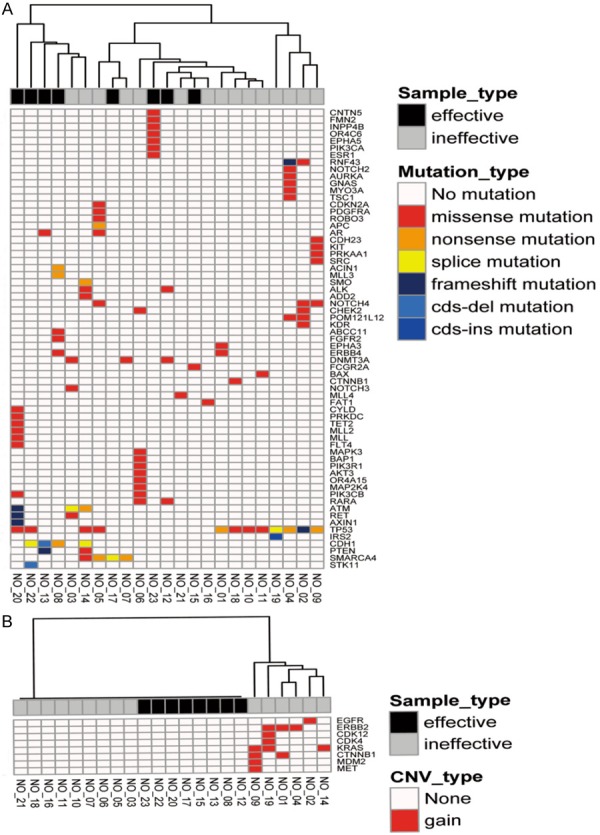
Heatmap of SNV and CNV. A. SNV detection heatmap of ctDNA genome in 23 patients with gastric cancer; B. CNV detection heatmap of ctDNA genome in 23 patients with gastric cancer.
General overview of ctDNA genomic analysis of 23 gastric cancer patients
To investigate whether genomic analysis of ctDNA of gastric cancer patients is feasible to be used to monitor advanced gastric cancer by NGS and predict prognosis, plasma samples from the 23 gastric cancer patients were subjected to DNA extraction at three different time points and NGS of all coding exons and selected introns of 1,021 cancer genes with a target region of about 1.1 Mb was performed at an average sequencing depth of 1094X (67X-2697X). DNA of paired peripheral blood lymphocytes of the same patient were sequenced as the germline control. On average, 4.57 mutations (range 1-11) were found in each sample. Among these mutant genes, Tumor Protein P53 (TP53) and Cadherin 1 (CDH1) were the most common mutant genes in this cohort of patients, being found in 57% (13/23) and 17% (4/23) of patients, respectively (Figure 2A), while 13% (3/23) of patients showed amplification of the Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2) and Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS) genes. Among several types of mutation, missense mutations were the most common type of mutation, accounting for up to 74% (76/103) of mutations, followed by 12% (12/103) of nonsense mutations, 6% (6/103) of splicing mutations, 6% (6/103) of frame shift mutations, 2% (2/103) of cds-del mutations o and 1% (1/103) of cds-ins mutations (Figure 2B).
Figure 2.
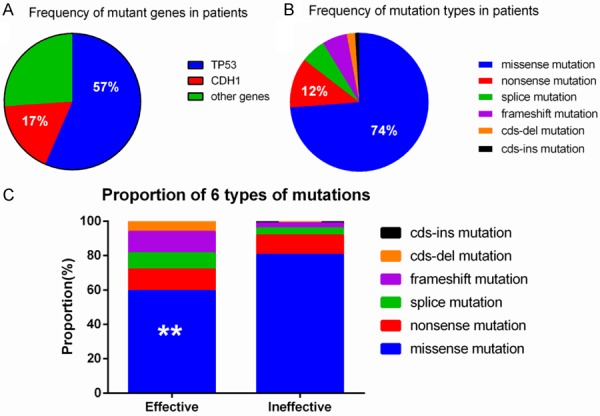
Distribution of mutant genes and mutation types. A. Proportion of mutant genes in 23 patients; B. Proportion of mutation types in 23 patients; C. Comparison of gene types in different treatment groups.
Genomic profiling of ctDNA of 23 gastric cancer patients at three different time points
Analysis of SNVs and CNVs in the effective and ineffective groups in the first assessment
All 23 patients received 2 assessments respectively, of which 37 SNVs and 7 CNVs were detected in the first assessment. As seen in Tables 2 and 3, the frequency of the most common mutant gene, TP53, showed significant differences between the effective and the ineffective group (P = 0.040), with the frequency of gene TP53 mutations being higher in the ineffective group. There were no statistical differences of the remaining 36 SNVs and 7 CNVs between the two groups.
Table 2.
Genes harboring SNVs in patients with effective and ineffective treatment
| Mutated Genes | Effective N = 13 | Ineffective N = 10 | P |
|---|---|---|---|
|
|
|
||
| n (%) | n (%) | ||
| TP53 | 3 | 7 | 0.040 |
| EPHA3 | 0 | 1 | 0.435 |
| CTNNB1 | 0 | 2 | 0.178 |
| ERBB4 | 0 | 1 | 0.435 |
| NOTCH4 | 0 | 1 | 0.435 |
| ATM | 3 | 0 | 0.229 |
| DNMT3A | 3 | 0 | 0.229 |
| SMARCA4 | 1 | 0 | 1.000 |
| PIK3CB | 1 | 1 | 1.000 |
| RARA | 1 | 0 | 1.000 |
| ACIN1 | 0 | 1 | 0.435 |
| MLL3 | 0 | 1 | 0.435 |
| ABCC11 | 0 | 1 | 0.435 |
| CDH1 | 3 | 1 | 0.604 |
| FGFR2 | 0 | 2 | 0.178 |
| ALK | 1 | 0 | 1.000 |
| ADD2 | 1 | 0 | 1.000 |
| PTEN | 1 | 0 | 1.000 |
| SMO | 1 | 0 | 1.000 |
| STK11 | 1 | 0 | 1.000 |
| IRS2 | 0 | 1 | 0.435 |
| MLL | 0 | 1 | 0.435 |
| FLT4 | 0 | 1 | 0.435 |
| FCGR2A | 0 | 1 | 0.435 |
| AR | 1 | 0 | 1.000 |
| FAT1 | 0 | 1 | 0.435 |
| PRKAA1 | 0 | 1 | 0.435 |
| SRC | 0 | 1 | 0.435 |
| KIT | 0 | 1 | 0.435 |
| CDH23 | 0 | 1 | 0.435 |
| PIK3CA | 0 | 1 | 0.435 |
| ESR1 | 0 | 1 | 0.435 |
| EPHA5 | 0 | 1 | 0.435 |
| OR4C6 | 0 | 1 | 0.435 |
| INPP4B | 0 | 1 | 0.435 |
| FMN2 | 0 | 1 | 0.435 |
| CNTN5 | 0 | 1 | 0.435 |
Table 3.
Genes harboring CNVs in patients with effective and ineffective treatment
| CNV Gene | Effective N = 13 | Ineffective N = 10 | P |
|---|---|---|---|
|
|
|
||
| n (%) | n (%) | ||
| ERBB2 | 1 | 2 | 0.560 |
| EGFR | 1 | 0 | 1.000 |
| KRAS | 1 | 2 | 0.560 |
| CDK12 | 0 | 1 | 0.435 |
| CDK4 | 0 | 1 | 0.435 |
| MDM2 | 0 | 1 | 0.435 |
| MET | 0 | 1 | 0.435 |
Analysis of the types of gene mutations in the effective and ineffective groups
The six most common types of mutations found were: missense mutations, nonsense mutation, splice mutation, frame shift mutation, cds-del, and cds-ins mutations. The proportions of different mutation types in the effective and the ineffective group were clearly shown in Figure 2C. The highest proportion of mutations in both groups were missense mutations, and there was a significant difference in its frequency between the two groups (P = 0.026). The next highest proportion was of nonsense mutations, followed in order by splice mutations, frame shift mutations, cds-del, and cds-ins mutations. There were no statistically significant differences in the frequency of the other types of mutations between the two groups.
Analysis of the number of individual mutations and copy number levels of the effective and the ineffective groups
We found that the number of gene mutations and the change in copy number levels were related to therapeutic effect. As shown in Figure 3A and 3B, in the effective group, the number of gene mutations and the copy number level decreased from that of the baseline time point. While Figure 3C and 3D show that in the ineffective group, the number of gene mutations increased and the copy number level increased simultaneously from that of the baseline time point. There was a significant difference in the number of gene mutations (P = 0.0006) among the patients of the ineffective group.
Figure 3.
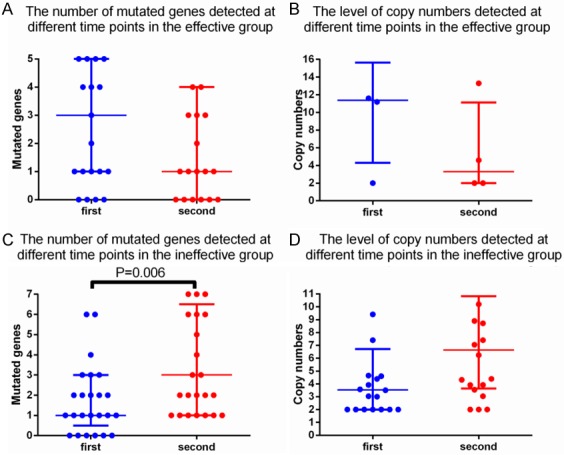
Changes in gene mutations and copy number changes in different therapeutic effects groups. A. Changes in the number of mutant genes at different detection points in the effective group; B. Changes in the level of copy number at different detection points in the effective group; C. Changes in the number of mutant genes at different detection points in the ineffective group; D. Changes in the level of copy number at different detection points in the ineffective group.
Analysis of the change of SNVs and CNVs and the therapeutic effect
We further divided the increase and decrease of the number of gene mutations and the change of copy number into an increase group and a decrease group. A chi-squared test was performed between the increase group and the decrease group, the effective group and the ineffective group. It was found that by grouping according to the increase and decrease of the number of gene mutations significant differences were found between the effective and the ineffective group (P = 0.038). No difference was found between the effective and ineffective group by grouping the changes of the copy number level (Tables 4, 5).
Table 4.
Changes in the number of mutated genes detected in the two groups
| Mutated genes change | Effective | Ineffective | P |
|---|---|---|---|
| increase | 5 | 14 | 0.038 |
| decrease | 16 | 11 |
Table 5.
Changes in the level of copy numbers detected in the two groups
| Copy number change | Effective | Ineffective | P |
|---|---|---|---|
| increase | 1 | 13 | 0.272 |
| decrease | 3 | 7 |
Correlation analysis between VAF and OS
The measured VAF was divided into a baseline VAF and a total VAF based on the test results of ctDNA at three different time points for each patient. In the current study, a reference to the relevant study [13], with a single test VAF>10% set to high mutation abundance, three tests for total VAF>30% set high mutation abundance.
The median follow-up time of this study was 25 months, no loss of follow-up, and the follow-up rate was 100%. In the baseline VAF analysis, the 1-year and 2-year survival rates of the high tumor burden group were only 50.0% and 37.50%, while the 1-year and 2-year survival rates of the low tumor burden group were 86.67% and 64.20%. The median survival time of the group was 15.5 months and 62 months, respectively. In the total VAF analysis, the survival rates of the high tumor burden group were only 42.86% and 28.57%, while the survival rates of the low tumor burden group were as high as 87.50% and 74.04%. The median survival time was 62 months and 8 months, respectively. Kaplan-Meier survival curves and Logrank test results showed that whether according to baseline VAF (P = 0.046<0.05, HR = 0.38, 95% CI: 0.07-0.91), or according to the total VAF analysis (P = 0.0071<0.01, HR = 0.28, 95% CI: 0.03 to 0.54), the overall survival time of patients with low tumor burden was significantly longer than that of patients with high tumor burden, as shown in Figure 4.
Figure 4.
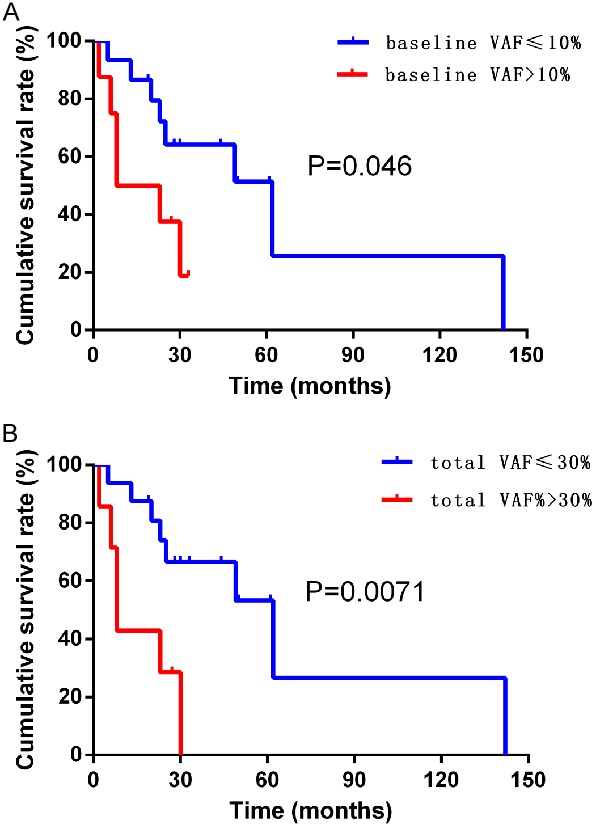
Survival analysis of different VAF. A. Overall survival curve for different baseline VAF; B. Overall survival curve for different total VAF.
Discussion
ctDNA is most suitable in detecting advanced tumors with high tumor burden. In view of the relatively low mutation rate and high heterogeneity of gastric cancer, the detected mutant genes cannot be easily classified, therefore patients with advanced gastric cancer were selected for this study. One patient had no obvious signs of metastasis in the imaging examination, but the patient’s tumor markers were persistently elevated and couldn’t be ruled out as potential progression. It was hoped that meaningful gene mutations will be detected in patients with advanced gastric cancer with high tumor burden.
Firstly, on one hand, as shown in Table 1, we did not find the correlation between VAF and the treatment effect. Tumor differentiation, baseline CEA levels, and two prognostic factors were associated with treatment efficacy in this study with statistical significance. On the other hand, as shown in Figure 1, we clustered genes and gene mutation types of the tumors according to therapeutic effect. No meaningful clustering results were found.
Secondly, we analyzed the specific genes and therapeutic effect and found that the TP53 gene was significantly associated with therapeutic effect in the first evaluation. TP53 is the most common mutated gene in human cancer and at least 50% of human malignant tumors change and play a key role in tumor development [14]. Experimental evidence indicated that TP53 status was associated with tumor response to toxic substances [15-17]. In this study, the TP53 gene was detected as the most frequently occurring gene and the mutated TP53 gene was frequently found in the ineffective treatment group. From this result, it can be inferred that the mutated TP53 gene may make the tumor insensitive to chemotherapeutic drugs, resulting in treatment failure. Meanwhile, type of gene mutation was also compared with the therapeutic effect. It found that the missense mutations accounted for the largest proportion of mutations, mostly in the ineffective treatment group. The difference in this type of mutation was statistically significant among the two groups, indicating that the occurrence of missense mutations was a predictive effect of poor treatment outcome.
Several studies have shown that as tumors progress, peripheral blood ctDNA levels gradually increased and subsequently declined after surgery or effective chemotherapy [4,18-21]. Therefore, it seems logical that ctDNA levels are used as surrogate markers of therapeutic response. In this study, we found that the changes in number of gene mutations and copy number levels have a fixed trend in different therapeutic effects. When the treatment was effective, the frequency of most gene mutations decreased, along with a decrease in copy numbers. On the contrary, when the treatment was ineffective, the frequency of most gene mutations increased, along with an increase in copy numbers. Some studies have shown that ctDNA usually provides the earliest measurement of therapeutic response for the detection of sensitive solid tumors [5,22]. Lipson et al. reported that changes in ctDNA levels were associated with the therapeutic outcome of three melanoma patients treated with nivolumab [23]. The conclusions of these studies were consistent with our results. Especially in the ineffective group; the number of gene mutations after treatment was significantly higher than that before treatment, which was statistically significant. According to the results detected in the effective group, the frequency of most gene mutations showed a decreasing trend. If the sample size is increased, we are likely to discover more information.
Thirdly, we analyzed the increase and decrease in the number of gene mutations and the change in the level of copy numbers, and found that the increase and decrease in the number of gene mutations was related to the therapeutic outcome. When the number of mutant genes increased, the treatment was effective, and the number of mutant genes decreased, indicating that the treatment effect was poor. In theory, the half-life of ctDNA is about 2 hours [2], and the protein lasts for weeks to months in the blood [24,25]. Therefore, the analysis of ctDNA should be able to quickly assess changes in the tumor within hours, rather than weeks to months [26]. In a 17/19 metastatic breast cancer study, the increase of ctDNA was associated with disease progression and progression was reported through ctDNA levels 5 months earlier than imaging studies were able to detect [4].
Finally, we performed a correlation analysis between the tested VAF and the OS. In the study, it was found that patients with high mutation abundance of gastric cancer had a shorter overall survival than patients with low mutation abundance, regardless of whether they were analyzed from baseline VAF or total VAF grouping. The difference between the two groups was statistically significant (Figure 4).
There are still some limitations of this study. First, as with most retrospective studies, this study was biased in the selection of cases. For example, the balance of the number of different groupings and the diversity of treatments can be biased. Then, the relatively small number of cases was another important limiting factor of our study, which may affect our analysis.
Conclusions
TP53 was a common mutant gene and its mutation frequency and type of mutation were related to the outcome of advanced gastric cancer treatment. The frequency of mutations of the gene was related to therapeutic outcome, especially in the ineffective group of this study. Changes in the number of gene mutations were negatively correlated with treatment outcomes. The mutation abundance level of gastric cancer was also negatively correlated with the overall survival of patients. ctDNA can be used as an effective tool to monitor the efficacy of chemotherapy and predict prognosis in advanced gastric cancer.
Acknowledgements
This work was supported by the Joint Project of the Natural Science and Health Foundation of Fujian Province, China (grant number 2015J01397).
This retrospective study was approved by the Ethical Committees of Fujian Medical University Union Hospital. Informed consent was signed for all enrolled patients.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Center MM, DeSantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19:1893–1907. doi: 10.1158/1055-9965.EPI-10-0437. [DOI] [PubMed] [Google Scholar]
- 2.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, Kinzler KW, Vogelstein B, Diaz LA Jr. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang JY, Hsieh JS, Chang MY, Huang TJ, Chen FM, Cheng TL, Alexandersen K, Huang YS, Tzou WS, Lin SR. Molecular detection of APC, K- ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J Surg. 2004;28:721–726. doi: 10.1007/s00268-004-7366-8. [DOI] [PubMed] [Google Scholar]
- 4.Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, Halsall D, Wallis M, Bentley D, Caldas C, Rosenfeld N. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199–1209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 5.Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, Kinzler KW, Oliner KS, Vogelstein B. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486:537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorensen BS, Wu L, Wei W, Tsai J, Weber B, Nexo E, Meldgaard P. Monitoring of epidermal growth factor receptor tyrosine kinase inhibitor-sensitizing and resistance mutations in the plasma DNA of patients with advanced non-small cell lung cancer during treatment with erlotinib. Cancer. 2014;120:3896–3901. doi: 10.1002/cncr.28964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–376. doi: 10.1007/s10555-016-9629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li J, Lupat R, Amarasinghe KC, Thompson ER, Doyle MA, Ryland GL, Tothill RW, Halgamuge SK, Campbell IG, Gorringe KL. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–1313. doi: 10.1093/bioinformatics/bts146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, Wilson RK, Ding L, Mardis ER. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–681. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, Anagnostou V, Fiksel J, Cristiano S, Papp E, Speir S, Reinert T, Orntoft MW, Woodward BD, Murphy D, Parpart-Li S, Riley D, Nesselbush M, Sengamalay N, Georgiadis A, Li QK, Madsen MR, Mortensen FV, Huiskens J, Punt C, van Grieken N, Fijneman R, Meijer G, Husain H, Scharpf RB, Diaz LA Jr, Jones S, Angiuoli S, Ørntoft T, Nielsen HJ, Andersen CL, Velculescu VE. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9 doi: 10.1126/scitranslmed.aan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tewari M, Krishnamurthy A, Shukla HS. Predictive markers of response to neoadjuvant chemotherapy in breast cancer. Surg Oncol. 2008;17:301–311. doi: 10.1016/j.suronc.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T. p53 status and the efficacy of cancer therapy in vivo. Science. 1994;266:807–810. doi: 10.1126/science.7973635. [DOI] [PubMed] [Google Scholar]
- 16.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 17.Weller M. Predicting response to cancer chemotherapy: the role of p53. Cell Tissue Res. 1998;292:435–445. doi: 10.1007/s004410051072. [DOI] [PubMed] [Google Scholar]
- 18.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat Methods. 2006;3:551–559. doi: 10.1038/nmeth898. [DOI] [PubMed] [Google Scholar]
- 19.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, Dawson SJ, Piskorz AM, Jimenez-Linan M, Bentley D, Hadfield J, May AP, Caldas C, Brenton JD, Rosenfeld N. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra168. doi: 10.1126/scitranslmed.3003726. [DOI] [PubMed] [Google Scholar]
- 20.Shinozaki M, O’Day SJ, Kitago M, Amersi F, Kuo C, Kim J, Wang HJ, Hoon DS. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin Cancer Res. 2007;13:2068–2074. doi: 10.1158/1078-0432.CCR-06-2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bidard FC, Madic J, Mariani P, Piperno-Neumann S, Rampanou A, Servois V, Cassoux N, Desjardins L, Milder M, Vaucher I, Pierga JY, Lebofsky R, Stern MH, Lantz O. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int J Cancer. 2014;134:1207–1213. doi: 10.1002/ijc.28436. [DOI] [PubMed] [Google Scholar]
- 22.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipson EJ, Velculescu VE, Pritchard TS, Sausen M, Pardoll DM, Topalian SL, Diaz LA Jr. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother Cancer. 2014;2:42. doi: 10.1186/s40425-014-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riedinger JM, Wafflart J, Ricolleau G, Eche N, Larbre H, Basuyau JP, Dalifard I, Hacene K, Pichon MF. CA 125 half-life and CA 125 nadir during induction chemotherapy are independent predictors of epithelial ovarian cancer outcome: results of a French multicentric study. Ann Oncol. 2006;17:1234–1238. doi: 10.1093/annonc/mdl120. [DOI] [PubMed] [Google Scholar]
- 25.Ito K, Hibi K, Ando H, Hidemura K, Yamazaki T, Akiyama S, Nakao A. Usefulness of analytical CEA doubling time and half-life time for overlooked synchronous metastases in colorectal carcinoma. Jpn J Clin Oncol. 2002;32:54–58. doi: 10.1093/jjco/hyf011. [DOI] [PubMed] [Google Scholar]
- 26.Kidess E, Heirich K, Wiggin M, Vysotskaia V, Visser BC, Marziali A, Wiedenmann B, Norton JA, Lee M, Jeffrey SS, Poultsides GA. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget. 2015;6:2549–2561. doi: 10.18632/oncotarget.3041. [DOI] [PMC free article] [PubMed] [Google Scholar]