

Abstract
Over the past few decades, atherogenic dyslipidaemia has become one of the most common phenotypic presentations of lipid abnormalities, being strongly and unequivocally associated with an increased risk of cardiovascular (CV) disease. Despite the excellent results achieved from statin and non-statin management of LDL cholesterol and CV events prevention, there still remains a significant residual risk, associated with the prevalence of non-LDL cholesterol lipid patterns characterised by elevated triglyceride levels, low HDL cholesterol, a preponderance of small and dense LDL particles, accumulation of remnant lipoproteins and postprandial hyperlipidaemia. These qualitative and quantitative lipid modifications are largely associated with insulin resistance, type 2 diabetes and obesity, the prevalence of which has grown to epidemic proportions throughout the world. In this review, we analyse the pathophysiology of this particular dyslipidaemia, its relationship with the development of atherosclerotic CV disease and, finally, briefly describe the therapeutic approaches, including changes in lifestyle and current pharmacological interventions to manage these lipid alterations aimed at preventing CV events.
Keywords: Apolipoprotein B, atherogenic dyslipidaemia, atherosclerosis, ezetimibe, fibrate, lipoprotein, omega-3 fatty acid, proprotein convertase subtilisin/kexin type 9, statin, triglycerides
We are witnessing an epidemic global increase in the prevalence of obesity and its clinical consequences (e.g. insulin resistance and diabetes). This epidemic has been potentiated and sustained by the widespread adoption of unhealthy lifestyles in broad swathes of the population and is characterised by a sedentary lifestyle and an imbalance between the type and characteristics of nutrition, dominated by an excess of calorie intake. Its effects have come to offset the decline in cardiovascular (CV) mortality achieved in recent years as a result of marked therapeutic advances.[1,2]
The prevalence of atherogenic dyslipidaemia (AD) has increased considerably. AD is characterised by the coexistence of profound qualitative and quantitative modifications in lipid metabolism. The excess of non-LDL particles is a distinctive feature, with an increase in triglyceride (TG)-rich lipoproteins (TRL), low HDL cholesterol levels, accumulation of lipoprotein remnants (i.e. small very LDL [VLDL] and intermediate-density lipoprotein [IDL]), a preponderance of numerous small and dense (sd) LDL particles and postprandial hyperlipidaemia.[3–5]
The management of AD requires therapeutic lifestyle modification coupled with pharmacological intervention aimed at reducing CV risk. Because of the range of cardiometabolic alterations these patients present with, trying to attenuate their CV risk can pose formidable management issues.[6,7]
Atherogenic Dyslipidaemia and its Relationship with Atherosclerotic Cardiovascular Disease
The increasing prevalence of obesity is directly associated with the increase in type 2 diabetes (T2D) and metabolic syndrome (MS), which, in turn, are associated with lipoprotein abnormalities described as AD. AD is causally linked to the development and progression of atherosclerotic CV disease (ASCVD).[8,9] The relationship between AD and ASCVD is supported by prospective longitudinal cohorts, clinical evidence and genetic linkage studies. As an example, the best predictor of risk of MI at the population level in the INTERHEART study was the apolipoprotein (apo) B100/apoA-I ratio, reflecting the correlation between all apoB (atherogenic lipoproteins) and HDL (representing the classically anti-atherogenic particles).[10] In addition, a huge registry of almost 140,000 patients hospitalised in the US due to acute coronary syndromes (ACS) showed that more than half had LDL cholesterol levels <2.59 mmol/l, whereas mean HDL cholesterol and TG values were <1.03 and >1.81 mmol/l, respectively.[11,12] In these patients the LDL cholesterol level was not reflecting the real burden of atherogenic lipoproteins; this was more aptly quantified by non-HDL cholesterol (total cholesterol minus HDL cholesterol), a better predictor of CV risk in these individuals.
The evidence goes beyond epidemiological studies. The relationship between AD and ASCVD has also been demonstrated in prospective randomised clinical trials using statins. Even when treated with statins, patients with the AD phenotype have a higher risk of CV events than those without AD.[13,14]
The Pravastatin or Atorvastatin Evaluation and Infection Therapy – Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial showed that among patients receiving high-intensity statins after an ACS, those with TG <1.69 mmol/l (adjusted by HDL cholesterol and LDL cholesterol levels) had a lower risk of coronary events (HR 0.80; 95% CI [0.66–0.97]; p=0.025) than those with TG exceeding this threshold.[15]
Similarly, in the Incremental Decrease in End Points through Aggressive Lipid Lowering (IDEAL) and Treating to New Targets (TNT) studies, even in patients who reached LDL cholesterol <1.81 mmol/l, the risk increased 63% (p<0.001) when comparing the highest quintile of TG levels with the lowest one.[16] Long-term (>20 years) follow-up of the Bezafibrate Infarction Prevention (BIP) study showed a significant association between elevated TG and all-cause mortality.[17] In addition, in a meta-analysis of prospective studies in patients treated with statins, increased TG concentrations were independently correlated with coronary disease and predicted recurrent ischaemic events in patients with a history of ACS treated with statins.[11,18,19]
Some studies have shown that the association between plasma TG concentrations and CV risk is attenuated when adjusted for other lipid parameters. In a meta-analysis by the Emerging Risk Factors Collaboration, which included data from 302,430 individuals, there was a significant association between TG concentrations and CV risk, but this association was attenuated after adjusting for HDL cholesterol and non-HDL cholesterol.[20] In some respects, the case can be made that because non-HDL cholesterol includes lipoproteins that can carry TGs, this likely represents an ‘over-adjustment’ of two highly inter-related risk factor covariates. However, in a more recent analysis, among approximately 46,000 high-risk patients on statin therapy whose LDL cholesterol was well controlled, TG >169 mmol/l was independently and significantly correlated with CV events even after adjusting for LDL cholesterol, non-HDL cholesterol and HDL cholesterol.[21]
TRL particles (precursors of LDL, including small VLDL and IDL) can be estimated in clinical practice as total cholesterol minus LDL cholesterol minus HDL cholesterol. TRLs are associated with increased CV risk.[22,23] Varbo et al. showed that each 1 mmol/l increase in TRLs is associated with a 2.8-fold increase in CV risk independent of the HDL cholesterol level.[24] Directly measured TRLs were correlated with an increased risk for ASCVD events in both the Framingham Heart Study and the Jackson Heart Study.[25] TRLs are also potently proinflammatory, which likely contributes to their overall atherogenic profile.[26] Highlighting the importance of TRLs, postprandial TG concentrations are a stronger predictor of CV events than fasting TG levels. Although most individuals are in a postprandial state during most hours of the day, changes in postprandial TG levels can have a significant effect on the development of atherosclerosis.[27–29]
In summary, there is convincing evidence that AD is highly atherogenic, although the true significance of each component in this context is incompletely characterised and understood. Human genetic evidence suggests that TRLs contribute causally to the development of ASCVD.[12] Gene variants leading to higher levels of plasma apoB-containing lipoproteins, including TRL, consistently increase ASCVD risk.[30]
Lipoprotein lipase (LPL) plays a critical role in the disposal of TGs carried with chylomicrons and apoB100-containing lipoproteins. LPL is tethered to vascular endothelial cells via glycosylphosphatidylinositol-anchored HDL-binding protein 1 and hydrolyses TGs within the core of TRLs (Figure 1).
LPL activity can be regulated by changes in nuclear expression of the gene for LPL, but it is also responsive to a variety of effector molecules. LPL is inhibited by apoCIII and activated by apoCII and apoA-V.[31] Angiopoietin-like protein (ANGPTL) 3 and ANGPTL4 exert inhibitory effects via distinct mechanisms. ANGPTL3 stimulates cleavage of LPL from GP1HBP by proprotein convertase subtilisin/kexin types 3 and 6, rendering it inactive.[32] LPL monomers are catalytically inactive; the active form is an LPL dimer. In contrast, ANGPTL4 competitively inhibits LPL by inducing the dissociation of its constituent dimers.[33] The severity of diabetes can also affect the dimeric integrity of this enzyme.[34]
The genetics of LPL and the effector molecules that regulate its activity support the conclusion that TGs are an independent risk factor for ASCVD. Loss-of-function mutations in apoCIII result in lower mean serum TG levels than in patients who express normal levels of this enzyme that are correlated with significant reductions in ASCVD risk.[35,36] Similarly, a variety of genetic polymorphisms giving rise to reduced activity of ANGPTL3 and ANGPTL4 result in lower mean TG levels and lower risk for ASCVD compared with wild-type controls.[37–39] Patients with loss-of-function mutations in apoA-V have higher TG concentrations and augmented risk for ASCVD and ischaemic stroke.[40,41] Consistent with these changes, gain-of-function mutations and loss-of-function mutations in LPL are correlated with lower TGs/lower ASCVD risk and higher TGs/higher ASCVD risk, respectively.[35]
Epidemiological data associate low HDL cholesterol with heightened ASCVD risk, although a more recent analysis has questioned this.[19,42–44] It was long believed that treating low serum HDL cholesterol concentrations would reduce residual risk. However, clinical outcomes trials targeting low HDL cholesterol with different pharmacological interventions failed to reduce CV endpoints, and, similarly, genetic studies do not support a protective role of HDL cholesterol in humans.[45–49] Together, these findings imply that HDL cholesterol may be considered a metabolic marker of increased CV risk rather than a therapeutic target. It will be some time before the HDL proteome and lipidome are understood well enough to tailor therapeutic interventions that affect ASCVD risk.[50,51]
The Study to Investigate CSL112 in Subjects With Acute Coronary Syndrome (AEGIS-II; NCT03473223) is a large Phase III trial testing the capacity of an infusible human apoAI preparation to reduce the risk of CV events in patients with a history of ACS. The trial will also evaluate the efficacy of CSL112 (apoA-I [human]) in inducing atherosclerotic plaque regression by promoting reverse cholesterol transport.
Abnormalities in Lipid Metabolism: The Perfect Storm
The aforementioned phenotypic characteristics of AD reflect an abnormal metabolism of TRL, conditioned by both genetic and acquired factors that also affect HDL and LDL particles.[52] In the setting of insulin resistance, insulin has reduced capacity to inhibit hormone-sensitive lipase in adipose tissue. This leads to a constitutive release of fatty acid from visceral adipose tissue stores. The liver can dispose of this fatty acid in multiple ways:
Figure 1: Regulation of Triglyceride-Enriched Lipoprotein Lipolysis.
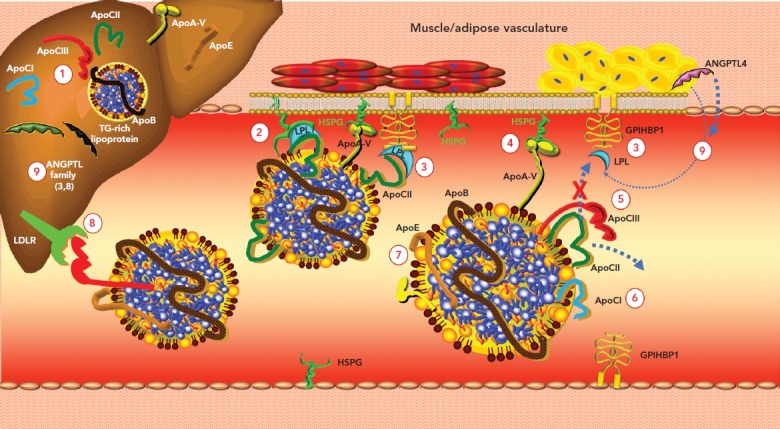
1. Apolipoprotein (apo) CIII stimulates hepatic triglyceride (TG)-rich lipoprotein production. 2. ApoCII is an essential cofactor and activator of lipoprotein lipase (LPL), which is anchored to heparan sulphate proteoglycan (HSPG) and glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1). These receptors are crucial for maintaining LPL activity. 3. GPIHBP1 seizes LPL in the interstitial space and shuttles it across endothelial cells to the luminal surface; it also tethers LPL to the capillary endothelium. 4. ApoA-V binds to HSPG and activates LPL. 5. ApoCIII inhibits LPL-mediated lipolysis by both displacing the LPL activator apoCII from the lipoprotein surface and blocking the interaction of apoCII with LPL (steric hindrance). 6. ApoCI inhibits LPL activity by preventing binding of LPL to TG-rich lipoproteins and by rendering LPL more susceptible to inhibition by angiopoietin-like protein (ANGPTL) 4. 7. ApoE2 can interfere with LPL. 8. ApoCIII inhibits receptor-mediated hepatic clearance of TG-rich lipoproteins. 9. ANGPTL3, ANGPTL4 and ANGPTL8 are members of a family of secreted proteins that inhibit LPL. Source: Sathiyakumar et al. 2018.[155] Reproduced with permission from Elsevier.
It can be oxidised in the mitochondrial matrix.
It can be reassimilated into TG and secreted in VLDL particles.
Some can be shunted toward gluconeogenesis by activation of phosphoenolpyruvate carboxykinase, which will exacerbate the hyperglycaemia of insulin resistance.
If all pathways become saturated, excess TG will be stored in the liver and manifest as hepatic steatosis.
In addition to the overproduction and increased hepatic secretion of VLDLs, there is reduced capacity to lipolyse TRLs because of decreased LPL activity. Insulin resistance can reduce nuclear expression of LPL, leading to increased production of apoCIII and decreased production of apoCII.[53] Secondary to both hepatic overproduction of TRL and their reduced catabolism, there is a considerable increase in TRLs in the serum.
As TRLs accumulate in serum, the activity of cholesterol ester transfer protein (CETP) increases. CETP catalyses the neutral lipid of exchange of TG out of TRLs for cholesterol from both HDLs and LDLs (Figure 2).[54] As the HDLs and LDLs become more enriched with TG, they become better substrates for lipolysis by hepatic lipase. Hepatic lipase catabolises HDL and promotes the wasting of apoA-I by the kidney. Hepatic lipase also converts large, buoyant LDLs into smaller (sdLDL) and more numerous ones, rendering the LDL fraction more atherogenic.[55] HDL cholesterol levels decrease via other mechanisms as well. There is an insulin response element in the gene for apoA-I, the primary apolipoprotein constituent of HDL particles.[56,57] As the liver becomes more insulin resistant, less apoA-I is produced and there is less HDL biogenesis. Adipocytes express the ATP-binding membrane cassette transport protein A1 (ABCA1). Insulin resistance downregulates expression of ABCA1 on the surface of adipocytes and reduces HDL formation by these cells.[58–60] Chylomicrons are enriched with apoA-I. Insulin resistance reduces the release of this apoA-I in serum by inhibiting LPL. In addition, within the milieu of insulin resistance or diabetes, HDL particle concentrations are not only quantitatively reduced, but also tend to be dysfunctional and thus are not able to perform their primary functions, including reverse cholesterol transport and inhibition of oxidative and inflammatory phenomena.[61]
Figure 2: Molecular Dynamics of Atherogenic Dyslipidaemia.
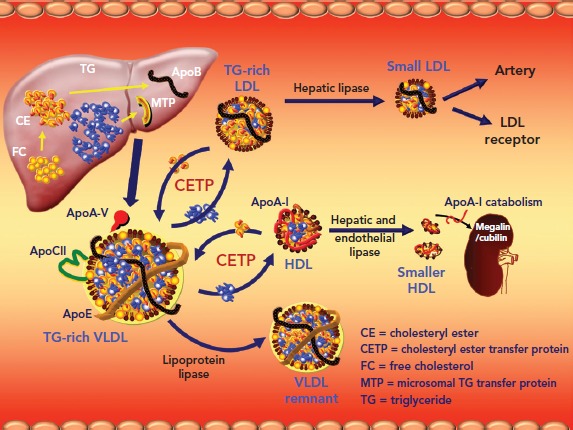
Hepatocytes produce and secrete triglyceride (TG)-rich VLDL. In the setting of insulin resistance with reduced lipoprotein lipase activity, cholesterol ester transfer protein is activated, which catalyses a 1:1 stoichiometric exchange of cholesterol from LDL and HDL particles for TGs from VLDLs of various sizes. As the LDL and HDL particles become more enriched with TG, they become better substrates for lipolysis by hepatic lipase. The result of hepatic lipase-driven lipolysis is to produce increased numbers of smaller, denser LDLs and catabolise HDL. As the HDL is catabolised, apolipoprotein (apo) A-I is released, bound to either cubulin or megalin in the renal ultrafiltrate, and eliminated via the urine.
The sdLDL particles are highly atherogenic.[62] There is accumulating evidence that smaller LDL particles are more atherogenic than larger, more buoyant ones. In addition, sdLDL:
Is more susceptible to oxidation (oxidised LDL is avidly scavenged by activated macrophages in the subendothelial space, giving rise to foam cell cells);[63]
Contains apoB100, which undergoes conformational alteration as the particle decreases in volume and size, resulting in lower affinity for, and clearance by, the LDL receptor;[64] and
Has increased affinity for proteoglycans in the subendothelial space, exacerbating lipoprotein retention.[5]
An increase in the hepatic content of TG also promotes an increase in the size of the TRL pool and augmentation of VLDL biosynthesis and secretion. All these observations help explain the extensive and more premature development of ASCVD in patients with insulin resistance and AD.[65–67]
In summary, changes in lipid metabolism in the setting of insulin resistance or diabetes are profound and include an excess of apoB, including remnants, HDL, which is decreased in quantity and function, and changes in LDL size and particle number. These metabolic changes, coupled with the pro-oxidative, proinflammatory and prothrombotic state of insulin resistance, predispose to the development of accelerated atherogenesis in ‘cardiometabolic’ patients.
Apolipoprotein B and Non-HDL Cholesterol as Risk Markers
ApoB measurement represents an estimation of all atherogenic lipoproteins, namely VLDL, IDL, LDL and lipoprotein(a) (Lp(a)), because they all contain a molecule of apoB100. In the same way, each chylomicron particle or its remnants contain a molecule of apoB48 (a truncated form of apoB100). In AD there is overproduction not only of VLDL, but also of apo B. In patients with cardiometabolic risk, the total number of sdLDL particles is increased, and hence apoB is elevated. Neither the sdLDL concentration nor the apoB level is reflected in LDL cholesterol measurements.[68] Among patients with insulin resistance, LDL cholesterol is frequently low or ‘normal’, despite increases in apoB. Non-HDL cholesterol is a simple and practical calculation (total cholesterol minus HDL cholesterol) that represents an estimation of the cholesterol concentration within all atherogenic lipoproteins. It is defined as a secondary treatment target in most international dyslipidaemia guidelines.[69–72] Using sophisticated techniques, the number of particles of each lipoprotein class and its subclasses can be quantified, but its application in daily clinical practice is an area of continuing investigation.[73]
Non-HDL cholesterol and apoB are better therapeutic targets in patients with AD.[74] In that sense, most guidelines established for patients at very high risk, include objectives of LDL cholesterol <1.81 mmol/l, non-HDL cholesterol <2.59 mmol/l and apoB <1.56 µmol/l, and in those at high risk of LDL cholesterol <2.59 mmol/l, non-HDL cholesterol <3.36 mmol/l and apoB <1.75 µmol/l. In addition, therapeutic effort should be made to reduce TG burden in serum through lifestyle modification and medication as indicated.[75]
Non-pharmacological Interventions: Therapeutic Changes in Lifestyle
Healthy Eating
Favourable therapeutic changes in lifestyle constitute the basic approach and the cornerstone of AD treatment. The greatest benefit is obtained with a reduction in saturated and trans-fats intake, along with an increase in consumption of mono- and polyunsaturated fats. It is essential to reduce the excess of carbohydrates in the diet, especially refined sugars.[76,77] The Mediterranean diet seems to be more effective than a low-fat diet; the Mediterranean diet has been shown to significantly reduce the total cholesterol:HDL cholesterol ratio and non-HDL cholesterol, and to reduce clinical events and CV mortality.[78,79]
Both low-fat and low-carbohydrate diets affect lipid levels. The low-fat diet has little effect in decreasing total cholesterol and LDL cholesterol, whereas the low-carbohydrate diet shows more favourable effects on TG and HDL cholesterol. In addition, the consumption of sea fish or omega-3 fatty acids has favourable effects.
In conclusion, lowering the dietary carbohydrate content or losing weight appears to attenuate AD, whereas reducing the total fat or saturated fat content has little effect.[80] Weight loss is associated with significant relief of insulin resistance.
Regular Physical Activity
The effects of physical activity on serum lipids have been widely studied. Regular aerobic exercise is associated with increased skeletal muscle and systemic tissue insulin sensitisation.[81,82] As this occurs, HDL cholesterol tends to increase and TGs and sdLDL decrease.[83,84] Regular physical activity is a key recommendation in the approach to AD.[85]
Unfortunately, despite the benefits, long-term adherence to lifestyle changes is often difficult to sustain over time.[86]
Current Pharmacological Interventions
Statins
Statins are first-line drugs in the treatment of AD. They comprise a pharmacological group that inhibit the rate-limiting step of cholesterol biosynthesis catalysed by 3¢-hydroxy-3¢-methylglutaryl coenzyme A. By decreasing intrahepatocyte concentrations of cholesterol, the statins activate the nuclear transcription factor sterol regulatory element binding protein-1c, which increases the cell surface expression of LDL receptors (LDLR). Thus, statins reduce circulating levels of LDL cholesterol by:
decreasing cholesterol biosynthesis and VLDL secretion; and
increasing the clearance of LDL particles from the circulation.
In addition, statins can reduce plasma TGs by 15–20% and increase HDL cholesterol by up to 15%.[87,88] However, the effect of statins on sdLDL has not been completely clarified.[89] A recent meta-analysis of six statin trials, including 802 subjects, demonstrated that statins significantly reduce circulating levels of apoCIII, which likely contributes to their modest TG-lowering capacity.[90] In addition to their lipid-lowering effects, the statins exert cholesterol-independent pleiotropic actions, which have been widely studied and contribute to the stabilisation of atherosclerotic plaques and reverse endothelial dysfunction, among other effects.[91] The beneficial effects of statins have been extensively demonstrated in both primary and secondary prevention studies because they incontrovertibly reduce risk for MI, ischemic stroke, revascularisation by both percutaneous transluminal coronary angioplasty and coronary artery bypass grafting, as well as cardiovascular and all-cause mortality.[92,93]
There is some concern that treatment of dyslipidaemia with statins increases the incidence of diabetes. As first reported by Ridker et al. in the Justification for the Use of statins in Prevention: an Intervention Trial Evaluating Rosuvastatin (JUPITER), rosuvastatin therapy is associated with an increased risk for new-onset diabetes.[94] A subsequent meta-analysis showed that statins increase the risk for diabetes by approximately 9%.[95] However, this does require some contextualisation. As shown in JUPITER, compared with placebo, statin therapy accelerates time to onset of diabetes by only 5.4 weeks and, for those with risk factors for diabetes, 134 vascular events or deaths were prevented for every 54 new cases of diabetes diagnosed.[96] Thus, the benefit:risk ratio of statin therapy is quite favourable.
Subsequent work showed that the greater the number of components of MS a patient has, the higher the risk for statin-induced diabetes.[97] In general, patients with MS have the highest risk for statin-accelerated diabetes. The mechanism(s) for this are as yet unknown. With low-dose statin therapy 1,000 patients have to be treated for 1 year in order to see one new case of diabetes; with moderate- to high-dose statin therapy, 500 patients have to be treated for 1 year to see one new case of diabetes.[98] Statin therapy should not be withheld out of concern that it may precipitate diabetes; it has been shown that diabetics derive every bit as much benefit from statin therapy as do non-diabetics.[99] Moreover, it has been suggested that patients with features of MS may derive particular benefit from statin therapy.[100]
Fibrates
Fibrates are weak agonists of the nuclear transcription factor peroxisome proliferator-activated receptor (PPAR) alpha and regulate the expression of genes that influence lipid metabolism. Fibrates were found to improve glucose homeostasis via activation of PPAR-alpha and increases in adiponectin levels.[101] Fibrates lower TGs by 25–50%, increase HDL cholesterol 5–30%, reduce LDL cholesterol up to 20% and decrease sdLDL particles, postprandial TGs and TRLs.[102,103]
In both the Helsinki Heart Study (primary prevention) and the Veterans Administration Low HDL Intervention Trial (secondary prevention), gemfibrozil significantly reduced risk of the primary composite endpoint.[104,105] However, because gemfibrozil reduces statin glucuronidation and can potentiate the risk of rhabdomyolysis, gemfibrozil should not be used in combination with a statin.[106,107] Fenofibrate is substantially safer than gemfibrozil when combined with a statin.[108] Fenofibrate therapy has been evaluated in two prospective randomised studies of patients with diabetes.
In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial, fenofibrate as monotherapy failed to reduce the primary composite endpoint, but did reduce the risk for MI, stroke, revascularisation and multiple microangiopathic endpoints, including retinopathy, nephropathy and neuropathy.[109] In a subgroup analysis of patients with TGs >2.26 mmol/l and HDL cholesterol <1.03 mmol/l, fenofibrate significantly reduced the primary composite endpoint by 27%. Although fenofibrate may increase serum creatinine concentrations, it has actually been shown to be nephroprotective and does not adversely affect renal function.[110,111] The Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial also failed to meet its primary endpoint in patients on statins randomised to either fenofibrate or placebo.[112] However, at baseline, both HDL cholesterol and TG levels were very near normal. In meta-analyses, fibrates show a reduction in CV morbidity and mortality, but not in total mortality.[113,114] In another meta-analysis, fibrates decreased major CV events by 13%, but this benefit was evident only in subjects with increased TG (>2.26 mmol/l).[115] It is in the setting of AD where fibrates are most effective.
As fibrates are weak PPAR-alpha agonists with limited efficacy and dose-related adverse events, a new generation of highly specific PPAR-alpha agonists, known as selective PPAR-alpha modulators, have been developed that preserve the beneficial effects of fibrates and eliminate the unwanted side effects. Recently, pemafibrate was introduced; it is >2,500-fold more potent than fibric acid, with a greater lipid-modifying efficacy and an improved safety and tolerability profile.[116] Pemafibrate is being evaluated in the CV outcomes trial, Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT), studying a high-risk diabetic population on statin therapy.[117]
Cholesterol Absorption Inhibition: Ezetimibe
Ezetimibe reduces the absorption of dietary and biliary cholesterol along the brush border of jejunal enterocytes. Ezetimibe inhibits the transmembrane sterol transporter Niemann–Pick C1-like protein.[118] This reduces the amount of cholesterol delivered to the liver and stimulates increased expression of LDLR along the hepatocyte cell surface.[119] As monotherapy, ezetimibe reduces LDL cholesterol by approximately 15–20%, but it is most commonly used in combination with a statin, where it provides additive benefit for reducing LDL cholesterol, non-HDL cholesterol and apoB and is safe.[120–122]
Recently, the IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE IT) demonstrated that ezetimibe provides an incremental reduction in CV events among patients with a previous ACS and being treated with a statin.[123] It has been successfully used in patients who do not tolerate statins and in those who are not at goal with adequate doses of statins.[119] Ezetimibe can be combined with any statin, at the usual single dose of 10 mg/day, with no significant side effects reported, except for occasionally mild elevation of liver enzymes or myalgia.
In a recent meta-analysis of eight studies including 80,790 diabetics and 85,555 non-diabetics, with a mean follow-up of 45 months, the risk for ASCVD events was significantly less with ezetimibe–statin combination therapy than statin monotherapy in both diabetic (relative risk 0.69; 95% CI [0.67–0.73]; p<0.00001) and non-diabetic (RR 0.68; 95% CI [0.52–0.90]; p=0.006) subjects.[124] In a post hoc analysis of IMPROVE IT, the diabetic subgroup on ezetimibe plus simvastatin achieved a significantly lower mean LDL cholesterol than the group on placebo plus simvastatin (1.27 versus 1.73 mmol/l, respectively; p<0.001), and the RR reduction for MI and stroke in diabetics was 24% and 39%, respectively. Thus, the benefit of adding ezetimibe to statin appeared to be enhanced in patients with diabetes.[125]
Omega-3 Fatty Acids
The omega-3 fatty acids (eicosapentaenoic acid [EPA] and docosahexaenoic acid [DHA]) are long-chain polyunsaturated fatty acids that inhibit the synthesis of VLDL and TG in the liver. Although the combination of EPA and DHA reduces serum TGs and VLDL, it also induces an elevation in LDL cholesterol that is proportional to the baseline TG level.[126,127] No recent study in the statin era has been able to demonstrate CV risk reduction using combinations of EPA and DHA.[128] The Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH) trial is currently testing whether or not a combination of EPA and DHA will affect the risk for ASCVD events in high-risk diabetic patients on a statin background.
The Japan EPA Lipid Intervention Study (JELIS) treated Japanese patients on low-dose statin therapy with 1.8 g EPA or placebo.[129] The addition of EPA did provide incremental risk addition beyond statin therapy, but patients were not blinded to therapy and the doses of statin used were quite low (oral simvastatin 5 mg once daily, or oral pravastatin 10 mg, once daily). In addition, the only endpoint that achieved statistical significance was unstable angina requiring hospitalisation. JELIS did not achieve significant reductions in endpoints such as non-fatal MI or stroke, CV mortality or need for revascularisation. Hence, given these limitations, this approach did not become very widely adopted.
Recently, the Reduction of Cardiovascular Events with Icosapent Ethyl–Intervention Trial (REDUCE-IT) showed that treatment of high-risk individuals (58% with diabetes) with 4 g EPA ethyl ester daily (mean TG 2.40 mmol/l, mean LDL cholesterol 1.68 mmol/l) resulted in relative reductions of 25% in the incidence of major adverse cardiac events (MACE) and 20% in CV mortality against a statin background. Entry criteria for the REDUCE-IT trial included age >45 years with established CV disease or age >50 years with diabetes and one or more additional risk factor, fasting TG 1.69–5.63 mmol/l, LDL cholesterol 1.06–2.59 mmol/l and a stable dose of statin for ≥4 weeks. This benefit was independent of both baseline TG and LDL cholesterol.[130] The precise mechanisms by which EPA exerts these benefits remains under active investigation, although it is likely that the inhibition of oxidative processes and the augmented production of downstream effectors such as resolvins and protectins play beneficial roles.[131]
Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors
Evolocumab and alirocumab, monoclonal antibodies (mAbs) that inhibit proprotein convertase subtilisin/kexin type 9 (PCSK9), have been approved for use and are available in a number of countries. They are indicated for use with or without combination statin therapy to reduce LDL cholesterol in patients with established ASCVD or heterozygous familial hypercholesterolaemia. PCSK9 regulates the expression of LDLRs by shuttling LDLR into the lysosome for proteolytic destruction.[132] Hence, when plasma PCSK9 is increased or exhibits augmented functionality, the density of hepatocyte surface LDLR is reduced, plasma LDL cholesterol increases and the risk for ASCVD increases.[133]
In contrast, when PCSK9 is inhibited by mAbs directed against it, cell surface expression of LDLR is increased and plasma LDL cholesterol decreases due to increased clearance.[134,135] The inhibition of PCSK9 with mAbs results in a profound decrease in LDL cholesterol of approximately 50–70%. These agents also induce significant reductions in non-HDL cholesterol, apoB and Lp(a).[136] The magnitude of the reduction in Lp(a) with the PCSK9 mAbs is proportional to the baseline level.[137,138] Patients with low Lp(a) typically experience no to small changes in this lipoprotein. The mechanism(s) by which these agents reduce Lp(a) is a matter of intense debate.[139,140] The PCSK9 mAbs are available for biweekly or monthly subcutaneous injection and have a favourable safety profile.
Two landmark studies have been conducted with CV endpoints including thousands of patients randomised to a PCSK9 inhibitor or placebo against statin backgrounds. The Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) study randomised 27,564 patients with established ASCVD to either evolocumab or placebo against a statin background. LDL cholesterol decreased 59% (mean level achieved in the active treatment group: 0.78 mmol/l). The risk for the primary (HR 0.85; 95% CI [0.79–0.92]; p<0.001) and secondary composite endpoints 20% (HR 0.80; 95% CI [0.73–0.88]; p<0.001) were substantially reduced after an average follow-up of 2.2 years.[141] Evolocumab significantly reduced CV risk in patients with diabetes and did not increase the risk of new-onset diabetes.[142] Evolocumab therapy was not correlated with an increased risk for cognitive impairment even when LDL cholesterol was reduced to less than 0.26 mmol/l.[143]
The Odyssey Outcomes study included 18,924 patients who were between 1 and 12 months after an ACS; 89% were treated with high-intensity statins but did not reach their LDL cholesterol goal (<1.81 mmol/l) and mean baseline LDL cholesterol was 2.25 mmol/l. Patients were randomised to alirocumab 75/150 mg twice weekly versus placebo.[144] LDL cholesterol decreased 54.7% in those treated with alirocumab, and the mean follow-up was 2.8 years. The primary endpoint, a combination of coronary death, non-fatal MI, fatal or non-fatal stroke and hospitalisation for unstable angina, decreased 15% (HR 0.85; 95% CI [0.78–0.93]; p=0.0003). Of great interest in this trial was the observation that alirocumab reduced the risk for the primary composite endpoint significantly more in diabetic patients than in patients with prediabetes or those who were normoglycaemic (2.3% absolute risk reduction versus 1.2% and 1.2%, respectively).[145]
In a variety of patient types with AD, alirocumab and evolocumab demonstrate excellent capacity on top of statins for reducing atherogenic lipoproteins other than LDL cholesterol.[146–148] Both evolocumab and alirocumab reduce serum concentrations of VLDL, remnant lipoproteins and LDL particle numbers.[149,150] In the FOURIER trial, the reduction in Lp(a) potentiated by evolocumab provided incremental risk reduction over and above LDL cholesterol reduction, an important advance in dyslipidaemia management.[137] Although a Mendelian randomisation study suggested that there may be a signal for increased risk of diabetes with a PCSK9 mAb, to date there is no evidence that either evolocumab or alirocumab increases the risk for impaired glucose tolerance, impaired fasting glucose or diabetes.[151–154]
Conclusion
Despite statin therapy, among patients with AD LDL cholesterol reduction is inadequate because significant residual risk remains. At least some of this risk is attributable to inadequate LDL cholesterol reduction. Clearly, however, mounting evidence suggests that inadequate reduction of other apoB-containing lipoproteins also contributes to residual risk. In the setting of insulin resistance, patients experience a large increase in apoB-containing lipoproteins characterised by elevated levels of VLDL, TRLs and LDL particles. These changes occur in response to increased hepatic TG and VLDL production, inhibition of LPL, activation of CETP and hepatic lipase and reduced clearance of apoB-containing lipoproteins. AD is also characterised by substantial changes in HDL metabolism at the level of multiple organs and cell types. The contributions of low HDL, HDL dysfunction and impaired reverse cholesterol transport to the net effect of AD are matters of continuing investigation. In order to better reduce residual risk among patients with AD and TGs >2.26 mmol/l, it is generally accepted that non-HDL cholesterol and apoB are better targets of therapy, and their aggressive reduction is a clinical priority.
Among the established therapies, statins are the first-line treatment. Ezetimibe and the PCSK9 mAbs constitute important additional therapies that should be used in patients who fail to reach their LDL cholesterol goal with statins alone or in statin-intolerant subjects. Fenofibrate can be considered in patients with AD, especially in those with high TG and low HDL cholesterol despite the use of statins in adequate doses. Although studies with CV outcomes have been inconsistent in demonstrating benefits with fibrates, post hoc analyses of the subgroups with AD (high TG and low HDL) consistently demonstrate ASCVD risk reduction. Based on the REDUCE-IT trial, EPA monotherapy constitutes an exciting, highly efficacious approach to reducing ASCVD events in patients with established ASCVD or T2D with additional CV disease risk factors and controlled LDL cholesterol on statin therapy but who still have TGs >1.69 mmol/l.
Acknowledgement
The authors thank Thomas Dayspring (Chief Academic Officer, True Health, Richmond, VA, US) for kindly rendering the figures.
References
- 1.Lahey R, Khan SS. Trends in obesity and risk of cardiovascular disease. Curr Epidemiol Rep. 2018;5:243–51. doi: 10.1007/s40471-018-0160-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zobel EH, Hansen TW, Rossing P, von Scholten BJ. Global changes in food supply and the obesity epidemic. Curr Obes Rep. 2016;5:449–55. doi: 10.1007/s13679-016-0233-8. [DOI] [PubMed] [Google Scholar]
- 3.Stone GW, Maehara A, Lansky AJ et al. A prospective natural-history study of coronary atherosclerosis. N Engl J Med. 2011;364:226–35. doi: 10.1056/NEJMoa1002358. [DOI] [PubMed] [Google Scholar]
- 4.Chapman MJ, Ginsberg HN, Amarenco P et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32:1345–61. doi: 10.1093/eurheartj/ehr112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ponte-Negretti CI, Isea-Perez JE, Lorenzatti AJ et al. Atherogenic dyslipidemia in Latin America: prevalence, causes and treatment: expert’s position paper made by the Latin American Academy for the Study of Lipids (ALALIP) endorsed by the Inter-American Society of Cardiology (IASC), the South American Society of Cardiology (SSC), the Pan-American College of Endothelium (PACE), and the International Atherosclerosis Society (IAS) Int J Cardiol. 2017;243:516–22. doi: 10.1016/j.ijcard.2017.05.059. [DOI] [PubMed] [Google Scholar]
- 6.Catapano AL, Reiner Z, De Backer G et al. ESC/EAS guidelines for the management of dyslipidaemias. The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) Atherosclerosis. 2011;217:3–46. doi: 10.1016/j.atherosclerosis.2011.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Catapano AL, Graham I, De Backer G et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias: the Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS) developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR) Atherosclerosis. 2016;253:281–344. doi: 10.1016/j.atherosclerosis.2016.08.018. [DOI] [PubMed] [Google Scholar]
- 8.Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365((9468)):1415–28. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- 9.Ginsberg HN. Insulin resistance and cardiovascular disease. J Clin Invest. 2000;106:453–8. doi: 10.1172/JCI10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yusuf S, Hawken S, Ounpuu S et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–52. doi: 10.1016/S0140-6736(04)17018-9. [DOI] [PubMed] [Google Scholar]
- 11.Do R, Willer CJ, Schmidt EM et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sachdeva A, Cannon CP, Deedwania PC et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in Get With The Guidelines. Am Heart J. 2009;157:111–7e2. doi: 10.1016/j.ahj.2008.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Guyton JR, Slee AE, Anderson T et al. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes) J Am Coll Cardiol. 2013;62:1580–4. doi: 10.1016/j.jacc.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Athyros VG, Tziomalos K, Karagiannis A, Mikhailidis DP. Dyslipidaemia of obesity, metabolic syndrome and type 2 diabetes mellitus: the case for residual risk reduction after statin treatment. Open Cardiovasc Med J. 2011;5:24–34. doi: 10.2174/1874192401105010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller M, Cannon CP, Murphy SA et al. Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724–30. doi: 10.1016/j.jacc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 16.Faergeman O, Holme I, Fayyad R et al. Plasma triglycerides and cardiovascular events in the Treating to New Targets and Incremental Decrease in End-Points through Aggressive Lipid Lowering trials of statins in patients with coronary artery disease. Am J Cardiol. 2009;104:459–63. doi: 10.1016/j.amjcard.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Klempfner R, Erez A, Sagit BZ et al. Elevated triglyceride level is independently associated with increased all-cause mortality in patients with established coronary heart disease: twenty-two-year follow-up of the Bezafibrate Infarction Prevention Study and Registry. Circ Cardiovasc Qual Outcomes. 2016;9:100–8. doi: 10.1161/CIRCOUTCOMES.115.002104. [DOI] [PubMed] [Google Scholar]
- 18.Sarwar N, Danesh J, Eiriksdottir G et al. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450–8. doi: 10.1161/CIRCULATIONAHA.106.637793. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz GG, Abt M, Bao W et al. Fasting triglycerides predict recurrent ischemic events in patients with acute coronary syndrome treated with statins. J Am Coll Cardiol. 2015;65:2267–75. doi: 10.1016/j.jacc.2015.03.544. [DOI] [PubMed] [Google Scholar]
- 20.Di Angelantonio E, Sarwar N, Perry P et al. Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toth PP, Philip S, Hull M, Granowitz C. Association of elevated triglycerides with increased cardiovascular risk and direct costs in statin-treated patients. Mayo Clin Proc. 2019;94:1670–80. doi: 10.1016/j.mayocp.2019.03.028. [DOI] [PubMed] [Google Scholar]
- 22.Toth PP. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc Health Risk Manag. 2016;12:171–83. doi: 10.2147/VHRM.S104369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reiner Ž. Hypertriglyceridaemia and risk of coronary artery disease. Nat Rev Cardiol. 2017;14:401–11. doi: 10.1038/nrcardio.2017.31. [DOI] [PubMed] [Google Scholar]
- 24.Varbo A, Benn M, Tybjaerg-Hansen A et al. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36. doi: 10.1016/j.jacc.2012.08.1026. [DOI] [PubMed] [Google Scholar]
- 25.Joshi PH, Khokhar AA, Massaro JM et al. Remnant lipoprotein cholesterol and incident coronary heart disease: the Jackson Heart and Framingham Offspring Cohort Studies. J Am H. eart Assoc. 2016;5:pii–e002765. doi: 10.1161/JAHA.115.002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varbo A, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low-grade inflammation and ischemic heart disease, whereas elevated low-density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128:1298–309. doi: 10.1161/CIRCULATIONAHA.113.003008. [DOI] [PubMed] [Google Scholar]
- 27.Masuda D, Yamashita S. Postprandial hyperlipidemia and remnant lipoproteins. J Atheroscler Thromb. 2017;24:95–109. doi: 10.5551/jat.RV16003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bansal S, Buring JE, Rifai N et al. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–16. doi: 10.1001/jama.298.3.309. [DOI] [PubMed] [Google Scholar]
- 29.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 30.Musunuru K, Kathiresan S. Surprises from genetic analyses of lipid risk factors for atherosclerosis. Circ Res. 2016;118:579–85. doi: 10.1161/CIRCRESAHA.115.306398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willer CJ, Sanna S, Jackson AU et al. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008;40:161–9. doi: 10.1038/ng.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu J, Afroza H, Rader DJ, Jin W. Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J Biol Chem. 2010;285:27561–70. doi: 10.1074/jbc.M110.144279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sukonina V, Lookene A, Olivecrona T, Olivecrona G. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc Natl Acad Sci USA. 2006;103:17450–5. doi: 10.1073/pnas.0604026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Puthanveetil P, Wang F et al. Severity of diabetes governs vascular lipoprotein lipase by affecting enzyme dimerization and disassembly. Diabetes. 2011;60:2041–50. doi: 10.2337/db11-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. doi: 10.1056/NEJMoa1307095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Natarajan P, Kohli P, Baber U et al. Association of APOC3 loss-of-function mutations with plasma lipids and subclinical atherosclerosis: the Multi-Ethnic BioImage Study. J Am Coll Cardiol. 2015;66:2053–5. doi: 10.1016/j.jacc.2015.08.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stitziel NO, Khera AV, Wang X et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054–63. doi: 10.1016/j.jacc.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dewey FE, Gusarova V, O’Dushlaine C et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–33. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helgadottir A, Gretarsdottir S, Thorleifsson G et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48:634–9. doi: 10.1038/ng.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Au A, Griffiths LR, Irene L et al. The impact of APOA5, APOB, APOC3 and ABCA1 gene polymorphisms on ischemic stroke: evidence from a meta-analysis. Atherosclerosis. 2017;265:60–70. doi: 10.1016/j.atherosclerosis.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 41.Chasman DI, Pare G, Zee RY et al. Genetic loci associated with plasma concentration of LDL-C, HDL-C, triglycerides, ApoA1, and ApoB among 6382 Caucasian women in genome-wide analysis with replication. Circ Cardiovasc Genet. 2008;1:21–30. doi: 10.1161/CIRCGENETICS.108.773168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castelli WP. Cholesterol and lipids in the risk of coronary artery disease – the Framingham Heart Study. Can J Cardiol. 1988. pp. 5A–10A. [PubMed]
- 43.Gordon DJ, Probstfield JL, Garrison RJ et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. doi: 10.1161/01.CIR.79.1.8. [DOI] [PubMed] [Google Scholar]
- 44.Bartlett J, Predazzi IM, Williams SM et al. Is isolated low high-density lipoprotein cholesterol a cardiovascular disease risk factor? New insights from the Framingham Offspring Study. Circ Cardiovasc Qual Outcomes. 2016;9:206–12. doi: 10.1161/CIRCOUTCOMES.115.002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barter PJ, Caulfield M, Eriksson M et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 46.Boden WE, Probstfield JL, Anderson T et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67. doi: 10.1056/NEJMoa1107579. [DOI] [PubMed] [Google Scholar]
- 47.HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25673 high-risk patients of ER niacin/laropiprant: trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. 2013;34:1279–91. doi: 10.1093/eurheartj/eht055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lincoff AM, Nicholls SJ, Riesmeyer JS et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N Engl J Med. 2017;376:1933–42. doi: 10.1056/NEJMoa1609581. [DOI] [PubMed] [Google Scholar]
- 49.Voight BF, Peloso GM, Orho-Melander M et al. Plasma HDL cholesterol and risk of myocardial infarction: a Mendelian randomisation study. Lancet. 2012;380:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Toth PP, Barylski M, Nikolic D et al. Should low high-density lipoprotein cholesterol (HDL-C) be treated? Best Pract Res Clin Endocrinol Metab. 2014;28:353–68. doi: 10.1016/j.beem.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 51.Toth PP, Barter PJ, Rosenson RS et al. High-density lipoproteins: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7:484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 52.Xiao C, Dash S, Morgantini C et al. Pharmacological targeting of the atherogenic dyslipidemia complex: the next frontier in CVD prevention beyond lowering LDL cholesterol. Diabetes. 2016;65:1767–78. doi: 10.2337/db16-0046. [DOI] [PubMed] [Google Scholar]
- 53.Bobik A. Apolipoprotein CIII and atherosclerosis: beyond effects on lipid metabolism. Circulation. 2008;118:702–4. doi: 10.1161/CIRCULATIONAHA.108.794081. [DOI] [PubMed] [Google Scholar]
- 54.Barter PJ, Brewer HB, Jr, Chapman MJ et al. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–7. doi: 10.1161/01.ATV.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- 55.Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–32. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- 56.Bays HE, Toth PP, Kris-Etherton PM et al. Obesity, adiposity, and dyslipidemia: a consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7:304–83. doi: 10.1016/j.jacl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 57.Mooradian AD, Haas MJ, Wong NCW. Transcriptional control of apolipoprotein A-I gene expression in diabetes. Diabetes. 2004;53:513–20. doi: 10.2337/diabetes.53.3.513. [DOI] [PubMed] [Google Scholar]
- 58.Chung S, Sawyer JK, Gebre AK et al. Adipose tissue ATP binding cassette transporter A1 contributes to high-density lipoprotein biogenesis in vivo. Circulation. 2011;124:1663–72. doi: 10.1161/CIRCULATIONAHA.111.025445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McGillicuddy FC, Reilly MP, Rader DJ. Adipose modulation of high-density lipoprotein cholesterol. Circulation. 2011;124:1602–5. doi: 10.1161/CIRCULATIONAHA.111.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang Y, McGillicuddy FC, Hinkle CC et al. Adipocyte modulation of high-density lipoprotein cholesterol. Circulation. 2010;121:1347–55. doi: 10.1161/CIRCULATIONAHA.109.897330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farbstein D, Levy AP. HDL dysfunction in diabetes: causes and possible treatments. Expert Rev Cardiovas Ther. 2012;10:353–61. doi: 10.1586/erc.11.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Packard C, Caslake M, Shepherd J. The role of small, dense low density lipoprotein (LDL): a new look. Int J Cardiol. 2000;74((Suppl 1)):S17–22. doi: 10.1016/S0167-5273(99)00107-2. [DOI] [PubMed] [Google Scholar]
- 63.Chait A, Brazg RL, Tribble DL, Krauss RM. Susceptibility of small, dense, low-density lipoproteins to oxidative modification in subjects with the atherogenic lipoprotein phenotype, pattern B. Am J Med. 1993;94:350–6. doi: 10.1016/0002-9343(93)90144-E. [DOI] [PubMed] [Google Scholar]
- 64.McNamara JR, Small DM, Li Z, Schaefer EJ. Differences in LDL subspecies involve alterations in lipid composition and conformational changes in apolipoprotein B. J Lipid Res. 1996;37:1924–35. [PubMed] [Google Scholar]
- 65.Ivanova EA, Myasoedova VA, Melnichenko AA, Grechko AV, Orekhov AN. Small dense low-density lipoprotein as biomarker for atherosclerotic diseases. Oxid Med Cell Longev. 2017;2017:1273042. doi: 10.1155/2017/1273042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Diffenderfer MR, Schaefer EJ. The composition and metabolism of large and small LDL. Curr Opin Lipidol. 2014;25:221–6. doi: 10.1097/MOL.0000000000000067. [DOI] [PubMed] [Google Scholar]
- 67.Julius U, Dittrich M, Pietzsch J. Factors influencing the formation of small dense low-density lipoprotein particles in dependence on the presence of the metabolic syndrome and on the degree of glucose intolerance. Int J Clin Pract. 2007;61:1798–804. doi: 10.1111/j.1742-1241.2007.01507.x. [DOI] [PubMed] [Google Scholar]
- 68.Sacks FM. The apolipoprotein story. Atheroscler Suppl. 2006;7:23–7. doi: 10.1016/j.atherosclerosissup.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 69.Jacobson TA, Ito MK, Maki KC et al. National Lipid Association recommendations for patient-centered management of dyslipidemia: part 1 – executive summary. J Clin Lipidol. 2014;8:473–88. doi: 10.1016/j.jacl.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 70.Rabar S, Harker M, O’Flynn N, Wierzbicki AS. Lipid modification and cardiovascular risk assessment for the primary and secondary prevention of cardiovascular disease: summary of updated NICE guidance. BMJ (Clin Res Ed) 2014;349:g4356. doi: 10.1136/bmj.g4356. [DOI] [PubMed] [Google Scholar]
- 71.Catapano AL, Graham I, De Backer G et al. 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J. 2016;37:2999–3058. doi: 10.1093/eurheartj/ehw272. [DOI] [PubMed] [Google Scholar]
- 72.Jellinger PS, Handelsman Y, Rosenblit PD et al. American Association of Clinical Endocrinologists and American College of Endocrinology guidelines for management of dyslipidemia and prevention of cardiovascular disease. Endocr Pract. 2017;23((Suppl 2)):1–87. doi: 10.4158/EP171764.APPGL. [DOI] [PubMed] [Google Scholar]
- 73.Kuller L, Arnold A, Tracy R et al. Nuclear magnetic resonance spectroscopy of lipoproteins and risk of coronary heart disease in the cardiovascular health study. Arterioscler Thromb Vasc Biol. 2002;22:1175–80. doi: 10.1161/01.ATV.0000022015.97341.3A. [DOI] [PubMed] [Google Scholar]
- 74.Barter PJ, Ballantyne CM, Carmena R et al. Apo B versus cholesterol in estimating cardiovascular risk and in guiding therapy: report of the thirty-person/ten-country panel. J Intern Med. 2006;259:247–58. doi: 10.1111/j.1365-2796.2006.01616.x. [DOI] [PubMed] [Google Scholar]
- 75.Brunzell JD, Davidson M, Furberg CD et al. Lipoprotein management in patients with cardiometabolic risk: consensus conference report from the American Diabetes Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2008;51:1512–24. doi: 10.1016/j.jacc.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 76.Van Horn L, McCoin M, Kris-Etherton PM et al. The evidence for dietary prevention and treatment of cardiovascular disease. J Am Diet Assoc. 2008;108:287–331. doi: 10.1016/j.jada.2007.10.050. [DOI] [PubMed] [Google Scholar]
- 77.Sacks FM, Lichtenstein AH, Wu JHY et al. Dietary fats and cardiovascular disease: a presidential advisory from the American Heart Association. Circulation. 2017;136:e1–23. doi: 10.1161/CIR.0000000000000510. [DOI] [PubMed] [Google Scholar]
- 78.Shai I, Schwarzfuchs D, Henkin Y et al. Weight loss with a low-carbohydrate, Mediterranean, or low-fat diet. N Engl J Med. 2008;359:229–41. doi: 10.1056/NEJMoa0708681. [DOI] [PubMed] [Google Scholar]
- 79.Guasch-Ferre M, Babio N, Martinez-Gonzalez MA et al. Dietary fat intake and risk of cardiovascular disease and all-cause mortality in a population at high risk of cardiovascular disease. Am J Clin Nutr. 2015;102:1563–73. doi: 10.3945/ajcn.115.116046. [DOI] [PubMed] [Google Scholar]
- 80.Musunuru K. Atherogenic dyslipidemia: cardiovascular risk and dietary intervention. Lipids. 2010;45:907–14. doi: 10.1007/s11745-010-3408-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu X, Yuan H, Niu Y et al. The role of AMPK/mTOR/S6K1 signaling axis in mediating the physiological process of exercise-induced insulin sensitization in skeletal muscle of C57BL/6 mice. Biochim Biophys Acta. 2012;1822:1716–26. doi: 10.1016/j.bbadis.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 82.Lim S, Choi SH, Jeong I-K et al. Insulin-sensitizing effects of exercise on adiponectin and retinol-binding protein-4 concentrations in young and middle-aged women. J Clin Endocrinol Metab. 2008;93:2263–8. doi: 10.1210/jc.2007-2028. [DOI] [PubMed] [Google Scholar]
- 83.Halverstadt A, Phares DA, Wilund KR et al. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism. 2007;56:444–50. doi: 10.1016/j.metabol.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 84.Trejo-Gutierrez JF, Fletcher G. Impact of exercise on blood lipids and lipoproteins. J Clin Lipidol. 2007;1:175–81. doi: 10.1016/j.jacl.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 85.Kodama S, Tanaka S, Saito K et al. Effect of aerobic exercise training on serum levels of high-density lipoprotein cholesterol: a meta-analysis. Arch Intern Med. 2007;167:999–1008. doi: 10.1001/archinte.167.10.999. [DOI] [PubMed] [Google Scholar]
- 86.Bassi N, Karagodin I, Wang S et al. Lifestyle modification for metabolic syndrome: a systematic review. Am J Med. 2014;127:1242, e1–10. doi: 10.1016/j.amjmed.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 87.Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- 88.Sirtori CR. The pharmacology of statins. Pharmacol Res. 2014;88:3–11. doi: 10.1016/j.phrs.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 89.Rizzo M, Berneis K. The clinical relevance of low-density-lipoproteins size modulation by statins. Cardiovasc Drugs Ther. 2006;20:205–17. doi: 10.1007/s10557-006-8283-x. [DOI] [PubMed] [Google Scholar]
- 90.Sahebkar A, Simental-Mendía LE, Mikhailidis DP et al. Effect of statin therapy on plasma apolipoprotein CIII concentrations: a systematic review and meta-analysis of randomized controlled trials. J Clin Lipidol. 2018;12:801–9. doi: 10.1016/j.jacl.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 91.Oesterle A, Laufs U, Liao JK. Pleiotropic effects of statins on the cardiovascular system. Circ Res. 2017;120:229–43. doi: 10.1161/CIRCRESAHA.116.308537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Baigent C, Keech A, Kearney PM et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90 056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 93.Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170 000 participants in 26 randomised trials. Lancet. 2010;376:1670–81. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ridker PM, Danielson E, Fonseca FA et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 95.Sattar N, Preiss D, Murray HM et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–42. doi: 10.1016/S0140-6736(09)61965-6. [DOI] [PubMed] [Google Scholar]
- 96.Ridker PM, Pradhan A, MacFadyen JG et al. Cardiovascular benefits and diabetes risks of statin therapy in primary prevention: an analysis from the JUPITER trial. Lancet. 2012;380:565–71. doi: 10.1016/S0140-6736(12)61190-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Waters DD, Ho JE, DeMicco DA et al. Predictors of new-onset diabetes in patients treated with atorvastatin: results from 3 large randomized clinical trials. J Am Coll Cardiol. 2011;57:1535–45. doi: 10.1016/j.jacc.2010.10.047. [DOI] [PubMed] [Google Scholar]
- 98.Preiss D, Seshasai SRK, Welsh P et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy. JAMA. 2011;305:2556–64. doi: 10.1001/jama.2011.860. [DOI] [PubMed] [Google Scholar]
- 99.Cholesterol Treatment Trialists’ (CTT) Collaboration. Efficacy of cholesterol-lowering therapy in 18 686 people with diabetes in 14 randomised trials of statins: a meta-analysis. Lancet. 2008;371:117–25. doi: 10.1016/S0140-6736(08)60104-X. [DOI] [PubMed] [Google Scholar]
- 100.Ballantyne CM, Olsson AG, Cook TJ et al. Influence of low high-density lipoprotein cholesterol and elevated triglyceride on coronary heart disease events and response to simvastatin therapy in 4S. Circulation. 2001;104:3046–51. doi: 10.1161/hc5001.100624. [DOI] [PubMed] [Google Scholar]
- 101.Simental-Mendia LE, Simental-Mendia M, Sanchez-Garcia A et al. Effect of fibrates on glycemic parameters: a systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol Res. 2018;132:232–41. doi: 10.1016/j.phrs.2017.12.030. [DOI] [PubMed] [Google Scholar]
- 102.Staels B, Dallongeville J, Auwerx J et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–93. doi: 10.1161/01.CIR.98.19.2088. [DOI] [PubMed] [Google Scholar]
- 103.Ferrari R, Aguiar C, Alegria E et al. Current practice in identifying and treating cardiovascular risk, with a focus on residual risk associated with atherogenic dyslipidaemia. Eur Heart J Suppl. 2016;18((Suppl_C)):C2–12. doi: 10.1093/eurheartj/suw009. [DOI] [PubMed] [Google Scholar]
- 104.Frick MH, Elo O, Haapa K et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med. 1987;317:1237–45. doi: 10.1056/NEJM198711123172001. [DOI] [PubMed] [Google Scholar]
- 105.Robins SJ, Collins D, Wittes JT et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA. 2001;285:1585–91. doi: 10.1001/jama.285.12.1585. [DOI] [PubMed] [Google Scholar]
- 106.Prueksaritanont T, Zhao JJ, Ma B et al. Mechanistic studies on metabolic interactions between gemfibrozil and statins. J Pharmacol Exp Ther. 2002;301:1042–51. doi: 10.1124/jpet.301.3.1042. [DOI] [PubMed] [Google Scholar]
- 107.Prueksaritanont T, Tang C, Qiu Y et al. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab Dispos. 2002;30:1280–7. doi: 10.1124/dmd.30.11.1280. [DOI] [PubMed] [Google Scholar]
- 108.Jones PH, Davidson MH. Reporting rate of rhabdomyolysis with fenofibrate + statin versus gemfibrozil + any statin. Am J Cardiol. 2005;95:120–2. doi: 10.1016/j.amjcard.2004.08.076. [DOI] [PubMed] [Google Scholar]
- 109.Keech A, Simes RJ, Barter P et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–61. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 110.Ansquer JC, Foucher C, Rattier S et al. Fenofibrate reduces progression to microalbuminuria over 3 years in a placebo-controlled study in type 2 diabetes: results from the Diabetes Atherosclerosis Intervention Study (DAIS) Am J Kidney Dis. 2005;45:485–93. doi: 10.1053/j.ajkd.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 111.Dalton RN, Crimet D, Ansquer JC. The Effect of Fenofibrate on Glomerular Filtration Rate (GFR) and Other Renal Function Tests. A Double-Blind Placebo-Controlled Cross-Over Study in Healthy Subjects. Philadelphia, PA: American Society of Nephrology; 2005. [Google Scholar]
- 112.Ginsberg HN, Elam MB, Lovato LC et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74. doi: 10.1056/NEJMoa1001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Saha SA, Arora RR. Fibrates in the prevention of cardiovascular disease in patients with type 2 diabetes mellitus – a pooled meta-analysis of randomized placebo-controlled clinical trials. Int J Cardiol. 2010;141:157–66. doi: 10.1016/j.ijcard.2008.11.211. [DOI] [PubMed] [Google Scholar]
- 114.Jun M, Foote C, Lv J et al. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010;375:1875–84. doi: 10.1016/S0140-6736(10)60656-3. [DOI] [PubMed] [Google Scholar]
- 115.Wang D, Liu B, Tao W et al. Fibrates for secondary prevention of cardiovascular disease and stroke. Cochrane Database Syst Rev. 2015;10:CD009580. doi: 10.1002/14651858.CD009580.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fruchart JC. Selective peroxisome proliferator-activated receptor alpha modulators (SPPARMalpha): the next generation of peroxisome proliferator-activated receptor alpha-agonists. Cardiovasc Diabetol. 2013;12:82. doi: 10.1186/1475-2840-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pradhan AD, Paynter NP, Everett BM et al. Rationale and design of the Pemafibrate to Reduce Cardiovascular Outcomes by Reducing Triglycerides in Patients with Diabetes (PROMINENT) study. Am Heart J. 2018;206:80–93. doi: 10.1016/j.ahj.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 118.Garcia-Calvo M, Lisnock J, Bull HG et al. The target of ezetimibe is Niemann–Pick C1-like 1 (NPC1L1) Proc Natl Acad Sci USA. 2005;102:8132–7. doi: 10.1073/pnas.0500269102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wierzbicki AS, Doherty E, Lumb PJ et al. Efficacy of ezetimibe in patients with statin-resistant and statin-intolerant familial hyperlipidaemias. Curr Med Res Opin. 2005;21:333–8. doi: 10.1185/030079905X28872. [DOI] [PubMed] [Google Scholar]
- 120.Toth PP, Catapano A, Tomassini JE, Tershakovec AM. Update on the efficacy and safety of combination ezetimibe plus statin therapy. Clin Lipidol. 2010;5:655–84. doi: 10.2217/clp.10.49. [DOI] [Google Scholar]
- 121.Morrone D, Weintraub WS, Toth PP et al. Lipid-altering efficacy of ezetimibe plus statin and statin monotherapy and identification of factors associated with treatment response: a pooled analysis of over 21,000 subjects from 27 clinical trials. Atherosclerosis. 2012;223:251–61. doi: 10.1016/j.atherosclerosis.2012.02.016. [DOI] [PubMed] [Google Scholar]
- 122.Toth PP, Morrone D, Weintraub WS et al. Safety profile of statins alone or combined with ezetimibe: a pooled analysis of 27 studies including over 22,000 patients treated for 6–24 weeks. Int J Clin Pract. 2012;66:800–12. doi: 10.1111/j.1742-1241.2012.02964.x. [DOI] [PubMed] [Google Scholar]
- 123.Cannon CP, Blazing MA, Giugliano RP et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 124.Miao XY, Liu HZ, Jin MM et al. A comparative meta-analysis of the efficacy of statin–ezetimibe co-therapy versus statin monotherapy in reducing cardiovascular and cerebrovascular adverse events in patients with type 2 diabetes mellitus. Eur Rev Med Pharmacol Sci. 2019;23:2302–10. doi: 10.26355/eurrev_201903_17279. [DOI] [PubMed] [Google Scholar]
- 125.Giugliano RP, Cannon CP, Blazing MA et al. Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and safety in patients with versus without diabetes mellitus: results from IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial) Circulation. 2018;137:1571–82. doi: 10.1161/CIRCULATIONAHA.117.030950. [DOI] [PubMed] [Google Scholar]
- 126.von Schacky C, Harris WS. Cardiovascular benefits of omega-3 fatty acids. Cardiovasc Res. 2007;73:310–5. doi: 10.1016/j.cardiores.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 127.Jain AP, Aggarwal KK, Zhang PY. Omega-3 fatty acids and cardiovascular disease. Eur Rev Med Pharmacol Sci. 2015;19:441–5. [PubMed] [Google Scholar]
- 128.Siscovick DS, Barringer TA, Fretts AM et al. Omega-3 polyunsaturated fatty acid (fish oil) supplementation and the prevention of clinical cardiovascular disease: a science advisory from the American Heart Association. Circulation. 2017;135:e867–84. doi: 10.1161/CIR.0000000000000482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yokoyama M, Origasa H, Matsuzaki M et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–8. doi: 10.1016/S0140-6736(07)60527-3. [DOI] [PubMed] [Google Scholar]
- 130.Bhatt DL, Steg PG, Miller M et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2018;380:11–22. doi: 10.1056/NEJMoa1812792. [DOI] [PubMed] [Google Scholar]
- 131.Kohli P, Levy BD. Resolvins and protectins: mediating solutions to inflammation. Br J Pharmacol. 2009;158:960–71. doi: 10.1111/j.1476-5381.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Reiner Z. PCSK9 inhibitors – past, present and future. Expert Opin Drug Metab Toxicol. 2015;11:1517–21. doi: 10.1517/17425255.2015.1075506. [DOI] [PubMed] [Google Scholar]
- 133.Abifadel M, Varret M, Rabes JP et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. doi: 10.1038/ng1161. [DOI] [PubMed] [Google Scholar]
- 134.Lüscher TF. LDL-cholesterol targets: perspectives for the use of PCSK9 inhibitors. Eur Heart J. 2016;37:1337–40. doi: 10.1093/eurheartj/ehw150. [DOI] [PubMed] [Google Scholar]
- 135.Stoekenbroek RM, Kastelein JJ, Huijgen R. PCSK9 inhibition: the way forward in the treatment of dyslipidemia. BMC Med. 2015;13:258. doi: 10.1186/s12916-015-0503-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Banerjee Y, Santos RD, Al-Rasadi K, Rizzo M. Targeting PCSK9 for therapeutic gains: have we addressed all the concerns? Atherosclerosis. 2016;248:62–75. doi: 10.1016/j.atherosclerosis.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 137.O’Donoghue ML, Fazio S, Giugliano RP et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–92. doi: 10.1161/CIRCULATIONAHA.118.037184. [DOI] [PubMed] [Google Scholar]
- 138.Gaudet D, Kereiakes DJ, McKenney JM et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from Phase 2 trials) Am J Cardiol. 2014;114:711–5. doi: 10.1016/j.amjcard.2014.05.060. [DOI] [PubMed] [Google Scholar]
- 139.Shapiro MD, Minnier J, Tavori H et al. Relationship between low-density lipoprotein cholesterol and lipoprotein(a) lowering in response to PCSK9 inhibition with evolocumab. J Am Heart Assoc. 2019;8:e010932. doi: 10.1161/JAHA.118.010932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Raal FJ, Giugliano RP, Sabatine MS et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: an analysis of 10 clinical trials and the LDL receptor’s role. J Lipid Res. 2016;57:1086–96. doi: 10.1194/jlr.P065334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sabatine MS, Giugliano RP, Keech AC et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–22. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 142.Sabatine MS, Leiter LA, Wiviott SD et al. Cardiovascular safety and efficacy of the PCSK9 inhibitor evolocumab in patients with and without diabetes and the effect of evolocumab on glycaemia and risk of new-onset diabetes: a prespecified analysis of the Fourier randomised controlled trial. Lancet Diabetes Endocrinol. 2017;5:941–50. doi: 10.1016/S2213-8587(17)30313-3. [DOI] [PubMed] [Google Scholar]
- 143.Giugliano RP, Mach F, Zavitz K et al. Cognitive function in a randomized trial of evolocumab. N Engl J Med. 2017;377:633–43. doi: 10.1056/NEJMoa1701131. [DOI] [PubMed] [Google Scholar]
- 144.Schwartz GG, Steg PG, Szarek M et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–107. doi: 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- 145.Ray KK, Colhoun HM, Szarek M et al. Effects of alirocumab on cardiovascular and metabolic outcomes after acute coronary syndrome in patients with or without diabetes: a prespecified analysis of the ODYSSEY OUTCOMES randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7:618–28. doi: 10.1016/S2213-8587(19)30158-5. [DOI] [PubMed] [Google Scholar]
- 146.Ray KK, Leiter LA, Müller-Wieland D et al. Alirocumab vs usual lipid-lowering care as add-on to statin therapy in individuals with type 2 diabetes and mixed dyslipidaemia: the ODYSSEY DM-DYSLIPIDEMIA randomized trial. Diabetes Obes Metab. 2018;20:1479–89. doi: 10.1111/dom.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lorenzatti AJ, Eliaschewitz FG, Chen Y et al. Randomised study of evolocumab in patients with type 2 diabetes and dyslipidaemia on background statin: primary results of the BERSON clinical trial. Diabetes Obes Metab. 2019;21:1455–63. doi: 10.1111/dom.13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rosenson RS, Daviglus ML, Handelsman Y et al. Efficacy and safety of evolocumab in individuals with type 2 diabetes mellitus: primary results of the randomised controlled BANTING study. Diabetologia. 2019;62:948–58. doi: 10.1007/s00125-019-4856-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Toth PP, Sattar N, Blom DJ et al. Effect of evolocumab on lipoprotein particles. Am J Cardiol. 2018;121:308–14. doi: 10.1016/j.amjcard.2017.10.028. [DOI] [PubMed] [Google Scholar]
- 150.Toth PP, Hamon SC, Jones SR et al. Effect of alirocumab on specific lipoprotein non-high-density lipoprotein cholesterol and subfractions as measured by the vertical auto profile method: analysis of 3 randomized trials versus placebo. Lipids Health Dis. 2016;15:28. doi: 10.1186/s12944-016-0197-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Schmidt AF, Swerdlow DI, Holmes MV et al. PCSK9 genetic variants and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105. doi: 10.1016/S2213-8587(16)30396-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.de Carvalho LSF, Campos AM, Sposito AC. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and incident type 2 diabetes: a systematic review and meta-analysis with over 96,000 patient-years. Diabetes Care. 2018;41:364–7. doi: 10.2337/dc17-1464. [DOI] [PubMed] [Google Scholar]
- 153.Colhoun HM, Ginsberg HN, Robinson JG et al. No effect of PCSK9 inhibitor alirocumab on the incidence of diabetes in a pooled analysis from 10 ODYSSEY Phase 3 studies. Eur Heart J. 2016;37:2981–9. doi: 10.1093/eurheartj/ehw292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Toth PP, Descamps O, Genest J et al. Pooled safety analysis of evolocumab in over 6000 patients from double-blind and open-label extension studies. Circulation. 2017;135:1819–31. doi: 10.1161/CIRCULATIONAHA.116.025233. [DOI] [PubMed] [Google Scholar]
- 155.Sathiyakumar V, Kapoor K, Jones SR et al. Novel therapeutic targets for managing dyslipidemia. Trends Pharmacol Sci. 2018;39:733–47. doi: 10.1016/j.tips.2018.06.001. [DOI] [PubMed] [Google Scholar]