

Abstract
Dopaminergic functions are important for various biological activities, and their impairment leads to neurodegeneration, a hallmark of Parkinson's disease (PD). Chronic manganese (Mn) exposure causes the neurological disorder manganism, presenting symptoms similar to those of PD. Emerging evidence has linked the transcription factor RE1-silencing transcription factor (REST) to PD and also Alzheimer's disease. But REST's role in dopaminergic neurons is unclear. Here, we investigated whether REST protects dopaminergic neurons against Mn-induced toxicity and enhances expression of the dopamine-synthesizing enzyme tyrosine hydroxylase (TH). We report that REST binds to RE1 consensus sites in the TH gene promoter, stimulates TH transcription, and increases TH mRNA and protein levels in dopaminergic cells. REST binding to the TH promoter recruited the epigenetic modifier cAMP-response element-binding protein–binding protein/p300 and thereby up-regulated TH expression. REST relieved Mn-induced repression of TH promoter activity, mRNA, and protein levels and also reduced Mn-induced oxidative stress, inflammation, and apoptosis in dopaminergic neurons. REST reduced Mn-induced proinflammatory cytokines, including tumor necrosis factor α, interleukin 1β (IL-1β), IL-6, and interferon γ. Moreover, REST inhibited the Mn-induced proapoptotic proteins Bcl-2–associated X protein (Bax) and death-associated protein 6 (Daxx) and attenuated an Mn-induced decrease in the antiapoptotic proteins Bcl-2 and Bcl-xL. REST also enhanced the expression of antioxidant proteins, including catalase, NF-E2–related factor 2 (Nrf2), and heme oxygenase 1 (HO-1). Our findings indicate that REST activates TH expression and thereby protects neurons against Mn-induced toxicity and neurological disorders associated with dopaminergic neurodegeneration.
Keywords: manganese, Parkinson disease, neurodegeneration, neurodegenerative disease, neurotoxin, dopaminergic neurons, neuron restrictive silencer factor (NRSF), neurotoxicity, RE1-silencing transcription factor (REST), Tyrosine Hydroxylase
Introduction
Dopaminergic neurons in the mammalian central nervous system (CNS)2 (1) regulate a variety of brain functions, including voluntary movement and a broad array of behavioral processes, such as mood, reward, addiction, and stress (2). The loss of dopaminergic neurons occurs naturally within the aging process, but their widespread loss is associated with neurological disorders, such as Parkinson's disease (PD) and manganism (3). Although some studies have reported that mitochondrial dysfunction, oxidative stress, neuroinflammation, transcriptional repression, and dysregulation of the dopamine synthesis enzyme tyrosine hydroxylase (TH) are involved in dopaminergic neuronal death, the mechanisms are not completely understood. TH is the rate-limiting enzyme in dopamine synthesis that is highly expressed in the substantia nigra pars compacta (SNpc), ventral tegmental area, hypothalamus, and brainstem (4–9). Numerous studies (10–12) suggest that chronic oxidative stress, inflammation, and the resultant apoptosis are also linked to various neurodegenerative disorders, including Alzheimer's disease (AD) (13), multiple sclerosis (14), amyotrophic lateral sclerosis (15), and PD (16).
Manganism is a condition caused by chronic exposure to manganese (Mn) that preferentially accumulates in the basal ganglia, leading to a variety of behavioral and motor disturbances with symptomatic resemblance to PD (17–19). At the molecular level, Mn induces oxidative stress that leads to damage of DNA, lipids, and proteins and promotes cellular injury (10, 11). Mn also induces inflammation characterized by high levels of proinflammatory cytokines in microglia and astrocytes, promoting neurodegeneration and blood–brain barrier degradation (20, 21). Mn-induced oxidative stress and inflammation induce apoptosis both in in vitro and in vivo settings (9, 16, 20, 21). Mn dysregulates proapoptotic/antiapoptotic molecules to signal a cascade of events leading to apoptosis in neuronal cell cultures (20, 22).
Molecular mechanisms of Mn-induced neuronal cell death have been extensively studied. Mn impaired TH activity and function by decreasing TH mRNA and protein levels both in vitro and in vivo (8, 9, 23–25). Mn disrupted TH activity via protein kinase C-δ and protein phosphatase 2A in dopaminergic neural cell lines (26). Because TH is a rate-limiting enzyme that plays a critical role in the production of dopamine (27), it is important to understand the regulatory mechanisms of TH expression for dopaminergic neuronal function and survival. Several transcription factors have been reported to regulate TH expression, including cAMP response element-binding protein (CREB) (28), nuclear receptor–related-1 protein (Nurr1) (29), pituitary homeobox 3 (Pitx3) (30), activating enhancer-binding protein 2 (AP-2) (31), hypoxia-inducible factor-1α (HIF-1α) (32), forkhead box O1 (FOXO1), and neuron-restrictive silencing factor/repressor element 1-silencing transcription factor (NRSF/REST) (33).
Recent studies have demonstrated that REST is increased in normal aging brains, preserving neuronal function and protecting against neurodegeneration through the repression of stress- and apoptosis-promoting genes (13). REST is an essential mammalian zinc finger transcriptional regulator (34) playing a variety of cellular functions, such as neurogenesis, differentiation, axonal growth, vesicular transport, and release, as well as ionic conductance (35–38). REST represses neuronal genes in non-neural cells by binding to a DNA sequence motif known as repressor element 1 (RE1; also known as NRSE) (36, 39, 40). REST forms a complex with co-repressors and epigenetic modifiers, including histone deacetylases (HDAC), mSin3a, and CoREST, to repress expression of target genes (41–43). However, accumulating evidence suggests that REST is highly complex and can also increase gene expression by acting as an activator of target genes. For example, REST splice variants, REST4/5 containing the N-terminal domain of REST (44, 45), have been shown to induce expression of glutamate synthetase, which is mediated by the transcription co-activator hBrm via the glucocorticoid receptor (46).
The loss of REST is associated with the pathogenesis of AD (13) and PD (47). Lewy bodies of PD brains contain REST protein aggregates, suggesting that abnormal protein trafficking and degradation may be related to the dysfunction of REST. Deletion of neuronal REST in mice treated with the PD neurotoxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) showed greater motor deficits and loss of dopaminergic neurons compared with the MPTP-treated group (8). These findings suggest that REST is protective in neurons against PD, warranting further studies of the protective mechanisms of REST in dopaminergic/catecholaminergic neurons.
The dual roles of REST as a repressor or as an activator in the regulation of gene expression are not completely understood. Studies have shown that variations within the RE1-binding sequence motifs modify REST–DNA-binding affinities that likely contribute to context-, tissue-, or cell-specific regulation of gene expression (48). RE1-binding sequence motifs can be classified into canonical/common, restricted, and unique motifs that facilitate varying binding affinity for REST. REST enhanced the expression of various neuronal genes by binding to the RE1 site in their promoters, including glutamate receptor 2 (GluR2), dynamin I, and N-methyl-d-aspartate (NMDA) receptor (49–51). Interestingly, REST can also bind to the RE1 motif in small noncoding dsRNAs to promote differentiation of adult hippocampal stem cells to mature neurons (52). These findings suggest that REST may regulate transcriptional activation or repression of neural genes depending on the cellular and microenvironmental context.
Although REST represses neural gene expression in non-neuronal cells, studies have demonstrated that it may act a positive transcriptional regulator of some genes, including B-cell lymphoma (Bcl)-2 (53). Thus, REST may serve as a critical mediator in protection against Mn toxicity through the up-regulation of TH expression and protecting against Mn-induced oxidative stress, inflammation, and apoptosis in dopaminergic neurons. In this study, we investigated the mechanism of REST in TH expression at the transcriptional level in dopaminergic neurons and tested whether REST is protective against Mn-induced oxidative stress and apoptosis. Our results demonstrated that full-length NRSF/REST increased TH gene expression by binding to an RE1-binding sequence motif that was newly identified in the human TH promoter region, co-interacting with an epigenetic modifier, CREB-binding protein (CBP)/p300. Overexpression of REST also attenuated Mn-induced neurotoxicity by regulating genes related to oxidative stress, inflammation, and apoptosis, including up-regulation of catalase and Bcl-2. Based on these results, REST protects against Mn neurotoxicity through the up-regulation of TH and attenuation of Mn-induced oxidative stress, inflammation, and apoptosis in dopaminergic neurons.
Results
REST positively regulates TH expression at the transcriptional level in dopaminergic neurons
Although REST functions as a repressor of neuronal genes in non-neural cells (36), studies have demonstrated that REST also acts as a positive transcriptional regulator of some genes, including Bcl-2 (53). Recently, loss of REST function has been implicated in the brains of PD and AD patients (13, 47), implicating that REST might play a critical role in the protection of aging brains. In this study, we have tested whether REST is protective against Mn-induced toxicity, and if it does, we aimed to understand its protective mechanisms of REST, including modulation of TH, the rate-limiting enzyme in dopamine synthesis. We used cath.-a-differentiated (CAD) neuronal cells as the experimental model. These cells are a variant of a CNS catecholaminergic cell line that expresses TH (54). REST overexpression with full-length human REST plasmid vectors in CAD cells resulted in a significant increase in the number of TH-positive cells compared with the control (Fig. 1A). Additionally, REST-expressing CAD neurons have higher TH levels (Fig. 1B), indicating that REST increases TH expression in CAD neurons.
Figure 1.

REST positively regulates TH expression levels at the transcriptional level. A, after REST transfection, neuronal cells were stained with anti-TH and anti-REST followed by flow cytometry as described under “Experimental procedures.” Cells stained with both TH and REST were also quantified. B, immunostaining images showing TH and REST expression in dopaminergic neurons expressing REST. C, LUHMES and CAD neurons were co-transfected with the human TH promoter vector and REST, followed by luciferase assay as described under “Experimental procedures.” D and E, after REST transfection, LUHMES and CAD neurons were extracted for total RNA and total protein, followed by qPCR (D) and Western blotting (E), respectively, as described under “Experimental procedures.” F, CAD neurons were co-transfected with human TH promoter and REST or dominant-negative REST (DN-REST), followed by luciferase assay. G, immunostaining images showing TH and REST expression in dopaminergic neurons expressing REST or DN-REST. H, after the transfection of REST or DN-REST, CAD neurons were extracted for total protein, followed by Western blotting. GAPDH and β-actin were used as loading controls of mRNA and protein, respectively. **, p < 0.01; ***, p < 0.001; #, p < 0.05; ##, p < 0.01 compared with EV; @@@, p < 0.001 compared to each other (Student's t test or one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
As REST regulates gene expression at the transcriptional level, we tested whether REST enhanced TH promoter activity along with its mRNA and protein levels. We used CAD neurons as well as human dopaminergic Lund human mesencephalic (LUHMES) cells. REST overexpression significantly increased TH promoter activity, mRNA, and protein levels in both LUHMES and CAD neurons (Fig. 1, C–E), indicating that REST up-regulates TH expression at the transcriptional level. Next, we tested whether inhibiting REST function can decrease TH expression by overexpressing a dominant-negative form (DN-REST), which expresses only the DNA-binding domain of REST (39) and thus can compete and block the endogenous REST function. The results showed that DN-REST decreased TH promoter activities (Fig. 1F), nuclear localization of REST (Fig. 1G), and TH protein levels in whole-cell lysates of CAD neurons (Fig. 1H).
REST binds to an RE1 cis-regulatory element in the TH promoter region
Because REST increased TH expression at the transcriptional level in CAD neurons, we further investigated whether REST binds to an RE1 cis-regulatory element on the TH promoter. We performed both a literature and database search for the RE1 consensus sequence within the TH promoter. We analyzed the 673-bp sequence of the human TH promoter region, using the PROMO and ENCODE database. A putative 21-bp RE1 consensus sequence is located −288 upstream of the TH transcription initiation site (Fig. 2A) in the JASPAR 2018 transcription factor database (55). To determine whether REST binds to this newly-identified binding site, we performed the in vivo chromatin immunoprecipitation (ChIP) assay. REST bound to this site of the TH promoter and increased the binding activity with REST overexpression in CAD neurons (Fig. 2B). Empty vector (EV) transfection showed some REST-binding activity to the TH promoter due to endogenous REST expression. We also employed EMSA and DAPA to determine whether REST forms a DNA–protein complex with the REST-binding site oligonucleotides in vitro. As shown in Fig. 2, C and D, the REST protein binds to its consensus site of the TH promoter. The interaction of the RE1 site with REST protein was specific for REST, as an excess of unlabeled RE1-specific oligonucleotides blocked the formation of this DNA–protein complex (Fig. 2C). The results from the DAPA also show that oligonucleotides interacted with REST protein at ∼150 kDa, indicating that the full-length REST bound to its consensus motifs (Fig. 2D).
Figure 2.

RE1 consensus site is critical for basal TH promoter activity and REST activation. A, novel RE1 consensus site (−288) in the human TH promoter was identified. B, after co-transfection of REST expression vectors and TH promoter vectors, ChIP assay was performed to determine REST binding to its consensus site in the TH promoter in vivo, followed by quantification of REST-bound DNA by real-time qPCR and agarose gel electrophoresis as described under “Experimental procedures.” C, EMSA was performed in nuclear extracts prepared from neurons transfected with REST as described under “Experimental procedures.” The red arrow shows the DNA–protein complex, and the blue arrow shows unbound oligos. D, DAPA was performed with nuclear extracts prepared from neurons transfected with REST, and the RE1 consensus sequence-bound protein was subjected to Western blotting to probe REST. As an input control, 10 μg of nuclear extract was used. E, two site-mutations of the RE1 consensus sequences are indicated in red. Neurons were transfected overnight with WT and RE1 mutants of TH promoter plasmids, followed by luciferase assay. F, after co-transfection of WT or mutant TH promoter and REST expression vectors, promoter activity was determined by luciferase assay. ***, p < 0.001 compared with EV or EV/WT; ###, p < 0.001 compared with WT (E) or EV/WT (F); @@@, p < 0.001 compared to each other (Student's t test or one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
Next, we performed mutagenesis on REST-binding sequences of the TH promoter to confirm whether this site is critical for REST-induced TH promoter activity. Mutation of the REST-binding sites completely abrogated TH promoter activity (Fig. 2E), and REST overexpression was unable to increase TH promoter activity (Fig. 2F).
REST interacts with CBP/p300 and the transcription factor CREB for TH expression
REST may recruit epigenetic modifiers to regulate its target gene expression (41, 56). To understand epigenetic modification of REST-enhanced TH transcription in CAD neurons, we tested whether epigenetic modifiers, such as histone acetyltransferase (HAT) and HDAC, as well as other transcription factors, modulated REST-induced TH up-regulation (Fig. 3, A–C). Co-transfection of REST with CBP further enhanced TH promoter activity in CAD neurons (Fig. 3, A and B). REST physically interacted with CBP in the nuclear fraction of CAD neurons by co-IP (Fig. 3D) and co-localized in the nucleus (Fig. 3E), indicating that REST interacts with CBP to activate TH promoter activity (Fig. 3A). We also found that REST interacted with the transcription factor CREB, which is known to increase TH (28). Further data reveal that p-CREB, an active form of CREB, interacts with REST in the nuclear region of CAD neurons (Fig. 3, D and F), indicating that REST, CREB, and CBP may form a complex to increase TH promoter activity. On the other hand, the epigenetic modifier HDAC, which is known to interact with REST to repress gene expression (41), abolished REST-induced TH promoter activity when HDAC1 and REST were co-expressed in CAD neurons (Fig. 3C). These findings indicate that basal cellular conditions with high levels of REST enhance TH expression, but activation of the HDAC epigenetic pathway would lead to REST-induced repression of TH.
Figure 3.

REST interacts with CBP and CREB in dopaminergic cells. A–C, CAD neurons were co-transfected overnight with the TH promoter vector and REST with CBP (A), CREB (B), or HDAC1 (C), followed by luciferase assay. D, CAD neurons transfected with various amounts of REST, followed by preparation of nuclear extract and co-IP for CBP, p-CREB, CREB, and REST as described under “Experimental procedures.” E and F, immunocytochemistry image showing REST, CBP, and CREB expression in CAD neurons. **, p < 0.01; ***, p < 0.001; #, p < 0.05 compared with EV; @@@, p < 0.001 compared to each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
REST attenuates Mn-induced repression of TH in CAD neuronal cells
As Mn decreases TH mRNA and protein levels in dopaminergic neurons of the SNpc of the mouse brain (9) as well as in TH-expressing cells (24), and REST increases TH expression in in vitro TH-expressing neuronal cultures, we next examined whether REST attenuates Mn-decreased TH expression in CAD neurons. Mn reduced promoter activities of both REST and TH in CAD neurons in a concentration-dependent manner (Fig. 4A). Mn also decreased both REST and TH mRNA (Fig. 4B) by qPCR and protein levels and quantification by flow cytometry (Fig. 4, C and D). Because REST binds to the TH promoter to increase TH, we hypothesized that REST may mediate Mn-reduced TH, and restoring REST may attenuate Mn-reduced TH expression. To test this, REST-overexpressing CAD neurons were treated with Mn, followed by an assessment of TH promoter activity, mRNA, and protein levels. REST overexpression attenuated Mn-reduced TH promoter activity, mRNA and protein levels (Fig. 4, E–H). Next, we tested whether DN-REST exacerbates Mn-induced reduction of TH levels. The results showed that inhibiting REST with DN-REST further decreased Mn-reduced TH mRNA and protein levels in CAD neurons (Fig. 4, I–K).
Figure 4.

REST attenuates Mn-induced TH repression. A, after transfection of REST or TH promoter vectors, cells were exposed to different amounts of Mn for 6 h, followed by cell lysis and luciferase assay to determine promoter activity, as described under “Experimental procedures.” B, cells were exposed to Mn (250 μm) for 6 h, followed by qPCR to determine mRNA levels of REST and TH. C and D, CAD neurons were treated with Mn (250 μm) for 12 h, followed by immunostaining with antibodies against TH and REST and flow cytometry analysis (C) and quantification (D) as described under “Experimental procedures.” E and F, after transfection of plasmid vectors, cells were exposed to Mn (250 μm), followed by the assessment of promoter activity (E), mRNA (F), protein (G) levels, and quantification (H) of TH and REST. I–K, after transfection of REST or DN-REST, cells were exposed to Mn (250 μm), followed by the assessment of mRNA (I), protein (J) levels, and protein quantification (K) of TH. GAPDH and β-actin were used as loading control of mRNA and protein, respectively. **, p < 0.01; ***, p < 0.001; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 compared with EV/Con; @, p < 0.05; @@, p < 0.01; @@@, p < 0.001 compared to each other (Student's t test or one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
REST protects CAD neurons against Mn-induced and oxidative stress
Studies have reported that REST protects aging neurons from oxidative stress and mitochondrial damage in AD by regulating the expression of multiple genes involved in oxidative stress and apoptosis (13). To determine whether REST protects CAD neurons from Mn-induced oxidative stress, we performed reactive oxygen species (ROS) and lipid peroxidation assays in Mn-treated REST-overexpressing CAD neurons. Overexpression of REST attenuated Mn-induced ROS (Fig. 5A) as well as malondialdehyde (MDA), a marker of lipid peroxidation resulting from oxidative stress (Fig. 5B).
Figure 5.

REST enhances the antioxidant defense system and protects dopaminergic cells against Mn-induced oxidative stress. CAD cells were transfected with the REST expression vector prior to experiments. A, cells were exposed to different amounts of Mn for 3 h; ROS was measured by a fluorometer using CM-H2DCFDA– as described under “Experimental procedures.” B and C, cells were exposed to Mn (250 μm) for 3 h, and MDA levels (B) and catalase activity (C) were measured as described under “Experimental procedures.” D–H, cells were exposed to Mn (250 μm), followed by measurement of catalase mRNA (D) and protein (E), Nrf2 and ubiquitinated (Ub)-Nrf2 protein (F), quantification (G), and HO-1 (H) by qPCR or Western blot analysis as described under “Experimental procedures.” GAPDH and β-actin were used as loading controls of mRNA and protein, respectively. *, p < 0.05; **, p < 0.01; ***, p < 0.001; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 compared with EV/Con; @, p < 0.05; @@, p < 0.01; @@@, p < 0.001 compared to each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
Since REST is also known to up-regulate antioxidant genes such as catalase (13), an antioxidant enzyme that catalyzes the breakdown of hydrogen peroxide (H2O2), we tested whether catalase is involved in REST protection against Mn toxicity. REST increased and attenuated Mn-induced reduction of catalase activity, mRNA, and protein levels in CAD neurons (Fig. 5, C–E), indicating that REST is an activator of catalase. REST also increased antioxidant protein nuclear factor erythroid 2–related factor 2 (Nrf2), possibly by inhibiting its ubiquitination for its proteasomal degradation (Fig. 5, F and G). Nrf2 increases an array of antioxidant-response element (ARE)-dependent genes (57, 58), including heme oxygenase 1 (HO-1), which is induced by ROS formation (59). REST attenuated Mn-induced repression of Nrf2-ARE by preventing Nrf2 degradation via ubiquitination, resulting in increased HO-1 expression (Fig. 5H).
REST protects CAD neurons against Mn-induced inflammation
Because Mn also promotes the production of inflammatory cytokines, leading to neuroinflammation and cell death, we determined whether REST could attenuate Mn-induced production of proinflammatory genes such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and interferon (IFN)-γ. REST attenuated Mn-produced TNFα protein and mRNA levels (Fig. 6, A and B). REST also inhibited Mn-induced proinflammatory cytokines IL-1β, IL-6, and IFNγ mRNA levels (Fig. 6, C–E).
Figure 6.

REST attenuates Mn-induced proinflammatory cytokine production. A, after REST transfection, cells were exposed to Mn (250 μm) for 12 h, and TNFα secretion was measured by ELISA as described under “Experimental procedures.” B–E, after REST transfection, cells were exposed to Mn (250 μm) for 6 h, and mRNA levels of proinflammatory cytokines TNFα (B), IL-1β (C), IL-6 (D), and IFNγ (E) were measured by qPCR as described under “Experimental procedures.” GAPDH was used as the loading control. **, p < 0.01; ***, p < 0.001 compared with EV/Con; @@, p < 0.01; @@@, p < 0.001 compared to each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
REST is protective against Mn-induced cell death in CAD neurons
Because REST exerts neuroprotection against AD and PD models (13, 47) and has been shown to regulate gene expressions associated with apoptosis for its neuroprotection (13), we tested whether REST could attenuate Mn toxicity by modulating the apoptotic signal. REST reduced Mn-induced apoptosis (Fig. 7A) in both early and late apoptotic stages (as shown by quadrants Q2 and Q3, respectively) (Fig. 7B). REST attenuated Mn-induced cytotoxicity, whereas REST alone did not alter cell viability (Fig. 7C).
Figure 7.
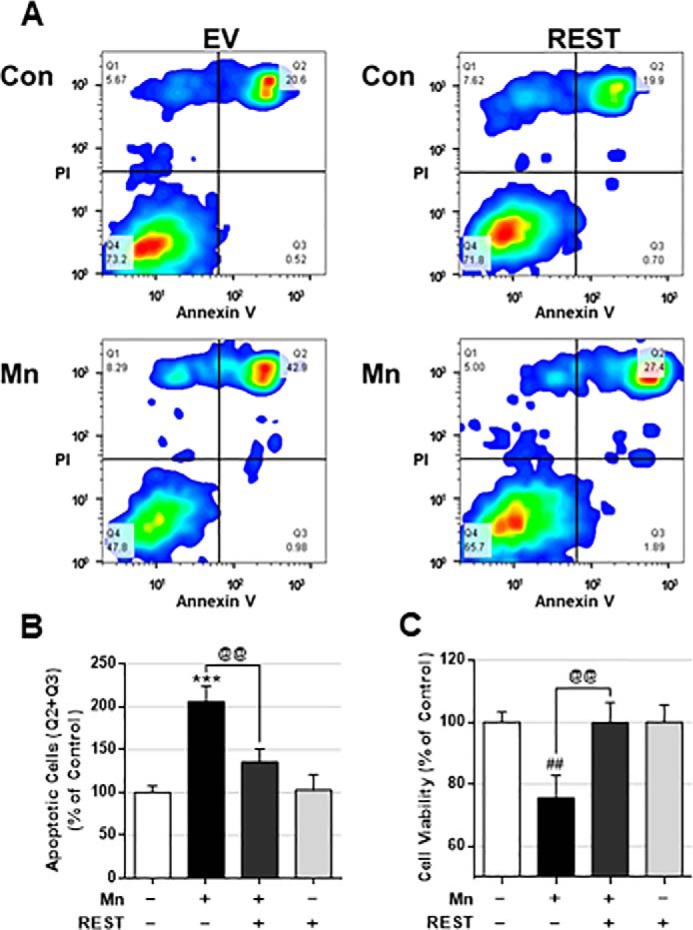
REST protects dopaminergic cells against Mn-induced cell death. A–C, after REST transfection, CAD neurons were exposed to Mn (250 μm) for 12 h, followed by annexin V and PI staining, and flow cytometry to determine apoptosis. B, early and late apoptotic cells (Q2 and Q3) were analyzed. C, after REST transfection, CAD neurons were exposed to Mn (250 μm) for 12 h, followed by MTT assay to determine cell viability as described under “Experimental procedures.” ***, p < 0.001; ##, p < 0.01 compared with EV/Con; @@, p < 0.01 compared to each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
To further dissect the molecular mechanisms by which REST induces protection against Mn toxicity, we tested whether REST modulates proapoptotic/antiapoptotic proteins by assessing proapoptotic (Bcl-2-associated X (Bax) and death domain-associated protein (Daxx)) and antiapoptotic (Bcl-2 and Bcl-xL) protein levels. The results showed that REST reduced Mn-induced proapoptotic Bax and Daxx protein levels (Fig. 8A). Moreover, REST increased antiapoptotic Bcl-2 and Bcl-xL protein levels and attenuated Mn-induced reduction of these antiapoptotic proteins (Fig. 8B), resulting in increased ratio of Bcl-2–to–Bax in REST-overexpressed CAD neurons (Fig. 8C). Because these Mn-induced dysregulations of proapoptotic and antiapoptotic proteins were directly associated with mitochondrial functions, including membrane potential (Δψm), we tested whether REST could attenuate Mn-induced impairment of mitochondrial membrane potential. The results showed that REST overexpression attenuated Mn-induced reduction of mitochondrial membrane potential in CAD neurons (Fig. 8D).
Figure 8.

REST regulates proapoptotic and antiapoptotic proteins involved in cell death and prevents Mn-induced mitochondrial damage. A and B, after REST transfection, cells were exposed to Mn (250 μm, 12 h), followed by detecting protein levels of proapoptotic Bax and Daxx (A), and antiapoptotic Bcl-2 and Bcl-xL (B). C, ratio of Bcl-2 and Bax. β-Actin was used as a loading control. D, after REST transfection, cells were exposed to Mn with varying concentrations for 1 h, and mitochondrial membrane potential (Δψm) was measured by fluorometer using TMRE reagent, as described under “Experimental procedures.” *, p < 0.05; ***, p < 0.001; #, p < 0.05; ##, p < 0.01; ###, p < 0.001 compared with EV/Con; @, p < 0.05; @@, p < 0.01; @@@, p < 0.001 compared to each other (one-way ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
REST protects CAD neurons against the PD neurotoxicant 1-methyl-4-phenylpyridinium (MPP+)-induced TH reduction
Because the PD neurotoxicant MPP+ induces decreases in TH levels (60), we tested whether MPP+ exerts similar effects as those of Mn in modulating REST and TH by assessing their promoter activities. The results showed that MPP+ decreased both REST and TH promoter activities in CAD neurons (Figs. 9, A and B, and 10). Moreover, REST overexpression attenuated MPP+-induced reduction of TH promoter activity in CAD neurons (Fig. 9C), suggesting that MPP+ exerts similar effects as those of Mn.
Figure 9.

Mn and MPP+ inhibit REST and TH in CAD neurons. A and B, neurons transfected with human REST (A) or TH (B) promoter vectors were treated with Mn (250 μm) or 1-methyl-4-phenylpyridinium (MPP+, 250 μm) for 6 h, followed by cell lysis and luciferase assay. C, neurons transfected with human TH promoter and REST expression vector were treated with Mn (250 μm) or MPP+ (250 μm) for 6 h, followed by cell lysis and luciferase assay. **, p < 0.01; ##, p < 0.01; ###, p < 0.001 compared with control or EV; @@, p < 0.01 compared to each other (ANOVA followed by Tukey's post hoc test; n = 3). The data shown are representative of three independent experiments.
Figure 10.

Schematic diagram of the proposed mechanism for REST-induced protection of dopaminergic neuronal function. REST targets TH at the transcriptional level by binding to the RE1 motif proximal to the TH promoter. REST activation is influenced by epigenetic modifiers, such as HATs and HDACs. REST interacts with co-activator CBP/p300 as well as the transcription factor CREB, further enhancing TH transcription, whereas HDAC blocks REST-induced TH activation. Mn represses REST and TH in dopaminergic cells and induces mitochondrial damage, cellular stress, and neuroinflammation and cell death, and REST overexpression protects dopaminergic neurons against Mn toxicity by attenuating Mn-reduced TH and Mn-induced oxidative stress and apoptosis by increasing antioxidative/antiapoptotic genes. These suggest that REST is an important regulator of the antioxidant system, inflammation, and apoptosis in dopaminergic cells.
Discussion
Our findings demonstrate for the first time that REST/NRSF activates transcription of the TH gene by increasing TH promoter activity, mRNA, and protein levels in dopaminergic LUHMES and catecholaminergic CAD neuronal cells. REST directly binds to an RE1/NRSE-binding consensus site motif in the human TH promoter to enhance TH expression. Moreover, REST activation afforded neuroprotection against Mn toxicity by attenuating Mn-induced TH repression, oxidative stress, inflammation, and apoptosis. These results indicate that REST exerts protective effects against Mn-induced neurotoxicity by up-regulating TH, as well as up-regulating antioxidant and antiapoptotic genes.
It has been previously reported that REST dysfunction is associated with PD pathogenesis (47), and neuronal REST deletion in the whole brain exacerbated MPTP toxicity in an experimental mouse model (8). Because REST was found as a repressor of multiple neuronal genes in non-neuronal cells in the brain (36), numerous studies have focused on REST's function as a repressor in various genes in concert with co-repressors, including Co-REST, mSin3a, and HDACs (56, 61, 62). However, considerable evidence indicates that an ensemble of target genes responds to REST (“transcriptionally-responsive” genes) in a cell-type–dependent and context-dependent manner (63). REST also activates genes, including catalase, Bcl-2, and dynamin-1 (13, 50). It has been previously reported that REST activates its target genes by recruiting other co-activators such as ten-eleven translocation hydroxylase 3 (TET3) and nuclear receptor–binding SET domain protein 3 (NSD3).
Our findings reveal that REST increased TH promoter activity, mRNA, and protein levels in dopaminergic neuronal cells. TH contributes to the biosynthesis of dopamine and catecholamines by catalyzing the conversion of the amino acid l-tyrosine to l-3,4-dihydroxyphenylalanine (27). Several studies demonstrated that REST functions as either a repressor or activator of gene transcription in a context-dependent manner, exhibiting complex regulatory effects (64, 65). Activation of the HDAC pathway can lead to interaction of HDAC1 with REST, resulting in abolished REST activation of TH promoter activity in dopaminergic neurons. In support of this, the REST-mediated repression of the corticotropin-releasing hormone gene was HDAC-dependent (64). Furthermore, MPTP treatment of neuronal REST conditional knockout (cKO) mice results in lower levels of TH in the striatum and decreased survival of dopaminergic neurons in the SNpc. Likewise, our in vitro studies show that MPP+, an active metabolite of MPTP, decreased REST and TH promoter activity (Fig. 9, A and B), whereas overexpression of REST attenuated MPP+-reduced TH promoter activity in dopaminergic neurons (Fig. 9C). Deletion of REST abrogated trichostatin A–induced neuroprotection in REST cKO mice after MPTP treatment with further reduction of TH and dopamine levels leading to greater locomotor deficits (66), indicating that REST is required for HDAC to decrease TH, and HDAC inhibition alone without REST is unable to protect the function of dopaminergic neurons.
Our findings reveal that REST increased TH expression by binding to a new REST consensus site in the human TH promoter (Fig. 2, A–C). It has been reported that REST repressed TH expression by binding to different REST-binding sites (NRSE-I, -II, and R) in the 3301 bp of the TH promoter in neural stem HB1.F3 cells (67), suggesting that REST regulation on either activation or repression of TH depends on its binding motifs as well as in a context- and cell-specific manner. Furthermore, some studies suggest that the REST isoform, lacking the C-terminal domain of the REST protein known as REST4/5 (55 kDa), activates transcription of genes, but our results indicate that the full-length REST (∼150 kDa) is responsible for increased TH expression in CAD neurons (Fig. 2D). REST also interacts with CBP/p300 as well as CREB to activate TH expression (28), suggesting that the full-length REST interacts with histone acetylation enhancer CBP and other transcription activators, forming a complex to increase TH transcription. Among the multiple toxicity mechanisms that are induced by Mn, oxidative stress, inflammation, and apoptosis in neurons have been well-established (10, 21, 68). Chronic exposure to Mn reduces Nrf2 levels, resulting in increased proinflammatory cyclooxygenase-2 and prostaglandin E2 levels in in vitro settings (69). REST exerts protective effects against toxic insults, such as MPTP, PD, and AD (8, 13, 47). Our findings reveal that REST increased Nrf2 levels, possibly by inhibiting its ubiquitination and thus promoting Nrf2 stabilization. Nrf2 is an inducible stress response transcription factor that binds to ARE in the promoter region of various genes to regulate the expression of antioxidant genes, such as HO-1, GSH S-transferase, and glutamate–cysteine ligase (58). REST regulates Nrf2 activation by modulating its binding activities or intracellular signaling molecules associated with Nrf2 in astrocytes and oligodendrocytes (62). Several studies suggest that the epigenetic modifier HAT may contribute to the activation of Nrf2 by increasing histone acetylation (62, 70). Taken together, REST-induced Nrf2 activation might be a critical mechanism by which REST mitigates Mn-induced oxidative stress in dopaminergic neurons.
Loss of REST exacerbates toxin-induced dopaminergic neurotoxicity in animal PD models (8) by inducing greater vulnerability to MPTP, resulting in severe impairment of locomotor activity (8). Deletion of neuronal REST also increased expression of the glial fibrillary acidic protein, a marker of astrocyte activation and inflammation, as well as expression of proinflammatory IL-1β in the brain (8). Given that inflammation is critically involved in Mn toxicity and REST deletion, REST overexpression-induced anti-inflammatory effects against Mn-induced proinflammatory cytokines, such as IL-1β, IL-6, TNFα, and IFNγ, might be important to mitigate the detrimental effects of neuroinflammation and to improve cell survival.
In addition to oxidative stress and inflammation, the apoptotic cell death pathway is implicated in a wide array of neurological disorders (71). Therefore, understanding the effects of REST on the transcription of genes related to these intracellular toxicity mechanisms is extremely important for cell survival and protection. In this context, our results show that REST enhanced antioxidant and antiapoptotic genes, indicating that REST modulates multiple levels of neuroprotection. Previous studies also revealed that REST binds to the promoter regions of several proapoptotic/antiapoptotic genes, including Bcl-2, Bax, and Daxx, which are dysregulated in AD brains (13). In addition, loss of REST binding to these antiapoptotic genes resulted in increased mRNA levels of proapoptotic Bax and Daxx and reduction of antiapoptotic Bcl-2 in neurons (13). In support of this, our findings also reveal that REST decreased Mn-induced proapoptotic Bax and Daxx, whereas it increased antiapoptotic Bcl-2 and Bcl-xL in dopaminergic neurons (Fig. 8). Enhanced expression of antiapoptotic genes, such as Bcl-2 and Bcl-xL, will lead to inhibition of apoptotic signaling and promote cell survival (72). In addition to antiapoptotic functions, Bcl-2 and Bcl-xL play a role in other noncanonical functions such as regulation of mitochondrial dynamics, metabolism, and synaptic activity of neurons (73). We found that REST protected dopaminergic neurons against Mn-induced mitochondrial damage (Fig. 8D), suggesting that REST also plays a crucial role in mitochondrial stability and integrity, possibly by regulating gene expression. In addition, DN-REST, which inhibits endogenous REST function, decreased TH expression as well as exacerbated Mn-reduced TH levels, suggesting that REST is a critical component in attenuation of Mn-reduced TH.
Taken together, our findings demonstrate for the first time that REST is a positive regulator of TH in dopaminergic neurons by binding to a newly-identified REST-binding consensus site in the TH promoter with collaboration of CBP/p300 and CREB. Because dopaminergic dysfunction and cell death are hallmarks of PD and critically involved in Mn-induced dopaminergic toxicity, understanding mechanisms by which REST induces protection in dopaminergic neurons by enhancing antioxidant, anti-inflammatory, and antiapoptotic action may help elucidate its molecular mechanisms of neuroprotection against many neurodegenerative diseases (Fig. 10). Given the myriad of protective effects invoked by REST in dopaminergic neurons, targeting REST through drug and therapeutic interventions may result in a promising avenue to treat neurodegenerative disorders associated with dopaminergic neurotoxicity.
Experimental procedures
Materials
Manganese(II) chloride (MnCl2), dimethyl sulfoxide (DMSO), and 3,4,5-dimethylthiazol-2,5-diphenyltetrazolium bromide (MTT) were purchased from MilliporeSigma. All cell culture media, including trypsin-EDTA, minimum essential media, Dulbecco's modified Eagle's medium (DMEM/F-12), and Opti-MEM, were obtained from Gibco. The chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), an ROS molecular probe, and tetramethylrhodamine ethyl ester (TMRE), a mitochondrial membrane potential probe, were purchased from Invitrogen. Antibodies for REST (07-579) and ChIP-validated REST (17-10456) were acquired from MilliporeSigma. Antibodies for TH (sc-25269), Bcl-2 (sc-7382), Bcl-xL (sc-8392), Bax (sc-7480), Daxx (sc-8043), catalase (sc-271803), Nrf2 (sc-271803), HO-1 (sc-136960), CBP/p300 (sc-48343), p-CREB (sc-81486), and β-actin (sc-47778) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). CREB antibody (9197S) was obtained from Cell Signaling Technology (Danvers, MA). Antibodies for horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG (ab6728), HRP-conjugated goat anti-rabbit IgG (ab97051), and goat anti-rabbit or anti-mouse antibodies conjugated with Alexa Fluor® 488, 568 or 647 were from Abcam (Cambridge, MA). Annexin-V staining buffer (420201), annexin-V binding buffer (422201), and FITC–annexin-V (640906) were purchased from Biolegend (San Diego, CA). Propidium iodide (PI, P4170) was obtained from MilliporeSigma. The TNFα standard tetramethylbenzidine enzyme-linked immunosorbent assay (ELISA) development kit for murine (900-T54) samples was acquired from PeproTech (Rocky Hill, NJ). All chemicals were prepared in phosphate-buffered saline (PBS), double-distilled H2O, or DMSO and diluted to working concentrations in Opti-MEM prior to use. Expression vectors REST-myc and its empty control vector (pCMV6-entry) were from OriGene Technologies (Rockville, MD). Control and expression vectors for CBP/p300, HDAC1, pHR′–NRSF–eGFP and TetO–FUW–DN-REST were obtained from Addgene (Watertown, MA). The human TH promoter vector was purchased from Active Motif. Catalase activity colorimetric/fluorometric kit (catalog no. K773-100) was obtained from BioVision (Milpitas, CA). MDA/lipid peroxidation/TBARS assay kit (catalog no. 10009055) was obtained from Cayman Chemical (Ann Arbor, MI).
Cell culture
Dopaminergic/catecholaminergic neuronal cell lines were used in the study. Mouse CAD (08100805) cell line was obtained from MilliporeSigma. LUHMES (CRL-2927) cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA). CAD neuronal cultures were maintained in DMEM/F-12 supplemented with 2 mm l-glutamine (Gibco), 8% fetal bovine serum, 1× GlutaMAXTM (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin. LUHMES cells were maintained in DMEM/F-12 with 1% N2 supplement (Gibco) and 40 ng/ml basic fibroblast growth factor (PeproTech). LUHMES cells were subcultured on culture flasks pre-coated with 50 μg/ml poly-l-ornithine. CAD cells were differentiated with serum-free media, whereas LUHMES cells were differentiated into morphologically and biochemically mature dopamine-like neurons following exposure to tetracycline, glial cell–derived neurotrophic factor, and dibutyryl-cAMP (PeproTech). Cells were dissociated using 0.025% trypsin, 0.1 g/liter EDTA (Gibco), then plated in 96-well plates for multiple assays or 6-well plates for flow cytometry, mRNA, or protein analysis. All cells were maintained at 37 °C in a 95% air, 5% CO2 incubator.
Transfections
Transfections were performed using Lipofectamine 3000 (Invitrogen) or by the GenePulser XcellTM electroporation system (Bio-Rad), according to manufacturer's instructions. For Lipofectamine transfection, cells were transfected with 0.1–1.0 μg of plasmid vectors per 5.0 × 105 cells. For electroporation, cells were transfected with 1.0–10.0 μg of plasmid vectors per 1.0 × 107 cells. Prior to electroporation, cells were grown until 90% confluency in growth media. The electroporation parameters were set for the exponential protocol at 180 V and 950-microfarad capacitance in 4-mm electroporation cuvettes, followed by gentle pipetting and incubation in growth media. Cells were transfected at least overnight with plasmid vectors, followed by further assays and analyses.
Measurement of promoter activity
CAD and LUHMES cells were transfected with the human TH promoter plasmid with Lipofectamine 3000 or by electroporation. The mutations on RE1 consensus binding sites (−288 to −268 position) in the human TH promoter plasmid was performed in this experiment. The human TH promoter plasmid was obtained from Active Motif (Carlsbad, CA), and the REST promoter plasmid was a gift from Dr. Yvon Trottier (INSERM, France). After overnight transfections, the effects of various compounds on promoter activities were determined with the Bright-Glo luciferase assay kit (Promega, Madison, WI) according to the manufacturer's instructions.
qPCR analysis
CAD and LUHMES cells were harvested after treatment with the designated compounds (three samples/group). Total RNA was extracted from samples using the RNeasy mini kit (Qiagen, Valencia, CA), and 2 μg of purified RNA was transcribed to cDNA with a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). Real-time qPCR was performed using the CFX96 real-time PCR detection system (Bio-Rad). The reaction mixture contained 1 μl of each cDNA template, 0.4 μm primers, and iQ SYBR Green Supermix (Bio-Rad). The total reaction volume was 25 μl. The following primers were used: mouse REST, 5′-ACT TTG TCC TTA CTC AAG CTC-3′ (forward) and 5′-CAT TTA AAT GGC TTC TCA CCT G-3′ (reverse); mouse TH 5′-CAC TAT GCC CAC CCC CAG-3′ (forward) and 5′-CGC CGT CCA ATG AAC CTT-3′ (reverse); mouse TNFα 5′-GGT CCC CAA AGG GAT GAG AAG TTC-3′ (forward) and 5′-CCA CTT GGT GGT TTG CTA CGA CG-3′ (reverse); mouse IL-1β 5′-CAA CCA ACA AGT GAT ATT CTC CAT G-3′ (forward) and 5′-GAT CCA CAC TCT CCA GCT GCA-3′ (reverse); mouse IL-6 5′-ATG GAT GCT ACC AAA CTG GAT-3′ (forward) and 5′-TGA AGG ACT CTG GCT TTG TCT-3′ (reverse); mouse IFNγ 5′-ATG AAC GCT ACA CAC TGC ATC-3′ (forward) and 5′-CCA TCC TTT TGC CAG TTC CTC-3′ (reverse); mouse catalase 5′-AGC GAC CAG ATG AAG CAG TG-3′ (forward) and 5′-TCC GCT CTC TGT CAA AGT GTG-3′ (reverse); mouse GAPDH 5′-CTC ATG ACC ACA GTC CAT GC-3′ (forward) and 5′-CAC ATT GGG GGT AGG AAC AC-3′ (reverse); human REST 5′-GTG AGC GAG TAT CAC TGG AGG-3′ (forward) and 5′-CCC ATT GTG AAC CTG TCT TGC-3′ (reverse); human TH 5′-GCG CAG GAA GCT GAT TGC TG-3′ (forward) and 5′-TGT CTT CCC GGT AGC CGC G-3′ (reverse); and human GAPDH 5′-ACA ACT TTG GTA TCG TGG AAG G-3′ (forward) and 5′-GCC ATC ACG CCA CAG TTT C-3′ (reverse). The qPCR parameters were set for 1 cycle at 95 °C for 10 min, 40 cycles at 95 °C for 15 s, and 60–65 °C for 1 min. GAPDH was utilized as an internal control. Following PCR, mRNA levels were analyzed using the Bio-Rad CFX Manager Version 3.1.
Western blot analysis
After treatment with the designated compounds, CAD and LUHMES cells were washed with ice-cold PBS. Cells were lysed by adding radioimmunoprecipitation assay (RIPA) buffer and protease inhibitor mixture and were then harvested. The protein concentration of the lysates was determined by bicinchoninic acid assay. Thirty μg of protein per sample was mixed with 4× Laemmli buffer and 5% β-mercaptoethanol in a 3:1 ratio and then heated at 95 °C for 10 min. The samples were run on 10% SDS-polyacrylamide gels and transferred to a nitrocellulose membrane for Western blot analysis. The primary antibodies were used at a 1:1000 dilution, and HRP-conjugated secondary antibodies were used at a 1:5000 dilution. Protein bands were detected with SuperSignalTM West Pico PLUS chemiluminescent substrate (Thermo Fisher Scientific, Rockford, IL) and quantified using the Molecular Imager ChemiDoc XRS+ System (Bio-Rad).
Immunocytochemistry
For immunostaining, cells were cultured on poly-l-lysine–coated glass coverslips in 6-well plates. After each experiment, cells were fixed using 4% paraformaldehyde in PBS, pH 7.4, for 10 min at room temperature, and then cells were washed three times with ice-cold PBS. Cells were permeabilized and then incubated with blocking buffer (1× PBS, 10% normal serum, 0.1% Tween 20) for 1 h at room temperature, washed, and incubated overnight with primary antibodies for TH, REST, CBP/p300, or CREB at 1:250 dilution at 4 °C. After overnight incubation, tissue sections were incubated with fluorescent-conjugated secondary antibodies Alexa Fluor® 488 and 568 (1:1,000 dilution) for 1 h at room temperature in the dark, then washed, and mounted on to slides with 4′,6-diamidino-2-phenylindole fluoromount solution for imaging analysis. Cellular localization and fluorescence intensity were assessed for each sample using a Ts2R fluorescence microscope (Nikon Instruments, Melville, NY).
Preparation of cytoplasmic and nuclear fractions
Cells were lysed in hypotonic buffer (10 mm HEPES-KOH, pH 7.9, 10 mm KCl, 1.5 mm MgCl2) containing 0.5% Nonidet P-40 and centrifuged at 2500 rpm for 5 min at 4 °C. The lysates containing cytoplasmic fractions were saved. The nuclei in the pellet were dissolved in hypertonic buffer (20 mm HEPES-KOH, pH 7.9, 0.4 m NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, and 25% glycerol) with periodic vortexing and incubation on ice for 30 min. The nuclear fractions were obtained after spinning at 20,000 × g for 10 min at 4 °C, and protein concentrations were determined by bicinchoninic acid assay.
ChIP assay
The ChIP assay was performed using the EZ-ChIP kit (Millipore) according to the manufacturer's instructions. Briefly, cross-linking was done by treating cells with formaldehyde for 10 min at room temperature. After washing with ice-cold PBS, cells were lysed in SDS lysis buffer containing protease inhibitor mixture. The cell lysates were sonicated and centrifuged at 15,000 × g for 10 min at 4 °C. The supernatant was mixed with ChIP dilution buffer, and 60 μl of protein G–agarose was added. After 1 h of incubation at 4 °C, the agarose beads were pelleted by spinning at 3000 × g for 1 min. Then, 1% of the supernatants were saved as inputs, and REST or rabbit IgG (negative control) antibodies were added to the remainder and incubated overnight at 4 °C. Protein G–agarose beads (60 μl) were added and incubated at 4 °C for 1 h. The agarose beads were pelleted and washed with low-salt and high-salt immune complex wash buffer. The free DNA obtained after reverse cross-linking of the protein–DNA complex was purified, and the real-time qPCR was carried out with the REST primers: 5′-TCG AAC CTG CAA AAG TGG GC-3′ (forward) and 5′-CCC CCA GCC CAT GTA ACA A-3′ (reverse). Following qPCR, immunoprecipitated DNA were quantified using the Bio-Rad CFX Manager 3.1. The end products of qPCR were also applied for the agarose gel electrophoresis.
DNA affinity purification assay (DAPA)
DAPA was performed according to the manufacturer's recommendation using a μMACS FactorFinder Kit (Miltenyi Biotec, Inc., Auburn, CA). Briefly, 1.5 μg of biotinylated oligonucleotides were incubated with 50 μg of nuclear extract in binding buffer for 20 min. The incubation was continued for another 10 min after the addition of 100 μl of streptavidin microbeads. The reaction mixture was applied onto the microcolumn that was already equilibrated with two 100-μl washes of binding buffer. After four washes of 100 μl each with low-salt and high-salt buffers, proteins were eluted using 30 μl of elution buffer and analyzed by Western blotting.
EMSA
EMSA was performed using a LightShift chemiluminescent kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Briefly, 5 μg of nuclear extract from cells were incubated with biotin-labeled oligonucleotides containing the REST consensus binding sites of the TH promoter for 20 min on ice. The DNA–protein complexes were resolved on 8% nondenaturing DNA polyacrylamide gels and transferred to nylon membranes. DNA–protein complexes were detected using a chemiluminescent nucleic acid detection module (Thermo Fisher Scientific). The primers pairs used for REST (TH promoter including the RE1 consensus site) were 5′-GCT GTC TCA GCC CCC CGC AGA GCA CGA GCC C-3′ and 5′-GGG CTC GTG CTC TGC GGG GGG CTG AGA CAG C-3′.
Site-directed mutagenesis
The RE1 consensus binding sequence in the TH promoter was mutated using PfuUltra II Fusion HS DNA polymerase kit (Agilent Technologies, Cedar Creek, TX) according to manufacturer's recommendations. The TH promoter (−673 to +97 bp) subcloned into the LightSwitchTM promoter reporter vector was used as the original template for mutation. The primer sets used were RE1 mutant #1 (RE1-mu1) 5′-GGC TGT CTC AGC CTC CCG CAG CGC ACG AGC C-3′ and 5′-GGC TCG TGC GCT GCG GGA GGC TGA GAC AGC C-3′, and RE1 mutant #2 (RE1-mu2) 5′-CTG GCT GTC TCA GCC TCC CTC AGA GCA CGA GCC-3′ and 5′-GGC TCG TGC TCT GAG GGA GGC TGA GAC AGC CAG-3′. The RE1 mutant clones were confirmed by Sanger sequencing.
Assays of oxidative stress, lipid peroxidation, and catalase activity
ROS, lipid peroxidation, and catalase activity were measured in accordance with the manufacturer's protocols. Generation of ROS as an indicator of oxidative stress was measured using the Life Technologies, Inc., CM-H2DCFDA ROS molecular probe. End-point product fluorescence was measured in each assay using the Spectramax® i3x multimode microplate reader from Molecular Devices (Sunnyvale, CA). Briefly, CAD neurons were washed with PBS and treated with Mn (250 μm, 3 h) at 37 °C. Cells were washed, and 2.5 μm CM-H2DCFDA was added for 10 min. End-point fluorescence was determined at an excitation/emission wavelength of 485/527 nm.
Lipid peroxidation was assessed in cell lysates by the measurement of MDA, an end product of lipid peroxidation, which reacts with thiobarbituric acid to form a complex. The reaction product fluorescence was determined at an excitation/emission wavelength of 530/550 nm in the fluorescence plate reader using the MDA standard curve.
Catalase activity was measured according to the manufacturer's protocol. Briefly, cells were extracted with mammalian cell lysis buffer and further analyzed using the assay kit. Catalase activity was calculated as the rate of breakdown in H2O2. OxiRedTM probe reacts with unconverted H2O2 and was measured at an excitation/emission wavelength of 535/587 nm in the fluorescence plate reader.
ELISA
TNFα release from CAD cells was determined using a murine TNFα ELISA kit (PeproTech) according to the manufacturer's instructions. Briefly, cell-free media (1 ml/well) were collected from samples after exposure to Mn (250 μm) for 12 h. ELISA was performed, and the concentration of secreted TNFα was determined using a multimode microplate reader (Molecular Devices) set to 450 nm, with wavelength correction set at 620 nm.
Flow cytometry
Cellular apoptosis was determined by an annexin V–FITC/PI staining using fluorescence-activated cell sorting (FACS). Mn (250 μm) was added; the cells were incubated for 12 h, and cells were dissociated via trypsinization, washed twice, and resuspended in a binding buffer. One hundred-μl aliquots containing 1.0 × 106 cells per sample were used for the experiment. Following resuspension, cells were stained sequentially with annexin V–FITC (5 μg/μl, Biolegend) and PI (5 μg/μl, Sigma) according to the manufacturer's instructions. After incubation for 15 min, 400 μl of binding buffer was added. Apoptotic cells were analyzed by a BD FACSCalibur 2.0 flow cytometer (BD Biosciences) using FlowJo X software Version 10 (Ashland, OR).
For protein staining, cells were washed twice and resuspended in cell staining buffer, and then stained with anti-TH and anti-REST antibodies at 1:100 dilution. After incubation for 1 h, cells were washed twice, followed by staining with fluorescent-labeled secondary antibodies for 30 min. Fluorescent cells were analyzed by a BD FACSCalibur 2.0 flow cytometer using FlowJo X software Version 10.
Cell viability assay
CAD neurons (2 × 104/well) were grown in 96-well plates then exposed to Mn (250 μm) for 12 h. After incubation for the designated time period, cells were washed twice with ice-cold PBS. Ten μl of MTT (5 mg/ml in PBS) was added, and cells were incubated for 3 h at 37 °C. After incubation, 100 μl of 0.1 n HCl was added, and the absorbance of the converted dye was measured at a wavelength of 570 nm using a microplate reader.
Δψm assay
Mitochondrial Δψm as an indicator of mitochondrial function was measured using TMRE from Life Technologies, Inc. End-point product fluorescence was measured in each assay using the Spectramax® i3x multimode microplate reader from Molecular Devices (Sunnyvale, CA). Briefly, CAD neurons were treated with Mn (50, 100, and 250 μm) for 1 h at 37 °C, followed by washing with PBS and incubation with 50 nm TMRE for 30 min. End-point fluorescence was determined at an excitation/emission wavelength of 549/575 nm.
Statistical analysis
All data were expressed as the mean ± S.E. of the mean. The statistical analyses were performed using either Student's t test or one-way analysis of variance (ANOVA), followed by Tukey's post hoc tests using the GraphPad Prism software Version 6.0 (San Diego, CA). A p value of less than 0.05 (p < 0.05) was considered statistically significant.
Author contributions
E. L. and E. P. conceptualization; E. P. data curation; E. P. software; E. P. formal analysis; E. P., D.-S. S., M. A., and E. L. validation; E. L. and E. P. investigation; E. P. visualization; E. P. and A. R. methodology; E. P. writing-original draft; E. P., A. R., D.-S. S., M. A., and E. L. writing-review and editing; E. L. resources; E. L. supervision; E. L. funding acquisition; E. L. project administration.
This work was supported by National Institutes of Health Research Grants R01 ES024756, U54 MD007582, NCI SC1 CA200519, R01 ES10563, R01 ES07331, and 1R21 ES025415. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CNS
- central nervous system
- PD
- Parkinson's disease
- qPCR
- quantitative RT-PCR
- TH
- tyrosine hydroxylase
- REST
- RE1-silencing transcription factor
- NRSF
- neuron-restrictive silencer factor
- CREB
- cAMP-response element-binding protein
- CBP
- CREB-binding protein
- DN-REST
- dominant-negative REST
- CAD
- cath.-a-differentiated
- LUHMES
- Lund human mesencephalic
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- EV
- empty vector
- Con
- control
- PI
- propidium iodide
- MTT
- 3,4,5-dimethylthiazol-2,5-diphenyltetrazolium bromide
- TMRE
- tetramethylrhodamine ethyl ester
- CM-H2DCFDA
- chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate
- MDA
- malondialdehyde
- EMSA
- electrophoretic mobility-shift assay
- DAPA
- DNA affinity purification assay
- ANOVA
- analysis of variance
- co-IP
- co-immunoprecipitation
- MPP+
- 1-methyl-4-phenylpyridinium
- HAT
- histone acetyltransferase
- HDAC
- histone deacetylase
- DMEM
- Dulbecco's modified Eagle's medium
- HRP
- horseradish peroxidase
- SNpc
- substantia nigra pars compacta
- MPTP
- 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- AD
- Alzheimer's disease
- ROS
- reactive oxygen species
- ARE
- antioxidant-response element
- TNF-α
- tumor necrosis factor-α
- IL
- interleukin
- IFN
- interferon
- cKO
- conditional knockout.
References
- 1. Chinta S. J., and Andersen J. K. (2005) Dopaminergic neurons. Int. J. Biochem. Cell Biol. 37, 942–946 10.1016/j.biocel.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 2. Schultz W. (1997) Dopamine neurons and their role in reward mechanisms. Curr. Opin. Neurobiol. 7, 191–197 10.1016/S0959-4388(97)80007-4 [DOI] [PubMed] [Google Scholar]
- 3. Bernheimer H., Birkmayer W., Hornykiewicz O., Jellinger K., and Seitelberger F. (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J. Neurol. Sci. 20, 415–455 10.1016/0022-510X(73)90175-5 [DOI] [PubMed] [Google Scholar]
- 4. Coulon J. F., Biguet N. F., Cavoy A., Delacour J., Mallet J., and David J. C. (1990) Gene expression of tyrosine hydroxylase in the developing fetal brain. J. Neurochem. 55, 1412–1417 10.1111/j.1471-4159.1990.tb03154.x [DOI] [PubMed] [Google Scholar]
- 5. Berod A., Hartman B. K., Keller A., Joh T. H., and Pujol J. F. (1982) A new double labeling technique using tyrosine hydroxylase and dopamine-β-hydroxylase immunohistochemistry: evidence for dopaminergic cells lying in the pons of the beef brain. Brain Res. 240, 235–243 10.1016/0006-8993(82)90219-0 [DOI] [PubMed] [Google Scholar]
- 6. Taylor J. M., Main B. S., and Crack P. J. (2013) Neuroinflammation and oxidative stress: co-conspirators in the pathology of Parkinson's disease. Neurochem. Int. 62, 803–819 10.1016/j.neuint.2012.12.016 [DOI] [PubMed] [Google Scholar]
- 7. Harischandra D. S., Ghaisas S., Zenitsky G., Jin H., Kanthasamy A., Anantharam V., and Kanthasamy A. G. (2019) Manganese-induced neurotoxicity: new insights into the triad of protein misfolding, mitochondrial impairment, and neuroinflammation. Front. Neurosci. 13, 654 10.3389/fnins.2019.00654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu M., Suo H., Liu M., Cai L., Liu J., Huang Y., Xu J., Wang Y., Zhu C., Fei J., and Huang F. (2013) NRSF/REST neuronal deficient mice are more vulnerable to the neurotoxin MPTP. Neurobiol. Aging 34, 916–927 10.1016/j.neurobiolaging.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 9. Pajarillo E., Johnson J. Jr, Kim J., Karki P., Son D. S., Aschner M., and Lee E. (2018) 17β-Estradiol and tamoxifen protect mice from manganese-induced dopaminergic neurotoxicity. Neurotoxicology 65, 280–288 10.1016/j.neuro.2017.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Malecki E. A. (2001) Manganese toxicity is associated with mitochondrial dysfunction and DNA fragmentation in rat primary striatal neurons. Brain Res. Bull. 55, 225–228 10.1016/S0361-9230(01)00456-7 [DOI] [PubMed] [Google Scholar]
- 11. Stredrick D. L., Stokes A. H., Worst T. J., Freeman W. M., Johnson E. A., Lash L. H., Aschner M., and Vrana K. E. (2004) Manganese-induced cytotoxicity in dopamine-producing cells. Neurotoxicology 25, 543–553 10.1016/j.neuro.2003.08.006 [DOI] [PubMed] [Google Scholar]
- 12. Lee E. S., Yin Z., Milatovic D., Jiang H., and Aschner M. (2009) Estrogen and tamoxifen protect against Mn-induced toxicity in rat cortical primary cultures of neurons and astrocytes. Toxicol. Sci. 110, 156–167 10.1093/toxsci/kfp081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lu T., Aron L., Zullo J., Pan Y., Kim H., Chen Y., Yang T. H., Kim H. M., Drake D., Liu X. S., Bennett D. A., Colaiácovo M. P., and Yankner B. A. (2014) REST and stress resistance in ageing and Alzheimer's disease. Nature 507, 448–454 10.1038/nature13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang P., Xie K., Wang C., and Bi J. (2014) Oxidative stress induced by lipid peroxidation is related with inflammation of demyelination and neurodegeneration in multiple sclerosis. Eur. Neurol. 72, 249–254 10.1159/000363515 [DOI] [PubMed] [Google Scholar]
- 15. Blasco H., Garcon G., Patin F., Veyrat-Durebex C., Boyer J., Devos D., Vourc'h P., Andres C. R., and Corcia P. (2017) Panel of oxidative stress and inflammatory biomarkers in ALS: a pilot study. Can. J. Neurol. Sci. 44, 90–95 10.1017/cjn.2016.284 [DOI] [PubMed] [Google Scholar]
- 16. Kim J., Pajarillo E., Rizor A., Son D. S., Lee J., Aschner M., and Lee E. (2019) LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS ONE 14, e0210248 10.1371/journal.pone.0210248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Newland M. C., Ceckler T. L., Kordower J. H., and Weiss B. (1989) Visualizing manganese in the primate basal ganglia with magnetic resonance imaging. Exp. Neurol. 106, 251–258 10.1016/0014-4886(89)90157-X [DOI] [PubMed] [Google Scholar]
- 18. Alsulimani H. H., Ye Q., and Kim J. (2015) Effect of Hfe deficiency on memory capacity and motor coordination after manganese exposure by drinking water in mice. Toxicol. Res. 31, 347–354 10.5487/TR.2015.31.4.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Menezes-Filho J. A., de Carvalho-Vivas C. F., Viana G. F., Ferreira J. R., Nunes L. S., Mergler D., and Abreu N. (2014) Elevated manganese exposure and school-aged children's behavior: a gender-stratified analysis. Neurotoxicology 45, 293–300 10.1016/j.neuro.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 20. Popichak K. A., Afzali M. F., Kirkley K. S., and Tjalkens R. B. (2018) Glial-neuronal signaling mechanisms underlying the neuroinflammatory effects of manganese. J. Neuroinflammation 15, 324 10.1186/s12974-018-1349-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirkley K. S., Popichak K. A., Afzali M. F., Legare M. E., and Tjalkens R. B. (2017) Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflammation 14, 99 10.1186/s12974-017-0871-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang K., Zhu Y., Wang X., Zhao X., Li S., and Teng X. (2017) Excess manganese-induced apoptosis in chicken cerebrums and embryonic neurocytes. Biol. Trace Elem. Res. 180, 297–305 10.1007/s12011-017-0992-4 [DOI] [PubMed] [Google Scholar]
- 23. Beck K. D., Knusel B., Pasinetti G., Michel P. P., Zawadzka H., Goldstein M., and Hefti F. (1991) Tyrosine hydroxylase mRNA expression by dopaminergic neurons in culture: effect of 1-methyl-4-phenylpyridinium treatment. J. Neurochem. 57, 527–532 10.1111/j.1471-4159.1991.tb03782.x [DOI] [PubMed] [Google Scholar]
- 24. Kumasaka M. Y., Yajima I., Ohgami N., Ninomiya H., Iida M., Li X., Oshino R., Tanihata H., Yoshinaga M., and Kato M. (2017) Manganese-mediated decrease in levels of c-RET and tyrosine hydroxylase expression in vitro. Neurotox. Res. 32, 661–670 10.1007/s12640-017-9783-0 [DOI] [PubMed] [Google Scholar]
- 25. Johnson J. Jr., Pajarillo E. A. B., Taka E., Reams R., Son D. S., Aschner M., and Lee E. (2018) Valproate and sodium butyrate attenuate manganese-decreased locomotor activity and astrocytic glutamate transporters expression in mice. Neurotoxicology 64, 230–239 10.1016/j.neuro.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang D., Kanthasamy A., Anantharam V., and Kanthasamy A. (2011) Effects of manganese on tyrosine hydroxylase (TH) activity and TH-phosphorylation in a dopaminergic neural cell line. Toxicol. Appl. Pharmacol. 254, 65–71 10.1016/j.taap.2010.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagatsu T., Levitt M., and Udenfriend S. (1964) Tyrosine hydroxylase. The initial step in norepinephrine biosynthesis. J. Biol. Chem. 239, 2910–2917 [PubMed] [Google Scholar]
- 28. Lim J., Yang C., Hong S. J., and Kim K. S. (2000) Regulation of tyrosine hydroxylase gene transcription by the cAMP-signaling pathway: involvement of multiple transcription factors. Mol. Cell. Biochem. 212, 51–60 10.1023/A:1007148719497 [DOI] [PubMed] [Google Scholar]
- 29. Sakurada K., Ohshima-Sakurada M., Palmer T. D., and Gage F. H. (1999) Nurr1, an orphan nuclear receptor, is a transcriptional activator of endogenous tyrosine hydroxylase in neural progenitor cells derived from the adult brain. Development 126, 4017–4026 [DOI] [PubMed] [Google Scholar]
- 30. Messmer K., Remington M. P., Skidmore F., and Fishman P. S. (2007) Induction of tyrosine hydroxylase expression by the transcription factor Pitx3. Int. J. Dev. Neurosci. 25, 29–37 10.1016/j.ijdevneu.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 31. Kim H. S., Hong S. J., LeDoux M. S., and Kim K. S. (2001) Regulation of the tyrosine hydroxylase and dopamine β-hydroxylase genes by the transcription factor AP-2. J. Neurochem. 76, 280–294 10.1046/j.1471-4159.2001.00044.x [DOI] [PubMed] [Google Scholar]
- 32. Lim J., Kim H. I., Bang Y., Seol W., Choi H. S., and Choi H. J. (2015) Hypoxia-inducible factor-1α upregulates tyrosine hydroxylase and dopamine transporter by nuclear receptor ERRγ in SH-SY5Y cells. Neuroreport 26, 380–386 10.1097/WNR.0000000000000356 [DOI] [PubMed] [Google Scholar]
- 33. Doan K. V., Kinyua A. W., Yang D. J., Ko C. M., Moh S. H., Shong K. E., Kim H., Park S. K., Kim D. H., Kim I., Paik J. H., DePinho R. A., Yoon S. G., Kim I. Y., Seong J. K., et al. (2016) FoxO1 in dopaminergic neurons regulates energy homeostasis and targets tyrosine hydroxylase. Nat. Commun. 7, 12733 10.1038/ncomms12733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen Z. F., Paquette A. J., and Anderson D. J. (1998) NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 20, 136–142 10.1038/2431 [DOI] [PubMed] [Google Scholar]
- 35. Paquette A. J., Perez S. E., and Anderson D. J. (2000) Constitutive expression of the neuron-restrictive silencer factor (NRSF)/REST in differentiating neurons disrupts neuronal gene expression and causes axon pathfinding errors in vivo. Proc. Natl. Acad. Sci. U.S.A. 97, 12318–12323 10.1073/pnas.97.22.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jones F. S., and Meech R. (1999) Knockout of REST/NRSF shows that the protein is a potent repressor of neuronally expressed genes in non-neural tissues. Bioessays 21, 372–376 [DOI] [PubMed] [Google Scholar]
- 37. Moreno-Gonzalez G., López-Colomé A. M., Rodríguez G., and Zarain-Herzberg A. (2008) Transcription of the chicken Grin1 gene is regulated by the activity of SP3 and NRSF in undifferentiated cells and neurons. Biosci. Rep. 28, 177–188 10.1042/BSR20080058 [DOI] [PubMed] [Google Scholar]
- 38. Shimojo M., and Hersh L. B. (2004) Regulation of the cholinergic gene locus by the repressor element-1 silencing transcription factor/neuron restrictive silencer factor (REST/NRSF). Life Sci. 74, 2213–2225 10.1016/j.lfs.2003.08.045 [DOI] [PubMed] [Google Scholar]
- 39. Chong J. A., Tapia-Ramírez J., Kim S., Toledo-Aral J. J., Zheng Y., Boutros M. C., Altshuller Y. M., Frohman M. A., Kraner S. D., and Mandel G. (1995) REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80, 949–957 10.1016/0092-8674(95)90298-8 [DOI] [PubMed] [Google Scholar]
- 40. Schoenherr C. J., and Anderson D. J. (1995) The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 267, 1360–1363 10.1126/science.7871435 [DOI] [PubMed] [Google Scholar]
- 41. Murai K., Naruse Y., Shaul Y., Agata Y., and Mori N. (2004) Direct interaction of NRSF with TBP: chromatin reorganization and core promoter repression for neuron-specific gene transcription. Nucleic Acids Res. 32, 3180–3189 10.1093/nar/gkh550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nomura M., Uda-Tochio H., Murai K., Mori N., and Nishimura Y. (2005) The neural repressor NRSF/REST binds the PAH1 domain of the Sin3 corepressor by using its distinct short hydrophobic helix. J. Mol. Biol. 354, 903–915 10.1016/j.jmb.2005.10.008 [DOI] [PubMed] [Google Scholar]
- 43. Watanabe H., Mizutani T., Haraguchi T., Yamamichi N., Minoguchi S., Yamamichi-Nishina M., Mori N., Kameda T., Sugiyama T., and Iba H. (2006) SWI/SNF complex is essential for NRSF-mediated suppression of neuronal genes in human nonsmall cell lung carcinoma cell lines. Oncogene 25, 470–479 10.1038/sj.onc.1209068 [DOI] [PubMed] [Google Scholar]
- 44. Palm K., Belluardo N., Metsis M., and Timmusk T. (1998) Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J. Neurosci. 18, 1280–1296 10.1523/JNEUROSCI.18-04-01280.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shimojo M., Lee J. H., and Hersh L. B. (2001) Role of zinc finger domains of the transcription factor neuron-restrictive silencer factor/repressor element-1 silencing transcription factor in DNA binding and nuclear localization. J. Biol. Chem. 276, 13121–13126 10.1074/jbc.M011193200 [DOI] [PubMed] [Google Scholar]
- 46. Abramovitz L., Shapira T., Ben-Dror I., Dror V., Granot L., Rousso T., Landoy E., Blau L., Thiel G., and Vardimon L. (2008) Dual role of NRSF/REST in activation and repression of the glucocorticoid response. J. Biol. Chem. 283, 110–119 10.1074/jbc.M707366200 [DOI] [PubMed] [Google Scholar]
- 47. Kawamura M., Sato S., Matsumoto G., Fukuda T., Shiba-Fukushima K., Noda S., Takanashi M., Mori N., and Hattori N. (2019) Loss of nuclear REST/NRSF in aged-dopaminergic neurons in Parkinson's disease patients. Neurosci. Lett. 699, 59–63 10.1016/j.neulet.2019.01.042 [DOI] [PubMed] [Google Scholar]
- 48. Bruce A. W., López-Contreras A. J., Flicek P., Down T. A., Dhami P., Dillon S. C., Koch C. M., Langford C. F., Dunham I., Andrews R. M., and Vetrie D. (2009) Functional diversity for REST (NRSF) is defined by in vivo binding affinity hierarchies at the DNA sequence level. Genome Res. 19, 994–1005 10.1101/gr.089086.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Brené S., Messer C., Okado H., Hartley M., Heinemann S. F., and Nestler E. J. (2000) Regulation of GluR2 promoter activity by neurotrophic factors via a neuron-restrictive silencer element. Eur. J. Neurosci. 12, 1525–1533 10.1046/j.1460-9568.2000.00040.x [DOI] [PubMed] [Google Scholar]
- 50. Yoo J., Jeong M. J., Lee S. S., Lee K. I., Kwon B. M., Kim D. S., Park Y. M., and Han M. Y. (2001) The neuron restrictive silencer factor can act as an activator for dynamin I gene promoter activity in neuronal cells. Biochem. Biophys. Res. Commun. 283, 928–932 10.1006/bbrc.2001.4857 [DOI] [PubMed] [Google Scholar]
- 51. Bai G., Zhuang Z., Liu A., Chai Y., and Hoffman P. W. (2003) The role of the RE1 element in activation of the NR1 promoter during neuronal differentiation. J. Neurochem. 86, 992–1005 10.1046/j.1471-4159.2003.01922.x [DOI] [PubMed] [Google Scholar]
- 52. Kuwabara T., Hsieh J., Nakashima K., Taira K., and Gage F. H. (2004) A small modulatory dsRNA specifies the fate of adult neural stem cells. Cell 116, 779–793 10.1016/S0092-8674(04)00248-X [DOI] [PubMed] [Google Scholar]
- 53. Zhao Y., Zhu M., Yu Y., Qiu L., Zhang Y., He L., and Zhang J. (2017) Brain REST/NRSF is not only a silent repressor but also an active protector. Mol. Neurobiol. 54, 541–550 10.1007/s12035-015-9658-4 [DOI] [PubMed] [Google Scholar]
- 54. Qi Y., Wang J. K., McMillian M., and Chikaraishi D. M. (1997) Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 17, 1217–1225 10.1523/JNEUROSCI.17-04-01217.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J. A., van der Lee R., Bessy A., Chèneby J., Kulkarni S. R., Tan G., Baranasic D., Arenillas D. J., Sandelin A., Vandepoele K., Lenhard B., et al. (2018) JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D1284 10.1093/nar/gkx1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Naruse Y., Aoki T., Kojima T., and Mori N. (1999) Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc. Natl. Acad. Sci. U.S.A. 96, 13691–13696 10.1073/pnas.96.24.13691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Alam J., Stewart D., Touchard C., Boinapally S., Choi A. M., and Cook J. L. (1999) Nrf2, a Cap'n'Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 274, 26071–26078 10.1074/jbc.274.37.26071 [DOI] [PubMed] [Google Scholar]
- 58. Loboda A., Damulewicz M., Pyza E., Jozkowicz A., and Dulak J. (2016) Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell. Mol. Life Sci. 73, 3221–3247 10.1007/s00018-016-2223-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Poss K. D., and Tonegawa S. (1997) Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10925–10930 10.1073/pnas.94.20.10925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin K. H., Li C. Y., Hsu Y. M., Tsai C. H., Tsai F. J., Tang C. H., Yang J. S., Wang Z. H., and Yin M. C. (2019) Oridonin, a natural diterpenoid, protected NGF-differentiated PC12 cells against MPP+- and kainic acid-induced injury. Food Chem. Toxicol. 133, 110765 10.1016/j.fct.2019.110765 [DOI] [PubMed] [Google Scholar]
- 61. Abrajano J. J., Qureshi I. A., Gokhan S., Zheng D., Bergman A., and Mehler M. F. (2009) REST and CoREST modulate neuronal subtype specification, maturation and maintenance. PLoS ONE 4, e7936 10.1371/journal.pone.0007936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abrajano J. J., Qureshi I. A., Gokhan S., Zheng D., Bergman A., and Mehler M. F. (2009) Differential deployment of REST and CoREST promotes glial subtype specification and oligodendrocyte lineage maturation. PLoS ONE 4, e7665 10.1371/journal.pone.0007665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hwang J. Y., and Zukin R. S. (2018) REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 48, 193–200 10.1016/j.conb.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Seth K. A., and Majzoub J. A. (2001) Repressor element silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) can act as an enhancer as well as a repressor of corticotropin-releasing hormone gene transcription. J. Biol. Chem. 276, 13917–13923 10.1074/jbc.M007745200 [DOI] [PubMed] [Google Scholar]
- 65. Perera A., Eisen D., Wagner M., Laube S. K., Künzel A. F., Koch S., Steinbacher J., Schulze E., Splith V., Mittermeier N., Müller M., Biel M., Carell T., and Michalakis S. (2015) TET3 is recruited by REST for context-specific hydroxymethylation and induction of gene expression. Cell Rep. 11, 283–294 10.1016/j.celrep.2015.03.020 [DOI] [PubMed] [Google Scholar]
- 66. Suo H., Wang P., Tong J., Cai L., Liu J., Huang D., Huang L., Wang Z., Huang Y., Xu J., Ma Y., Yu M., Fei J., and Huang F. (2015) NRSF is an essential mediator for the neuroprotection of trichostatin A in the MPTP mouse model of Parkinson's disease. Neuropharmacology 99, 67–78 10.1016/j.neuropharm.2015.07.015 [DOI] [PubMed] [Google Scholar]
- 67. Kim S. M., Yang J. W., Park M. J., Lee J. K., Kim S. U., Lee Y. S., and Lee M. A. (2006) Regulation of human tyrosine hydroxylase gene by neuron-restrictive silencer factor. Biochem. Biophys. Res. Commun. 346, 426–435 10.1016/j.bbrc.2006.05.142 [DOI] [PubMed] [Google Scholar]
- 68. Stanwood G. D., Leitch D. B., Savchenko V., Wu J., Fitsanakis V. A., Anderson D. J., Stankowski J. N., Aschner M., and McLaughlin B. (2009) Manganese exposure is cytotoxic and alters dopaminergic and GABAergic neurons within the basal ganglia. J. Neurochem. 110, 378–389 10.1111/j.1471-4159.2009.06145.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Qi Z., Mi C., Wu F., Yang X., Sang Y., Liu Y., Li J., Yang H., Xu B., Liu W., Xu Z., and Deng Y. (2019) The effect of manganese exposure on GnRH secretion via Nrf2/mGluR5/COX-2/PGE2/signaling pathway. Toxicol. Ind. Health 35, 211–227 10.1177/0748233719825720 [DOI] [PubMed] [Google Scholar]
- 70. Zhang Z., Guo Z., Zhan Y., Li H., and Wu S. (2017) Role of histone acetylation in activation of nuclear factor erythroid 2-related factor 2/heme oxygenase 1 pathway by manganese chloride. Toxicol. Appl. Pharmacol. 336, 94–100 10.1016/j.taap.2017.10.011 [DOI] [PubMed] [Google Scholar]
- 71. Isaev N. K., Genrikhs E. E., Oborina M. V., and Stelmashook E. V. (2018) Accelerated aging and aging process in the brain. Rev. Neurosci. 29, 233–240 10.1515/revneuro-2017-0051 [DOI] [PubMed] [Google Scholar]
- 72. Cheng E. H., Wei M. C., Weiler S., Flavell R. A., Mak T. W., Lindsten T., and Korsmeyer S. J. (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8, 705–711 10.1016/S1097-2765(01)00320-3 [DOI] [PubMed] [Google Scholar]
- 73. Aouacheria A., Baghdiguian S., Lamb H. M., Huska J. D., Pineda F. J., and Hardwick J. M. (2017) Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem. Int. 109, 141–161 10.1016/j.neuint.2017.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]