
Fig. 3. Structure of YGTG.
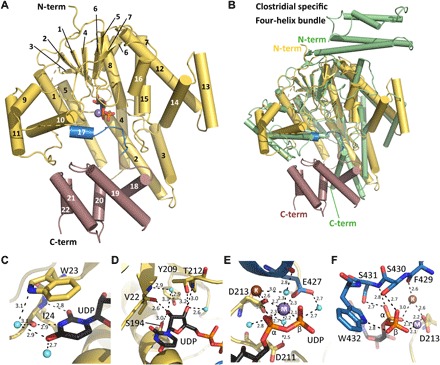
(A) The YGTG structure displays a typical GT-A glycosyltransferase fold (yellow) comprising a seven-stranded β sheet surrounded by α helices and a C-terminal five α-helical bundle (violet). UDP (black sticks) and Mn2+ (purple sphere) are bound centrally. Helix 17 and the loop that encloses the ligand binding site are highlighted in blue. (B) Structural superposition of YGTG [colored as in (A)] with TcdB from C. difficile [Protein Data Bank (PDB) ID: 2BVL, green] showing the GT-A glycosyltransferase fold. While the N-terminal four-helix bundle of TcdB is absent in YGTG, four helices at the C terminus of YGTG are not conserved in clostridial toxins. (C to F) Closeup views of the active site showing the binding mode of UDP (black) with Mn2+ and K+ ions represented as spheres (purple and brown, respectively) and YGTG represented as a cartoon with relevant residues in sticks and colored as in (A). (C) The uridine base moiety is coordinated by a π-stacking interaction with Trp23 and by several direct and water-mediated hydrogen bonds (dashed lines). (D) The ribose moiety is coordinated by a series of hydrogen bonds with its hydroxyl groups. (E) The diphosphate from UDP is stabilized via interactions with the Mn2+ ion. Mn2+ is coordinated with aspartate side chains of the DXD motif, Glu427, and water molecules. Note the presence of a K+ ion in close vicinity. (F) The C-terminal loop of the GT-A fold encloses the nucleotide-binding pocket comprising direct interactions between the diphosphate moiety and Ser430, Ser431, and Trp432.