

Abstract
The photolysis of diazoalkanes is a timely strategy to conduct carbene‐transfer reactions under mild and metal‐free reaction conditions, and has developed as an important alternative to conventional metal‐catalyzed carbene‐transfer reactions. One of the major limitations lies within the rapidly occurring side reaction of the carbene intermediate with remaining diazoalkane molecules that result in the use of an excess of the reaction partner and thus impacts on the reaction efficiency. Herein, we describe a protocol that takes advantage of the in situ generation of donor–acceptor diazoalkanes by Bamford–Stevens reaction. Following this strategy, the concentration of the diazoalkane reaction partner can be minimized to reduce unwanted side reactions and to now conduct photochemical carbene transfer reactions under stoichiometric reaction conditions. We have explored this approach in the C−H and N−H functionalization and cyclopropanation reaction of N‐heterocycles and could demonstrate the applicability of this method in 51 examples.
Keywords: carbenes, C−H functionalization, cyclopropanation, N−H functionalization, photochemistry
Cyclopropanation: Bamford–Stevens reactions in the presence of blue light open up a pathway to overcome typical limitations of photochemical carbene‐transfer reactions. Following this methodology, the C−H functionalization of indole heterocycles can be performed in high efficiency under stoichiometric conditions (see scheme).

The photolysis of diazomethane in ethereal solution was studied by Meerwein and co‐workers in 1942 and is probably one of the first examples for the direct, yet unselective, functionalization reaction of unreactive C−H bonds.1, 2 The limitations of this methodology lie within the high reactivity of the carbene intermediate and thus unselective downstream reaction, which led to the development of metal‐catalyzed carbene‐transfer reactions. The advances in this research have guided the development of highly efficient catalysts, based on precious metals, such as Rh, Ir, Pd, or Au, and today there several protocols that allow highly efficient and site‐selective C−H functionalization reactions.2, 3
In recent years, the visible‐light photolysis of diazoalkanes attracted the interest of organic synthetic chemists to conduct metal‐free carbene‐transfer reactions,4, 5, 6, 7, 8, 9 and first reports already accentuate its high potential in photochemical, metal‐free cycloaddition,6 rearrangement,6b, 7 olefination,8 X−H,6a, 6e, 9 and C−H functionalization reactions.6a Despite these advances, applications are mainly limited to the use of a large excess of one reaction partner due to rapidly occurring side reactions of the carbene intermediate with remaining unreacted diazoalkane molecules, for example, in diazine (3) formation (Scheme 1).
Scheme 1.
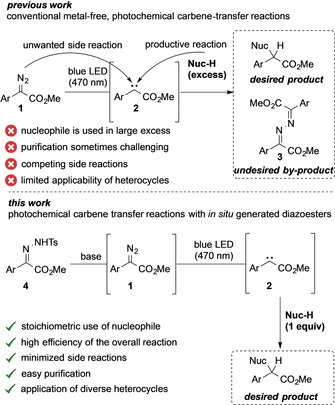
Photochemical carbene‐transfer reactions: conventional vs. in situ approach.
Stoichiometric photochemical carbene‐transfer reactions are highly attractive and this approach should allow highly atom‐economic transformations under mild reaction conditions. To address this challenge, we sought to study the in situ generation of diazoalkanes from bench‐stable precursors. For this purpose, we decided to study the reaction of tosyl hydrazones (4) under basic reaction conditions,10 while illuminating the reaction mixture with visible light at ambient temperature. This approach should allow the in situ generation of diazoalkanes,11 which should undergo carbene‐transfer reactions under photochemical conditions with significantly improved side‐reaction profile compared to the direct photolysis of diazoalkanes. Against this background, we have conducted a systematic study on photochemical carbene‐transfer reactions by using an in situ protocol for the generation of the diazoalkane. Following this approach, we now open up a new pathway towards carbene transfer reactions under stoichiometric reaction conditions.
We initiated our investigations by studying the Bamford–Stevens reaction of tosyl hydrazone (4 a) with and without the addition of N‐methyl indole (5) as a nucleophile and both in the dark and under irradiation with visible light at 470 nm. When subjecting tosyl hydrazone 4 a to basic conditions in dichloromethane solvent at room temperature, we could indeed observe the formation of the desired methyl phenyldiazoacetate 1 (Scheme 2 a). When performing this reaction under irradiation with blue LEDs under otherwise identical conditions, methyl phenyldiazoacetate 1 was only formed in small amounts (<10 %) and the decomposition of the diazoester to the diazine 3 was observed (Scheme 2 b).
Scheme 2.

a) Reaction of tosyl hydrazone 4 a and Cs2CO3 (1.5 equiv) in the dark; b) Reaction of tosyl hydrazone 4 a and Cs2CO3 (1.5 equiv) under irradiation with 470 nm LEDs (3 W); c) Reaction of tosyl hydrazone 4 a and Cs2CO3 (1.5 equiv) in the presence of N‐methyl indole (5, 1.0 equiv) under irradiation with 470 nm LEDs (3 W); Color coding: orange tosyl hydrazone 4 a, blue methyl phenyldiazoacetate 1, grey diazine 3, brown N‐methyl indole 5, green C−H functionalization product 6 a.
Next, we performed this experiment in the presence of N‐methyl indole 5. We could observe that tosyl hydrazone 4 a is rapidly consumed and the desired C−H functionalization product 6 a is formed at the same time. Notably, only small amounts of diazoester 1 could be observed by 1H NMR spectroscopy of the reaction mixture (<7 %) and as a consequence the carbene can only undergo minimal side reactions (Scheme 2 c). In the absence of light, no C−H functionalization reaction was observed under otherwise identical conditions (Table 1, entry 6). This data indicates that the C−H functionalization of indole heterocycles only takes place in the presence of light via a carbene intermediate.
Table 1.
Reaction optimization.
|
| ||||
|---|---|---|---|---|
|
Entry[a] |
R |
Base |
Solvent |
Yield [%] |
|
1 |
CO2Me |
Cs2CO3 |
DCM |
86 |
|
2 |
CO2Me |
K2CO3 |
DCM |
31 |
|
3 |
CO2Me |
KOtBu |
DCM |
36 |
|
4 |
CO2Me |
NaOtBu |
DCM |
59 |
|
5 |
CO2Me |
NEt3 |
DCM |
49 |
|
6[b] |
CO2Me |
Cs2CO3 |
DCM |
no rct. |
|
7[c] |
methyl phenyldiazoacetate (1) |
DCM |
38 |
|
|
8[d] |
CO2Me |
Cs2CO3 |
DCM |
66 |

[a] Tosyl hydrazone 4 a (0.2 mmol, 1.0 equiv) and Cs2CO3 (0.3 mmol, 1.5 equiv) were suspended in 1.0 mL of DCM. Then indole (0.2 mmol, 1.0 equiv) and another 1.0 mL of DCM were added to the reaction mixture and irradiated with one 3 W LED for 5–6 hours. [b] Reaction in the dark. [c] Reaction with methyl phenyldiazoacetate (1.0 equiv) instead of 4 a and N‐methyl indole 2 (1.0 equiv). [d] Reaction with 4 a (1.0 g, 3 mmol, 1.0 equiv), 5 (3 mmol, 1.0 equiv), Cs2CO3 (4.5 mmol, 1.5 equiv) in 10 mL DCM using 50 W blue LED.
In further experiments, we investigated different solvents, reaction times, and bases in this transformation to further optimize the C−H functionalization reaction of indole.12 However, in the case of tertiary amines or alkoxide bases, only significantly reduced reaction yields were observed, which might be reasoned by the high basicity and rapidly occurring Bamford–Stevens reaction (Table 1). Among all other bases and solvents tested, the combination of Cs2CO3 and dichloromethane (DCM) solvent proved to be optimal and the C−H functionalization product 6 a was obtained in 86 % isolated yield under stoichiometric reaction conditions. Importantly, when performing the reaction of methyl phenyldiazoacetate and N‐methyl indole under stoichiometric conditions, the desired product was obtained only in significantly decreased yield (Table 1, entry 7). We also studied the application of this methodology in gram scale, and could obtain the desired C−H functionalization product in 66 % yield without further optimization (Table 1, entry 8).
Next, we embarked on the application of this method in the C−H functionalization of N‐heterocycles. For this purpose, we studied different N‐protected indole derivatives (5) and tosyl hydrazones (4) under the optimized reaction conditions. Different substitution patterns around the indole heterocycle were well tolerated and the C−H functionalization products were obtained in high isolated yield. Notably, even indole heterocycles bearing strongly electron‐withdrawing substituents, such as a nitrile or an ester group, reacted under the present reaction conditions to the corresponding C−H functionalization products in good yield (Scheme 3, 6 d and e). When comparing to currently available protocols that rely on metal‐catalyzed carbene‐transfer reactions, electron‐deficient indole heterocycle often react under either reduced yield or have not been reported at all.13 Quite unexpected, even the substitution of the indole heterocycle in the fourth position was tolerated, and the C−H functionalization reaction proceeded in good yield (Scheme 3, 6 g). Next, different N‐substituted indole derivatives were studied, which gave the desired C−H functionalization product in good isolated yield. A range of functional groups, including esters, nitriles, trifluoromethoxy, olefins, or halogens, are tolerated under the present reaction conditions and in all cases the desired C−H functionalization product was obtained in good‐to‐high isolated yield (Scheme 3, 6 i–w).
Scheme 3.

Investigation of the substrate scope with N‐protected indole heterocycles.
In further investigations, we studied different tosyl hydrazones under the present reaction conditions. Ester‐substituted hydrazones readily underwent C−H functionalization reaction with N‐methyl indole in good yield (Scheme 3, 6 x–ad). Importantly, the present method is particularly suited to conduct carbene‐transfer reactions with electron‐rich aryldiazoacetates (Scheme 3, 6 z and aa), which are challenging substrates in conventional photochemical carbene‐transfer reactions due to the electron‐donating nature of the aryl substituent. Halogen‐ or trifluoromethoxy‐substituted tosyl hydrazones react similarly and the product of C−H functionalization can be obtained in moderate to good yield (Scheme 3, 6 ab–ad). The limitation of the present methodology lies within the use of electron‐poor diazoacetates, because the required ketoesters can only be prepared by multistep synthesis. Notably, when using trifluoromethyl‐ (Scheme 3, 7) or donor‐substituted tosyl hydrazones (Scheme 3, 8 a and b) the C−H functionalization reaction did not occur.
We next focused on the application of different N‐heterocycles, which underwent smooth C−H functionalization by using this in situ generation protocol. Control experiments with methyl phenyldiazoacetate revealed only diminutive amounts of the C−H functionalization product (up to 13 % yield). Noteworthy, under the present reaction conditions, the photochemical approach towards C−H functionalization of 7‐azaindole heterocycles is superior over currently available methods using specialized RhII catalyst, excess amount of the expensive heterocycle and continuous‐addition techniques.14 Similarly, the direct C−H functionalization of indazole heterocylcles still remains a challenge in organic synthesis.15 When switching to the electron‐rich N‐methyl pyrrole the product of C−H functionalization of the C2−H bond was obtained in moderate yield (Scheme 4).
Scheme 4.

C−H functionalization of different N‐heterocycles under stoichiometric conditions.
Encouraged by the above‐described observations on efficient C−H functionalization reactions of indole heterocycles, we became intrigued in studying the N−H functionalization of unprotected N‐heterocycles, such as indole and carbazole.16 For this purpose, we studied the reaction of indole and 3‐methyl indole with tosyl hydrazone 4 a under the optimized, stoichiometric reaction conditions, yet the desired N−H functionalization product was obtained in only low yield. When switching to carbazole heterocycles, the desired N−H functionalization products could be obtained in good isolated yield. Similarly, tetrahydroquinoline or thienopiperidine reacted smoothly to the desired N−H functionalization product. The reaction with the latter provides a direct access to biologically important molecules and enables the direct synthesis of an analogue of clopidogrel in good isolated yield. In comparison to our previously reported method for N−H functionalization reactions of carbazole heterocycles,9 the present method now allows the use of secondary amines and stoichiometric reaction conditions and thus improves on the reaction efficiency (Scheme 5).
Scheme 5.

N−H functionalization under stoichiometric conditions.
In further studies, we embarked on studying cyclopropanation reactions of heterocycles. For this purpose, we investigated the influence of the N‐protecting group of indole heterocycles in the reaction with tosyl hydrazone 4 a under stoichiometric reaction conditions. Both a Boc and a pivaloyl protecting were tolerated under the present conditions and the desired cyclopropanation products were obtained in good yield. Boc‐protected indole gave the desired cyclopropanation product (15) only as a 1:1 mixture of both diastereoisomers. Contrarily, the pivaloyl protected indole and derivatives thereof reacted in a highly diastereoselective reaction to the corresponding cyclopropanes (16 a–d). It is important to note that the yields of the in situ protocol compare well with the yields using light mediated cyclopropanation reactions of protected indole heterocycles with aryldiazoacetates; however, the in situ protocol now allows a stoichiometric use of the important heterocyclic reaction partner. Under the present reaction conditions, sulfur‐containing heterocycles react selectively under cyclopropanation reaction, and no product arising from C−H functionalization was observed. The desired cyclopropanes 17 and 18 were obtained in moderate yield, which is comparable to present synthesis methods using expensive AuI catalysts (Scheme 6).17
Scheme 6.

Cyclopropanation of heterocycles under stoichiometric conditions.
In summary, we could demonstrate that photochemical carbene‐transfer reactions can now be conducted under stoichiometric reaction conditions by using a protocol that provides an in situ access to aryldiazoacetates from tosyl hydrazones under Bamford–Stevens conditions at ambient temperature. The in situ generation of diazoesters is pivotal to reduce the concentration of the diazoester in the reaction solution and to thus reduce unwanted side reactions. This strategy now allows efficient metal‐free C−H and N−H functionalization and cyclopropanation reactions of heterocycles under mild and operationally simple reaction conditions and avoids the use of toxic diazoalkane reaction partners.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—project no. 408033718.
S. Jana, F. Li, C. Empel, D. Verspeek, P. Aseeva, R. M. Koenigs, Chem. Eur. J. 2020, 26, 2586.
Contributor Information
Sripati Jana, http://www.koenigslab.rwth-aachen.de.
Prof. Dr. Rene M. Koenigs, Email: rene.koenigs@rwth-aachen.de.
References
- 1. Meerwein H., Rathjen H., Werner H., Ber. Dtsch. Chem. Ges. A/B 1942, 75, 1610–1622. [Google Scholar]
- 2.Selected review articles on the functionalization of C−H bonds:
- 2a. Girard S. A., Knauber T., Li C.-J., Angew. Chem. Int. Ed. 2014, 53, 74–100; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 76–103; [Google Scholar]
- 2b. Li B., Dixneuf P. H., Chem. Soc. Rev. 2013, 42, 5744–5767; [DOI] [PubMed] [Google Scholar]
- 2c. Gensch T., Hopkinson M. N., Glorius F., Wencel-Delord J., Chem. Soc. Rev. 2016, 45, 2900–2936; [DOI] [PubMed] [Google Scholar]
- 2d. Moselage M., Lie J., Ackermann L., ACS Catal. 2016, 6, 498–525; [Google Scholar]
- 2e. Zhu S.-F., Zhou Q.-L., Natl. Sci. Rev. 2014, 1, 580–603; [Google Scholar]
- 2f. Shang R., Ilies L., Nakamura E., Chem. Rev. 2017, 117, 9086–9139. [DOI] [PubMed] [Google Scholar]
- 3.Selected articles on precious metal-catalyzed C−H functionalization reactions:
- 3a. Davies H. M. L., Manning J. R., Nature 2008, 451, 417–424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Ford A., Miel H., Ring A., Slattery C. N., Maguire A. R., McKervey M. A., Chem. Rev. 2015, 115, 9981–10080; [DOI] [PubMed] [Google Scholar]
- 3c. Doyle M. P., Duffy R., Ratnikov M., Zhou Z., Chem. Rev. 2010, 110, 704–724; [DOI] [PubMed] [Google Scholar]
- 3d. Xia Y., Qiu D., Wang J., Chem. Rev. 2017, 117, 13810–13889. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Ciszewski L. W., Rybicka-Jasinska K., Gryko D., Org. Biomol. Chem. 2019, 17, 432–448; [DOI] [PubMed] [Google Scholar]
- 4b. Candeias N. R., Afonso C. A. M., Curr. Org. Chem. 2009, 13, 763–787; [Google Scholar]
- 4c. Galkina O. S., Rodina L. L., Russ. Chem. Rev. 2016, 85, 537–555; [Google Scholar]
- 4d. Empel C., Koenigs R. M., Synlett 2019, 30, 1929–1934. [Google Scholar]
- 5.Selected references:
- 5a. Wang Z., Herraiz A. G., del Hoyo A. M., Suero M. G., Nature 2018, 554, 86–91; [DOI] [PubMed] [Google Scholar]
- 5b. Mimieux Vaske Y. S., Mahoney M. E., Konopelski J. P., Rogow D. L., McDonald W. J., J. Am. Chem. Soc. 2010, 132, 11379–11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Jurberg I., Davies H. M. L., Chem. Sci. 2018, 9, 5112–5118; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b. Hommelsheim R., Guo Y., Yang Z., Empel C., Koenigs R. M., Angew. Chem. Int. Ed. 2019, 58, 1203–1207; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1216–1220; [Google Scholar]
- 6c. He F., Koenigs R. M., Chem. Commun. 2019, 55, 4881–4884; [DOI] [PubMed] [Google Scholar]
- 6d. Zhang X., Du C., Zhang H., Li X.-C., Wang Y.-L., Niu J.-L., Song M.-P., Synthesis 2019, 51, 889–898; [Google Scholar]
- 6e. Zhang Z., Yadagiri D., Gevorgyan V., Chem. Sci. 2019, 10, 8399–8404; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6f. Guo Y., Nguyen T. V., Koenigs R. M., Org. Lett. 2019, 21, 8814–8818; [DOI] [PubMed] [Google Scholar]
- 6g. Chidley T., Jameel I., Rizwan S., Peixoto P. A., Pouysegu L., Quideau S., Williams W. S., Murphy G. K., Angew. Chem. Int. Ed. 2019, 58, 16959–16965; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 17115–17121. [Google Scholar]
- 7.
- 7a. Jana S., Koenigs R. M., Asian J. Org. Chem. 2019, 8, 683–686; [Google Scholar]
- 7b. Yang Z., Guo Y., Koenigs R. M., Chem. Eur. J. 2019, 25, 6703–6706; [DOI] [PubMed] [Google Scholar]
- 7c. Yang J., Wang J., Huang H., Qin G., Jiang Y., Xiao T., Org. Lett. 2019, 21, 2654–2657; [DOI] [PubMed] [Google Scholar]
- 7d. Yang Z., Guo Y., Koenigs R. M., Chem. Commun. 2019, 55, 8410–8413; [DOI] [PubMed] [Google Scholar]
- 7e. Jana S., Yang Z., Pei C., Xu X., Koenigs R. M., Chem. Sci. 2019, 10, 10129–10134; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7f. He F., Pei C., Koenigs R. M., Chem. Commun. 2020, 56, 599–602. [DOI] [PubMed] [Google Scholar]
- 8. Xiao T., Mei M., He Y., Zhou L., Chem. Commun. 2018, 54, 8865–8868. [DOI] [PubMed] [Google Scholar]
- 9. Empel C., Patureau F. W., Koenigs R. M., J. Org. Chem. 2019, 84, 11316–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selected references:
- 10a. Fulton J. R., Aggarwal V. K., de Vicente J., Eur. J. Org. Chem. 2005, 1479–1492; [Google Scholar]
- 10b. Xia Y., Wang J., Chem. Soc. Rev. 2017, 46, 2306–2362; [DOI] [PubMed] [Google Scholar]
- 10c. Jia M., Ma S., Angew. Chem. Int. Ed. 2016, 55, 9134–9166; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9280–9313; [Google Scholar]
- 10d. Sun C.-L., Shi Z.-J., Chem. Rev. 2014, 114, 9219–9280; [DOI] [PubMed] [Google Scholar]
- 10e. Shao Z., Zhang H., Chem. Soc. Rev. 2012, 41, 560–572; [DOI] [PubMed] [Google Scholar]
- 10f. Barluenga J., Valdes C., Angew. Chem. Int. Ed. 2011, 50, 7486–7500; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7626–7640. [Google Scholar]
- 11.Selected references:
- 11a. Mykhailiuk P. K., Koenigs R. M., Chem. Eur. J. 2019, 26, 89–101;31415714 [Google Scholar]
- 11b. Morandi B., Carreira E. M., Angew. Chem. Int. Ed. 2010, 49, 938–941; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 950–953; [Google Scholar]
- 11c. Hock K. J., Spitzner R., Koenigs R. M., Green Chem. 2017, 19, 905–909; [Google Scholar]
- 11d. Hock K. J., Mertens L., Hommelsheim R., Spitzner R., Koenigs R. M., Chem. Commun. 2017, 53, 6577–6580; [DOI] [PubMed] [Google Scholar]
- 11e. Mykhailiuk P. K., Eur. J. Org. Chem. 2015, 7235–7239; [Google Scholar]
- 11f. Mykhailiuk P. K., Org. Biomol. Chem. 2017, 15, 7296–7301; [DOI] [PubMed] [Google Scholar]
- 11g. Mykhailiuk P. K., Angew. Chem. Int. Ed. 2015, 54, 6558–6561; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6658–6661. [Google Scholar]
- 12.For details, see the Supporting Information.
- 13.Selected articles:
- 13a. Delgado-Rebollo M., Prieto A., Pérez P. J., ChemCatChem 2014, 6, 2047–2052; [Google Scholar]
- 13b. Özüduru G., Schubach T., Boyson M. M., Org. Lett. 2012, 14, 4990–4993; [DOI] [PubMed] [Google Scholar]
- 13c. Gnad F., Poleschak M., Reiser O., Tetrahedron Lett. 2004, 45, 4277–4280; [Google Scholar]
- 13d. Cai Y., Zhu S.-F., Wang G.-P., Zhou Q.-L., Adv. Synth. Catal. 2011, 353, 2939–2944. [Google Scholar]
- 14. Sarkar M., Daw P., Ghatak T., Bera J. K., Chem. Eur. J. 2014, 20, 16537–16549. [DOI] [PubMed] [Google Scholar]
- 15. Hock K. J., Knorrscheidt A., Hommelsheim R., Ho J., Weissenborn M. J., Koenigs R. M., Angew. Chem. Int. Ed. 2019, 58, 3630–3634; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 3669–3673. [Google Scholar]
- 16. Arredondo V., Hiew S. C., Gutman E. S., Premachandra I. D. U. A., van Vranken D., Angew. Chem. Int. Ed. 2017, 56, 4156–4159; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4220–4223. [Google Scholar]
- 17. Xu G., Liu K., Sun J., Org. Lett. 2018, 20, 72–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary