

Abstract
Transcription factor STAT3 has been shown to regulate genes that are involved in stem cell self-renewal and thus represents a novel therapeutic target of great biological significance. However, many small molecule agents with potential effects in cancer therapy lack aqueous solubility and high off-target toxicity, hence impeding efficient bioavailability and activity. This work, for the first time, reports a prodrug-based strategy for selective and safer delivery of STAT3 inhibitors designed towards metastatic and drug resistant breast cancer. We have synthesized a novel lipase-labile SN-2 phospholipids-pro-drug from a clinically investigated STAT3 inhibitor, nifuroxazide (Pro-nifuroxazide), which can be regioselectively cleaved by the membrane abundant enzymes in cancer cells. Pro-nifuroxazide self-assembled to sub 20 nm nanoparticles and the cytotoxic ability was screened in ER (+)-MCF-7 and ER(−)-MD-MB231 cells at 48–72h using MTT proliferation assay. Results indicated that pro-nifuroxazide NP are multifold more effective towards inhibiting cancer cells in a time dependent manner compared to parent nifuroxazide. A remarkable improvement in the local concentration of drug to as high as ~240 folds when assembled into nanoparticles is presumably the reason for this functional improvement. We introduced molecular dynamics (MD) simulations to generate Pro-nifuroxazide nano-assembly, a model assembly from triggerable anti-cancer drug, to provide molecular insights correlating physico-chemical and anti-cancer properties. In silico properties of Pro-nifuroxazide including size and chemistry of nanoparticles and membrane interactions with individual molecules could be validated by in vitro functional activities in cells of breast cancer origin. The in vivo anti-cancer efficiencies of Pro-nifuroxazide nanoparticles in nude mice xenografts with MCF-7 revealed remarkable growth inhibition as high as 400% for Pro-nifuroxazide nanoparticle. Histopathological analysis corroborated these findings showing significantly high nuclear fragmentation and retracted cytoplasm. Immuno-staining on tumor section demonstrated significantly lower level of pSTAT-3 by Pro-nifuroxazide nanoparticle treatment establishing the inhibition of STAT-3 phosphorylation. Our strategy for the first time proposes a translatable prodrug agent self-assembled into nanoparticles and demonstrate remarkable enhancement in IC50, induced apoptosis and reduced stem like cancer cell population through STAT-3 inhibition and reduced phosphorylation.
Keywords: prodrug, nanoparticle, self-assembly, dissipative particle dynamics, cancer therapy
Graphical Abstract
INTRODUCTION
In cancer, a small population of self-renewing, tumorigenic cancer stem cells (CSCs), show resistance to treatment and cause tumor recurrence. Cancer stem cells are known to overexpress signal transducer and activator of transcription factor 3 (STAT-3). Strategies that block STAT3 may prove efficacious for sustained cancer treatment. Several therapeutic approaches are being pursued including inhibitors of STAT3 function, agents that block either dimerization or DNA binding by STAT3 and strategies to reduce STAT3 expression. Among these, STAT-3 inhibition approach can be highly effective for cancer treatment as normal cells can tolerate a reduction in STAT3 function, but cancer cells require constitutive STAT3 signaling for its survival.
In this work, we considered drug repurposing strategy to achieve enhanced therapeutic efficacy and minimize huge cost associated with the drug development process. A nitro furan-based drug nifuroxazide has been found as an effective inhibitor of STAT3 function. Patented in 1966, nifuroxazide has been initially used as an oral antibiotic to treat traveler’s diarrhea or colitis. Independent studies show effectiveness of this agent as an inhibitor for STAT-3 by constitutive phosphorylation in multiple myeloma (MM) by the reduction of Jak kinase autophosphorylation, which leads to down-regulation of the STAT-3 target gene Mcl-1. With the long history of use in humans and expiry of original patent rights, this agent qualifies as a generic ‘biosimilar’ that may offer inexpensive option for cancer therapy, expanding patient access to affordable treatments. Originally marketed by SmithKline Beecham, it is also known that this agent is poorly absorbed from the gastrointestinal tract. Due to its poorly absorbable nature, the potential of this agent cannot be fully realized, and a few known instances of off target toxicity greatly limit its application. Other small molecule inhibitors regulating STAT-3 expression are known in the literature, however, majority of them lack aqueous solubility hence impeding efficient bioavailability. The administration of these poorly soluble bioactive agents usually requires a vehicle or carrier, facilitating improved dispersion, selectivity, and activity.1–5 Prodrugs are often designed to improve bioavailability of an active drug, and to improve its selective interaction with desired cells via site specific triggerability.6–10 Triggerable pro-drugs ensure that even their entry to off-target cells do not cause any adverse effect. This eventually reduces the side effects of the parent drug, especially critical in treatments such as chemotherapy.
A nanoparticle-enabled delivery approach can be used as a possible solution to enrich payload molecules at the site of delivery and can be engineered to transport therapeutics and imaging agents.11–14 Various carbon based nanoparticles have been used to deliver drugs and drug combinations but require specific targeting ability to improve on efficacy and reduction of side effects.15 A nano-delivery of pro-drug molecule could be an answer to off-target toxicity and side effects by combining the site-specific enrichment and activations by localized trigger. In nanomedicine, the hydrophobicity of drug favors its incorporation into many nanoparticle formulations, including into the phospholipid outer membrane of lipid-based particles. Although direct drug-encapsulation is an effective tactics for delivery, previous in vivo pharmacokinetic studies have shown that even hydrophobic drugs included in the nanoparticle lipid membrane were significantly lost in circulation en route to the target cells, with the premature release of the drug arising faster and to a greater extent. To address this issue, we hypothesized that a phospholipid prodrug approach that couples the active pharmaceutical ingredient (API) through the SN2 acyl position (i.e., stereospecific hydroxyl group of the second carbon of glycerol) would present a stable membrane complex in the nanoparticle during circulatory transit to the target site. Subsequent transfer of the monolayer components into the target cell membrane through fusion-triggered mechanism would allow cell surface or cytosolic phospholipases to enzymatically cleave the SN2 ester and release the drug, allowing it to diffuse into the cytosol for effect.16–18 The objectives of the present work were: a) to develop and characterize an SN2 lipase-labile prodrug of nifuroxazide (Pro-nifuroxazide) and self-assembled nanoparticles; b) characterize prodrug derived nanoparticles using simulation and analytical methods and demonstrate the activation in the presence of lipase; c) demonstrate the anti-proliferative efficacy of the agent in vitro in human breast cancer cells; d) to demonstrate the efficacy advantage of the prodrug derived nanoparticles in vivo in a rodent model; e) to microscopically characterize the impact of these agents on apoptosis and cell proliferation through STAT-3 inhibitory pathway.
Computational techniques, in particular molecular dynamics (MD) simulations, could provide molecular insights that may help rationally manipulate self-assembled structures of prodrugs even before performing the actual preparation. Our approach offers an opportunity to study assembled structure of a phospholipid prodrug via coarse-grained dissipative particle dynamics (DPD) simulations. We investigate the process of self-assembly of nanoparticle structures of Pro-nifuroxazide by DPD19,20 simulations. The simulation results demonstrated that the self-assembly morphologies of the Pro-nifuroxazide can lead to ~12 nm sized particles. In addition, both the molecular structure and the formation mechanisms of the Pro-nifuroxazide nanoparticles are systematically disclosed through the performed simulations. A three-layer core-shell-shell nanoparticle structure is also observed in the simulation. Furthermore, potential of mean force calculation of membrane insertion free energy of the prodrug indicates improved prodrug-cell interactions that may facilitate cellular internalization as demonstrated by the in vitro experimental studies. Owing to such a structure, a greater than 200-fold increase in local drug concentration is achieved with this approach, resulting in induced anti-cancer activities in in vitro and in vivo. Numerous therapeutics agents incorporated into nanoparticles are significantly lost during systemic circulation before reaching to the target. Earlier10–13, it has been successfully demonstrated that by using a phospholipid-based prodrug system, the pharmacokinetics and volume of distribution of systemic drug delivery can be altered with improved bio-availability with lower toxicity. Here we demonstrated that phospholipid prodrugs in conjunction with fusion-triggered drug delivery may prevent premature drug diffusional loss during circulation and increase target cell bioavailability.
Results and Discussion.
Although nanoparticle delivery of therapeutic agents has been found to be effective in preclinical animal models,20–26 simultaneous pharmacokinetic tracking of individual nanoparticle27,28 components has shown that a major fraction of the drug can be prematurely lost in blood29,30 before the nanoparticles reach the target tissue. To improve stability of the drug and to prevent its premature release from nanoparticles during circulation to target tissues, an SN2 lipase labile phospholipid prodrug31–35 has been designed. The phospholipid prodrug is constructed by coupling the active pharmaceutical ingredient (API, i.e., nifuroxazide) through the SN2 acyl position (i.e., stereospecific hydroxyl group of the second carbon of glycerol). The hydrolytic cleavage of the SN2 ester bond signifies a critical step to trigger second messengers, e.g., the conversion of liberated arachidonic acid into eicosanoids, which exert a wide range of physiological and pathological effects along with other activations.36,37 Enzyme triggered drug release is well described in many literature reports for pro-drug therapies.38–40 It is hypothesized that the resulting phospholipid prodrug could stably self-assemble into a ‘prodrug nanoparticles’ positioning the drug in a protective hydrophobic environment during circulatory transit to the target site. Once in the proximity to the cell, contact-mediated streaming of Pro-nifuroxazide into the outer leaflet of the target cell membrane would be facilitated. Translation of the Pro-nifuroxazide from the outer leaflet into the inner leaflet would lead to the rapid distribution of the prodrug throughout the intracellular membranes followed by active drug liberation by target cell phospholipase(s).
Computer simulation offers a powerful technique to visualize, characterize, and optimize different features and designs of such complex molecular systems such as the structure of a Pro-nifuroxazide nanoparticle. A molecular level simulation technique will allow to characterize and rationally optimize the experiments in a more efficient manner prior to their actual preparations. To enhance the efficiency of the self-assembly simulations, a coarse-grained (CG) DPD19,20 model (Figure 1A, Figure S1) has been employed where each chemical group is represented as a single interaction site. Previously, CG DPD simulations have been employed to successfully investigate the structure of lipid bilayers, amphiphiles and polymers.41–47
Figure 1.
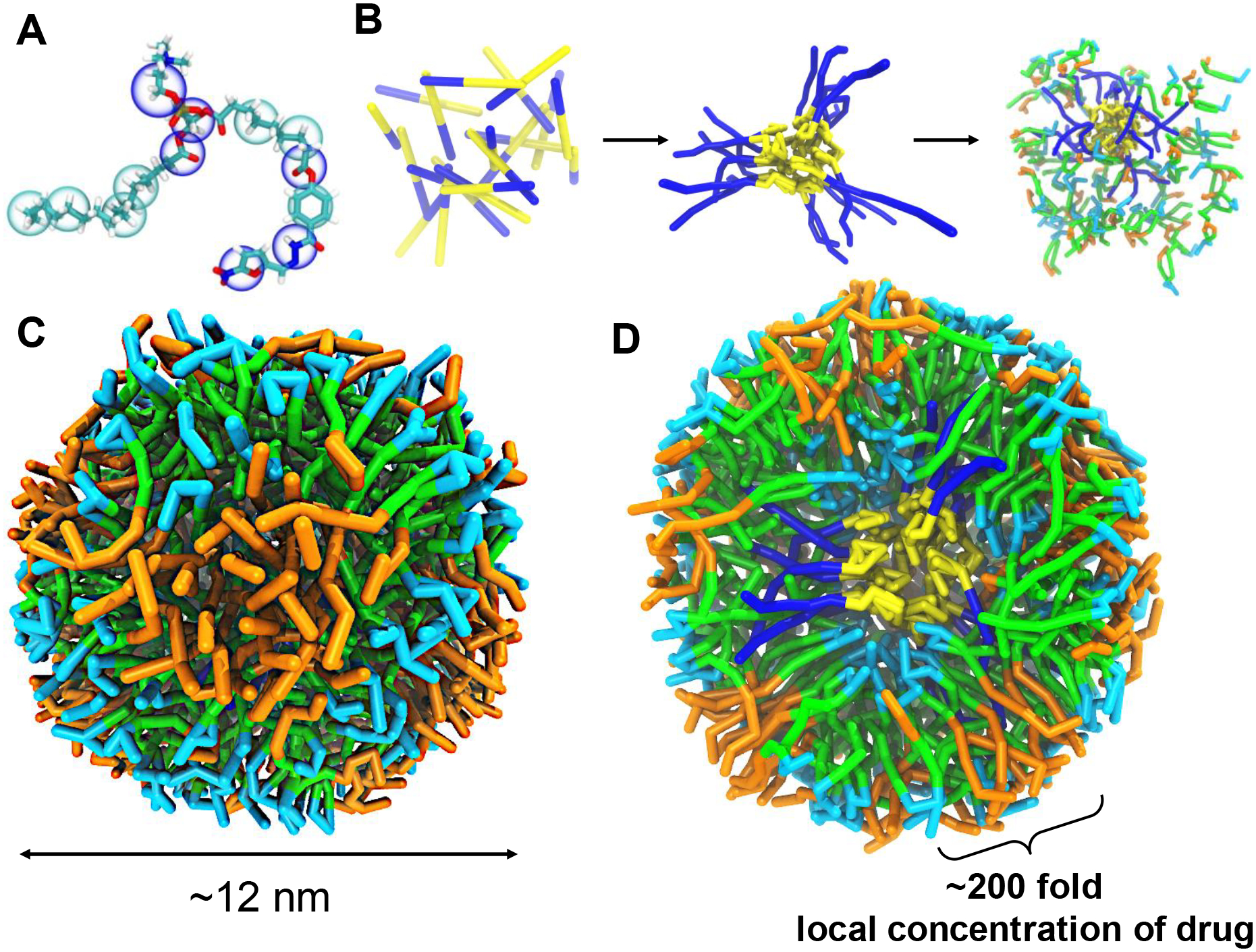
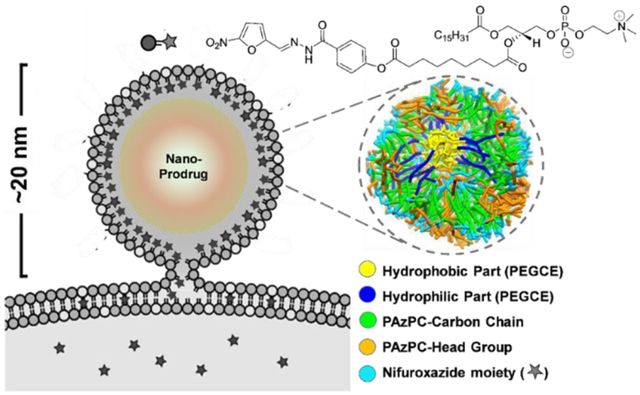
Pro-nifuroxazide nanoparticle structure resulted from coarse-grained (CG) dissipative particle dynamics (DPD) simulations of the self-assembly process. (A) CG representation of Pro-nifuroxazide. (B) Steps for the simulations before the self-assembly of Pro-nifuroxazide molecules. (C) A representative structure of a Pro-nifuroxazide nanoparticle. (D) Cross-section of the Pro-nifuroxazide nanoparticle. Color code: hydrophobic carbon chain of PEGCE (yellow), hydrophilic polyethylene glycolic part of PEGCE (blue), carbon chain of PAzPC in outer shell (green), PAzPC head group (light blue), and nifuroxazide moiety (orange).
We present a concerted approach combining simulation and experimental procedures to realize the preparation of a model nano-assembly using a repurposed drug, nifuroxazide, for cancer therapy. Nifuroxazide is an FDA-approved oral nitrofuran antibiotic, commonly used to treat colitis and associated diarrhea. The drug has been acknowledged to demonstrate anti-neoplastic properties via the active inhibition of pathways modulated in CSCs.48–50
Structure of prodrug nanoparticle modeled by coarse-grained dissipative particle dynamics simulations.
The self-assembly process was simulated in two steps using coarse-grained model parameters (Table S1), so as to mimic the experimental steps involved in preparation of the nanoparticles (Figure 1B, Figure S2). In the first step, CG DPD simulations were carried out to allow polyethylene glycol cetyl ether (PEGCE) chains to self-assemble into small core particles within the hydrophobic environment of cyclohexane as a solvent. In the second step, simulations were performed to capture self-assembly of prodrug molecules around the PEGCE core particle, during which the solvent was switched to water and the PEGCE core was fixed. These self-assembly simulations resulted in a three-layered, core-shell-shell structure for the Pro-nifuroxazide nanoparticles. The hydrophilic chains of PEGCE formed the core of the nanoparticle. Hydrophobic components, composed of PEGCE carbon chains and PAzPC lipid tails, form the middle layer. Hydrophilic PAzPC lipid head groups form the outer layer of the nanoparticles. With an average diameter of 12 ± 2 nm, the self-assembled Pro-nifuroxazide nanoparticles were found to be stable during the performed simulations (Figure 1C). Their core-shell-shell structure was maintained throughout the simulations, once the particles were fully developed. The hydrophilic chains of PEGCE formed the core (~1 nm in thickness) of the nanoparticle. Hydrophobic components, composed of PEGCE carbon chains and PAzPC lipid tails, form the middle layer (~4 nm in thickness). Hydrophilic PAzPC lipid head groups form the outer layer (~1 nm in thickness) of the nanoparticles. Each of these islands could be easily accessed from the particle surface (Figure 1D; Figure S3), yielding highly concentrated regions of drugs at the nanoparticle’s surface. Compared to a free nifuroxazide solution at the same molar concentration, the designed prodrug nanoparticle results in a remarkable increase of >200 times in local drug concentration.
In its prodrug form, the nifuroxazide is covalently conjugated to the oxidized lipid, and thus, amphiphilic and behaves similarly to a modified lipid molecule to assemble into a nanoparticle. For simulation studies, the ratio between different components, i.e., PEGCE and Pro-nifuroxazide, was optimized to reach a stable assembly. Number of molecules for each component in the simulation was derived based on initial prodrug molar concentration and resulted yield. Averaged size of the nanoparticle observed from simulation agrees with the experimental.
The ~240-fold difference in drug concentration was obtained nominally from the concentration of the prodrug in a nanoparticle divided by the experimentally measured nifuroxazide free form (non-prodrug, without nanoparticle formation) concentration. In its prodrug form, the nifuroxazide is covalently linked to the oxidized lipid, which co-assembles into a nanoparticle with PEGCE. With this approach, every single lipid tail will comprise one single drug molecule and thereby generates a high localized concentration of the drug.
The different nifuroxazide structures are the nifuroxazide as a small molecule (free form) and in its prodrug form. In its free form, the nifuroxazide molecules are freely dissolved in water, and thus, limited by its solubility. In its prodrug form, the nifuroxazide is covalently conjugated to the oxidized lipid, and thus, amphiphilic and behaves similarly to a modified lipid molecule to assemble into a nanoparticle.
Synthesis of Pro-nifuroxazide and release of API with external stimulation.
Computer simulations paved the path for the preparation of prodrug nanoparticles from Pro-nifuroxazide with optimized amount, ratio and medium of preparation. Firstly, Pro-nifuroxazide was synthesized by conjugating nifuroxazide with PAzPC followed by adding this molecule to micellar assembly of PEGCE in a molar ratio of 15:1, respectively in a mixture of water: organic solvent. Replacement of all the organic solvents allowed the preparation of prodrug nanoparticles. The synthesis of Pro-nifuroxazide involved a straightforward carbodiimide mediated coupling of nifuroxazide and 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAzPC). In a typical procedure, PAzPC (0.05 mM), EDC (0.1 mM) and DMAP (catalytic amount) were mixed in anhydrous CHCl3 solution (1 ml) and was allowed to stir for 15 min at ambient temperature. To this mixture nifuroxazide (0.05 mM) was added and then was stirred for 24 h at room temperature. The completion of the reaction was monitored by thin layer chromatography (silica). The resultant organic solution was then washed several times with water and extracted with excess dichloromethane to afford the desired compound as a pale-yellow solid in 56% yield. The compound was isolated and purified by preparative thin layer chromatography (prep-TLC), and the structure was confirmed by 1H-NMR (Figure 2G-a), 13C-NMR (Figure 2G-b), LRMS, and HRMS (Figure S4) spectroscopic analyses.
Figure 2.
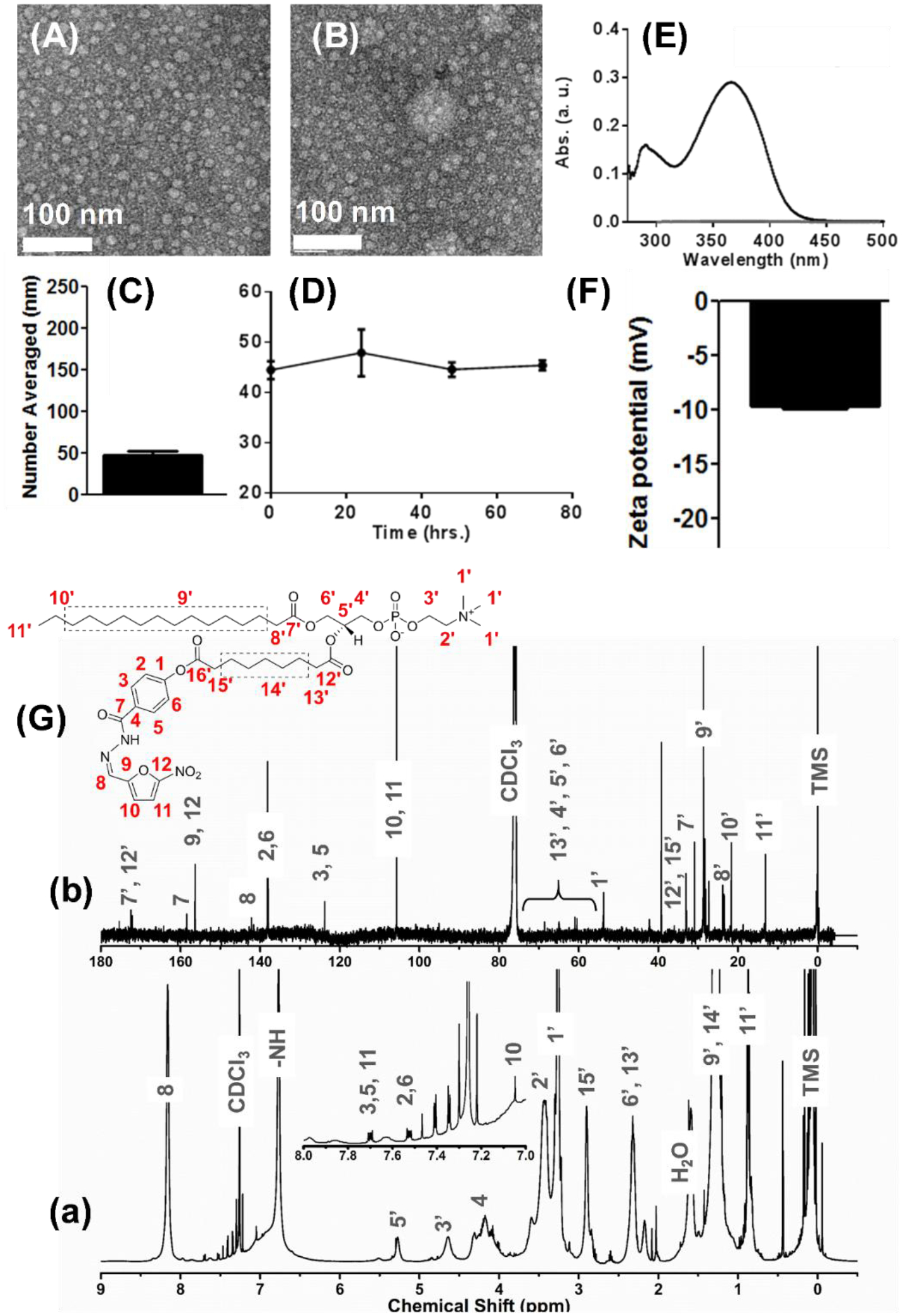
Physicochemical characterization of different chemistries and formulations of Nifuroxazide. (A-B) Anhydrous morphology of Pro-nifuroxazide nanoparticles (NPs) obtained using transmission electron microscopy in negative staining mode (TEM, uranyl acetate) from different section of electron grids. (C) Hydrodynamic diameter of Pro-nifuroxazide NP and (D) stability of the self-assembled Pro-nifuroxazide nanoparticle (pH 7.4). (E) UV-vis spectrum of free nifuroxazide molecules in water revealing characteristic absorbance of the drug molecule. (F) Zeta potential; (G) nuclear magnetic resonance (NMR) spectra of the Pro-nifuroxazide (a) 1H and (b) 13C traces (CDCl3).
Multiple family members of different types of phospholipases, serine hydrolases or other hydrolases are abundant in cancer cells. The presence of these enzymes creates a rich repertoire of potential activating agents within target cells. Phospholipase A2 (PLA2) enzymes are members of a superfamily that catalyze the hydrolysis of the SN2 ester bond in a variety of phospholipids, thereby releasing a free fatty acid and a lyso-phospholipid. The hydrolytic cleavage of the SN2 ester bond signifies a critical step to trigger second messengers, e.g., the conversion of liberated arachidonic acid into eicosanoids, which exert a wide range of physiological and pathological effects. To further assess the potential susceptibility of Pro-nifuroxazide toward acidic pH and enzyme-based liberation, the prodrug (500 μl) was incubated for 2h with 0.28 mg PLA2 or phosphate buffered saline at pH 4.5. The samples were continuously agitated on a nutator to avoid any settling. The mixture was worked up and analyzed by HRMS to observe signature peaks of cleaved and liberated nifuroxazide. For both occasions, HRMS confirmed the release of nifuroxazide from the prodrug under enzymatic or condition of low pH (Figure S4). Cytoplasmic pH in normal cells is between 6.8 to 7.2 but in cancerous cells and tumors it can drop up to pH 4.5 along with presence of abundant PLA2.51 Thus, presence of both conditions can work independently and need not to be dependent on each other. Though PLA2 are found in extracellular space too, the possibility of cleaving Pro-nifuroxazide by PLA2 in extracellular space will be much less compare to in membrane or intracellular space due to significantly low level. This selectivity will lead to significant activation of nifuroxazide only in intracellular space.
Synthesis and characterization of prodrug nanoparticles.
Pro-nifuroxazide nanoparticles were co-self-assembled via solvent evaporation (H2O: THF = 4:1) in the presence of PEGCE. Extensive physicochemical characterizations of the nanoparticles were subsequently carried out (Figure 2) to confirm the integrity of the structure, morphology and robustness of the system. Transmission electron microscopy (TEM) measurements revealed spheroidal particles of expected dehydrated state size (10 ± 2 nm, Figure 2A–B). Hydrodynamic diameter of Pro-nifuroxazide nanoparticles was determined by dynamic light scattering (DLS) experiment. It is critical to find out the hydrodynamic size of the nanoparticles for systemic applications as it may directly influence their bio-distributive properties. The resultant number averaged hydrodynamic diameter of prodrug nanoparticles was 38 ± 12 nm (Figure 2C). As evident from the simulation studies, pro-nifuroxazide NPs was found to be 12 ± 2 nm, which is corroborated well with the results obtained from anhydrous state TEM and hydrated state DLS values. The discrepancy in hydrated and anhydrous state diameter values can be explained due to the possible loss of water molecules of hydration during the sample preparation for TEM studies. Lack of hydration layer around the nanoparticles in simulation studies in silico, resulted in comparable size as obtained by TEM. UV-Vis spectroscopy was used to confirm the incorporation of nifuroxazide within the nanoparticles. (Figure 2E; Figure S5B, C). Characteristic absorbance signature nifuroxazide (Figure 2E) was used for the comparison (Figure S5C). The electrophoretic potential of Pro-nifuroxazide nanoparticles was found to be −10 ± 1 mV (Figure 2F). The discrepancy between hydrodynamic and anhydrous diameter was noticed which suggests the ‘soft’ nature of these particles and due to the flattening of the particles over the TEM grid. Hydrodynamic size measurements revealed that the nanoparticles were remarkably stable, without any noticeable aggregation in aqueous media at neutral and pH of 4.5 at 37°C (Figure 2D). Nanoparticles incubated in aqueous media containing 10% FBS, a widely used serum in cell culture and biological fluids, also demonstrated a stable profile. The stability of nanoparticles in different conditions can also be correlated with the negative electrophoretic potential (Figure 2F) values which result in particle repulsion and prevent aggregation or clumping. Drug and pro-drug nanoparticles were also investigated for stability via DLS measurements along 72h at pH 7.4 (Figure S5A). UV-vis absorbance was found to decrease with decrease in concentration of free available nifuroxazide (Figure S5B) with no specific change in absorbance pattern of the spectrum. It indicates the possibility of decreasing UV-Vis absorption in case of nanoparticle incorporation of nifuroxazide, which was achieved by converting it to pro-nifuroxazide NP and found to show very low absorption intensity (Figure S5C). The composition of each level has been illustrated in Figure 1 to indicate a tri-layered structure.
To verify the benefit of this tri-layered assembly, two more control nanoparticles were prepared and tested for their stability. nanoparticle with nifuroxazide encapsulated in lipid assembly with PEGCE core (Nifuroxazide NP) or Pro-nifuroxazide assembly without PEGCE core (Pro-nifuroxazide NP (no-core)). It was observed that where all other formulations showed destabilization of nanoparticles with high aggregation and increased hydrodynamic diameter in serum, pH 4.5 and pH 12 medium (Figure S6A, B), Pro-Nifuroxazide NPs found to be unstable only at pH 4.5 (Figure S6C). This observation is indicative of the overall stability of the colloidal system due to the presence of this multi-layered prodrug–solid-core system. Furthermore, the instability of the system is presumably indicating the hydrolysis of SN2 ester linkages to liberate nifuroxazide from Pro-Nifuroxazide. Stability of Pro-Nifuroxazide NPs in water, serum and pH 12 at 37°C indicate its long-term storage stability while destabilization at pH 4.5 supports that the active drug can be released either at a low pH environment or enzymatically.
Response of medium on drug release.
It was noted that none of the nanoparticles showed any burst release, but extent of sustained release varied vastly depending on suspension medium (Figure S7). It was found that Pro-nifuroxazide NPs were most stable formulation in water and serum mediums while under pH 4.5 a maximum drug release was reported, probably due to cleavage of drug molecule from pro-drug under low pH condition. Pro-nifuroxazide NP (no-core) were also responsive to serum incubation plausibly due to lack of core in nanoparticle which plays role in stabilizing the nanoassembly. Nifuroxazide NPs were most vulnerable among all in all the conditions due to no need of any trigger to break the assembly beyond change in medium temperature and found to be release maximum drug molecules in water medium itself making it least desirable nanoparticle for nanomedicine studies.
Protein interaction properties.
Systemic delivery of nanoparticles counters protein association abilities as one of the drawbacks to lose their efficiency. Protein interaction with different nanoparticles could be corroborated with a decrease in electrophoretic potential. After incubation with Nifuroxazide NP and Pro-Nifuroxazide NP (no-core) a significant decrease in zeta potential was indicative of the formation of protein corona while Pro-Nifuroxazide NPs remained largely unaffected (Figure S8). Similar trends were obtained from protein assay on protein associated with different nanoparticles (Figure S9). The amount of unbound protein was found maximum in case of Pro-Nifuroxazide NPs indicating minimum protein association.
It was found that even after seven days of incubation with 10% FBS, Pro-Nifuroxazide NPs were found to be significantly negative change with no significant change, indicating high stability. Other formulations either without core (Pro-Nifuroxazide NP (no-core)) or without pro-drug (Nifuroxazide NP) found to be highly associative with FBS proteins and decreasing their negative potentials in same order (Figure S8B, C). Prodrug protection of head region hydroxyl moiety of PAzPC likely prevents associative interaction with high protein molecules from FBS.
Size of the nanoparticle predicted by molecular dynamic simulation studies were calculated based on assuming the plausible number of PEGCE and Pro-nifuroxazide molecules would constitute a single particle in silico conditions and found to be ~12 nm. A hydrodynamic diameter distribution of ~30 nm from dynamic light scattering techniques was found representing number averaged distance between centers of two adjacent nanoparticles in hydrous state. This value does not signify only the structural boundaries of nanoparticles but also comprises layer of hydration present on nanoparticles and distances encountered due to repulsive forces between two similar charge surface nanoparticles. This further explains our observation that these nanoparticles were slightly bigger in suspended state which was condensed in size to reach ~20 nm in TEM due to loss of water of hydration and distances involved due to repulsive forces.
Increased membrane binding affinity shown by potential of mean force calculation.
Chemical linking of nifuroxazide to an oxidized lipid (PAzPC) not only enables the molecule to self-assemble into stable NPs, but also enhances the cell membrane uptake of an individual prodrug compared to the original nifuroxazide molecule (Figure 3A). The latter was justified by the potential of mean force calculations using atomistic simulations of membrane binding of the drug and prodrug molecules (Figure 3B–C). In these calculations, membrane insertion free energies of a nifuroxazide drug and the nifuroxazide moiety of a single prodrug were compared (Figure 3C).
Figure 3.
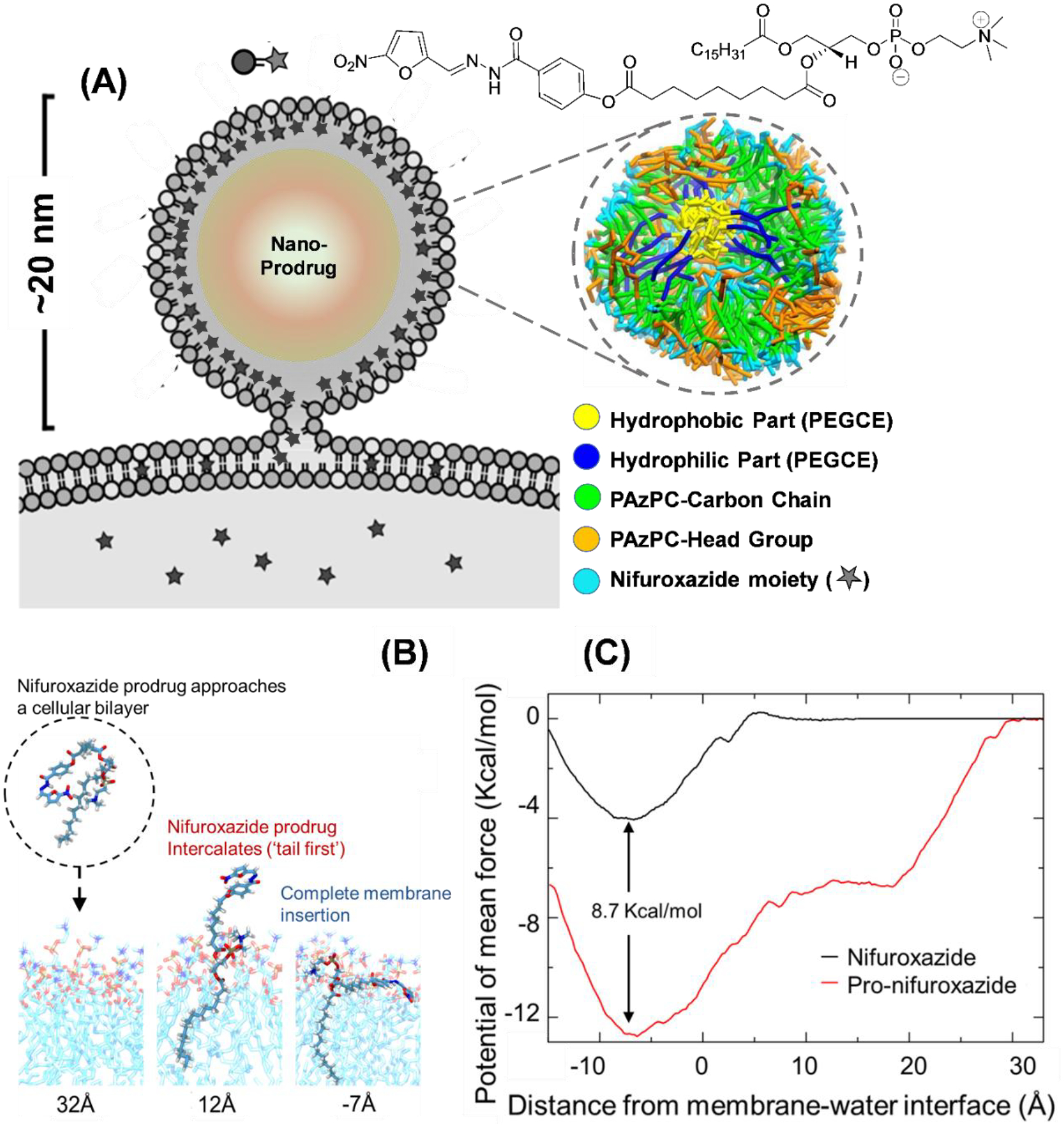
(A) Schematic representation of a prodrug nanoparticles interacting with cell membrane and cross section of simulated Pro-nifuroxazide nanoparticle structure with segregation of various components of the Pro-nifuroxazide and PEGCE molecules. (B) Conformations of Pro-nifuroxazide as it approaches the membrane. Distances below the images indicate the position of the nifuroxazide group relative to the membrane interface (z=0). Strong interaction of the PAzPC lipid tail with membrane increases the prodrug-membrane binding range and affinity. (C) Potential of mean force curves for individual nifuroxazide (black) or Pro-nifuroxazide (red) molecules inserting into a POPC membrane. The reaction coordinate (the abcissa) is the distance between center of mass of the nifuroxazide moiety and membrane interface. The free energy curves were calculated from −15 to 35 Å, that is, a range between close positioning of the drug moiety to the membrane center at z = −20 Å, and its complete dissociation from the membrane (35 Å).
A larger membrane affinity, −8.7 ± 0.5 kcal / mol, of the nifuroxazide moiety in a prodrug form has been found compared to the parent drug. Such an increase in membrane binding affinity is due to the strong interaction between the membrane and the PAzPC group of the Pro-nifuroxazide. As observed in the simulations, the Pro-nifuroxazide started to interact with the membrane via the PAzPC lipid tail when the nifuroxazide moiety was 30 Å away. As the PAzPC hydrophobic tail inserted into the membrane,21 it escorted the nifuroxazide moiety to membrane (Figure 3B). Optimal membrane partitioning of the nifuroxazide moiety in the Pro-nifuroxazide is the same as the free nifuroxazide drug, that is, ~7 Å below the membrane-water interface, similar to aromatic protein side chains, such as tyrosine and tryptophan.52
Figure 3A shows a typical nanoparticle structure, in its cross section, observed from simulations. As shown in atomistic potential of mean force calculation, the drug component of the prodrug prefers to bind to the hydrophobic and hydrophilic interface of a cell membrane. Therefore, in the nanoparticle, the drug component mainly binds to the phospholipid head group region and occasionally to the core interfaces, as both of these two regions are interfaces between hydrophobic and hydrophilic environments and their cell membrane interactions.53–55
Pro-nifuroxazide enhances cell growth inhibition in vitro.
Experimentally, functional superiority of the prodrug over the free drug was estimated by in vitro cancer cell growth regression in model cell lines of human breast origin, MCF-7 and MDA-MB231. A 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetra-zolium bromide (MTT) assay was performed on MCF-7 (Figure 4A) and MDA-MB231 (Figure 4B, Figure S11A) after 72 h treatment with the nifuroxazideor the Pro-nifuroxazide at concentrations ranging from 0.5 to 20 μM. It was noticed that conversion of nifuroxazide to Pro-nifuroxazide improved the cancer cell growth regression; the inhibition was 90% for Pro-nifuroxazide compared to 60% when treated with drug in MDA-MB231 cells. Similarly, an increase of 25% was observed in MCF-7 cells with the growth inhibition of 95% when treated with Pro-nifuroxazide compared to the drug-treated cells. This was achieved along with improving the IC50 in MCF-7 from 5 ± 0.5 to 2 ± 0.2 μM (Figure 4C) and 14 ± 2 to 10 ± 2 μM in MDA-MB231, respectively (Figure 4C, Figure S11B).
Figure 4.
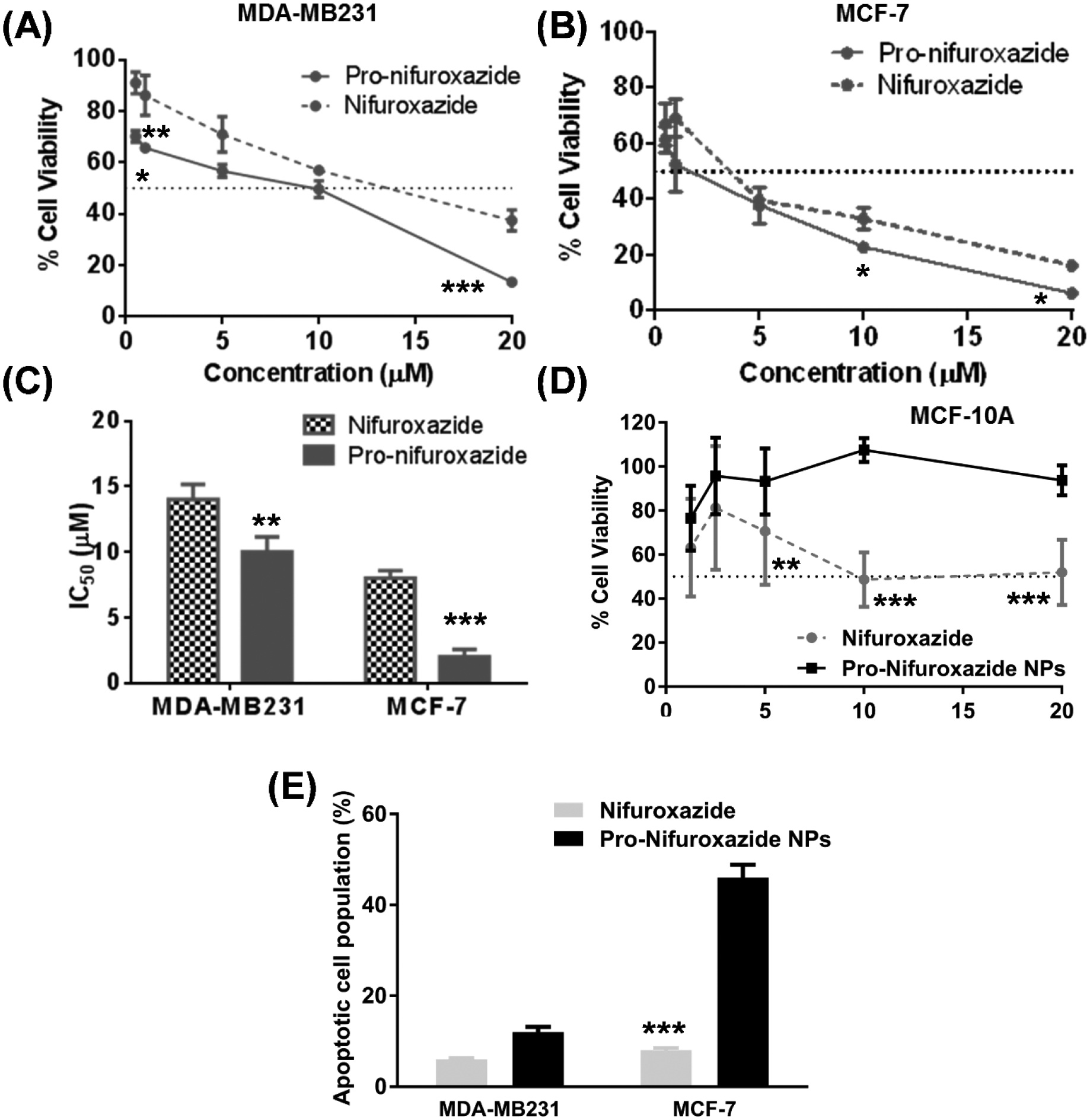
MTT assay performed on (A) MDA-MB-231 and (B) MCF-7 cells after 72 h treatment of nifuroxazide and Pro-nifuroxazide at concentrations ranging from 0.5 to 20 μM and (C) comparison of IC50 values; (D) selective low response of Pro-nifuroxazide nanoparticles demonstrated in non-cancerous MCF-10A breast cells; (E) summary of apoptotic cell population (20 μM). Biostatistical analysis was performed using ONE Way ANOVA with post Bonferroni test. Here *, ** and *** represent p values <0.05, 0.01 and 0.001, respectively.
Selectivity of prodrug and prodrug nanoparticle for cancer cell.
Selective growth regression of cancer cells has been a major goal for efficient, side-effect free therapy.56,57 Agents under investigation were subjected to cell viability measurements in non-cancerous cells (MCF-10A) of human breast origin.58,59 It was found that where nifuroxazide itself could inhibit cell growth by ~50%, Pro-nifuroxazide nanoparticles resulted in only 10% inhibition (Figure 4D) following a 48 h treatment at a concentration of 20 μM. This result suggests that the conversion of the API to Pro-nifuroxazide nanoparticles improved the selectivity of the agent towards cancer cells, producing little or negligible effect against non-cancerous population of cell.
Enhanced functional efficacy of prodrug nanoparticles over molecular prodrug in vitro.
As described above, the simulation studies indicated a remarkable improvement in the local concentration of drug to as high as ~240 folds when assembled into nanoparticles. This property should also reflect in their functional advantage. An in vitro study was performed to evaluate this. It was noticed that the conversion of Pro-nifuroxazide to Pro-nifuroxazide nanoparticles improved the cancer cell growth regression by improving IC50 in MDA-MB231 from 10 ± 2 to 6 ± 1 μM (Figure S11A), and in MCF-7 from 2 ± 0.5 to 1 ± 0.2 μM (Figure S11B), respectively (Figure S11C). To investigate the effect of treatments on cancer cell growth, bright field images were obtained from MCF-7 (Figure S11D–F) and MDA-MB231 (Figure S11G–I) cells treated with the drug (Figure S11E, H) or Pro-nifuroxazide nanoparticles (Figure S11F, I). Improved regression was observed for Pro-nifuroxazide nanoparticle compared to nifuroxazide at equivalent concentrations (20 μM).
Cell internalization mechanism of Pro-nifuroxazide NPs.
Drug efficacy is known to be affected by their mode of cellular entry and in turn highly dependent on specific chemistry of nanostructures. Among the most possible cell entry mechanisms of Pro-Nifuroxazide NPs, endocytosis was found not a major mode of transportation while electron microscopically condition of membrane fusion could be captured in cell-TEM experiments. It was found that use of endocytosis blockers (NaN3/DOG (1), Nystatin (2), Dynasore (3) and CPM (4)) could reduce the cell viability similar to cells treated with Pro-Nifuroxazide NP alone indicating no major role of endocytosis in cellular entry of Pro-Nifuroxazide NPs. Though use of high sucrose concentration found to be effective and improved cell viability indicating the involvement of some other energy dependent processes (Figure 5A). To verify the possibility of membrane fusion based cellular entry, a high electron density derivative of Pro-Nifuroxazide NP, Iodine labeled Pro-Nifuroxazide NPs (I-Pro-nifuroxazide NPs) was prepared and incubated with MCF-7 cells before fixing them to image under TEM machine. Highly informational nanoparticle fusing with cell membrane images could be captured to indicate the significant role of membrane fusion in cellular entry of Pro-Nifuroxazide NPs (Figure 5B–D). Cellular internalization of Pro-Nifuroxazide NP was also visualized by performing confocal imaging on MCF-7 cells incubated with rhodamine loaded Pro-Nifuroxazide NPs (Rh-Pro-nifuroxazide NPs) and found to be distributed in intra-cellular cytoplasmic space specifically (Figure 5E–H).
Figure 5.
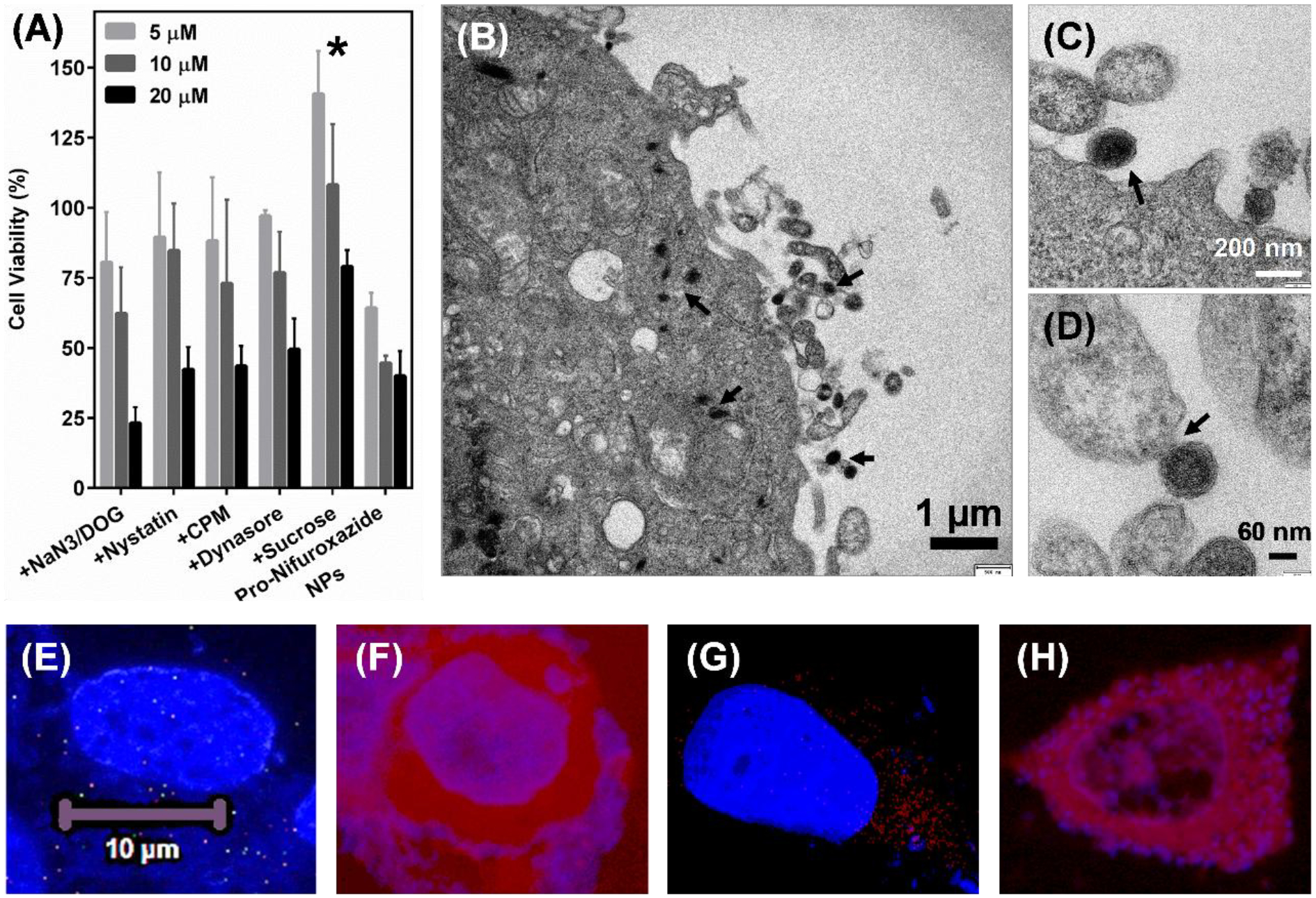
Cell internalization mechanism of Pro-Nifuroxazide NPs. (A) Inhibitor pre-incubation study performed on MCF-7 cells after Pro-Nifuroxazide treatment at 20, 10 and 5 μM concentration of Nifuroxazide or Pro-Nifuroxazide NPs alone. (B-D) Cell TEM performed on I-Pro-nifuroxazide NPs) after 30 min of incubation showing position of membrane fusion as mode of cellular entry. Cell incubated with rhodamine alone (E, F) and Rh-Pro-nifuroxazide NPs (G, H). Here E and G represent DAPI stained cellular nucleus while F and H shows Rh distributed in intracellular space. Cells were incubated with rhodamine and Rh-Pro-nifuroxazide NPs for 4h. Here * represents p values <0.05 and 0.001, respectively.
Mechanistic response of nifuroxazide prodrug and prodrug nanoparticle.
The induction of apoptosis by Pro-nifuroxazide nanoparticle was analyzed as sub-G0/G1 cell population in cell cycle study. Apoptotic path induction leads to cell shrinkage, membrane blebbing and fragmentation of genomic DNA into oligo-nucleosomal subunits.60,61 Incubation of apoptosis-induced cells with propidium iodide (PI) allows very low PI intercalation in genomic DNA due to its apoptosis mediated fragmentation. It leads to low fluorescence cell population in cell cycle analysis. Thus, apoptotic cell population can be categorized as PI stained population with lowest fluorescence. The mechanism of nifuroxazide on cancer cell growth regression was estimated by PI staining assay on MCF-7 (Figure S12A–C) and MDA-MB231 (Figure S12D–F) cells. Treatments with nifuroxazide (Figure S12B and E) or Pro-nifuroxazide nanoparticles (Figure S12C and F) produced different levels of apoptotic cell population. The calculated apoptotic cells percentage were compared for nifuroxazide and Pro-nifuroxazide nanoparticles. It was found that Pro-nifuroxazide nanoparticles improved the percentage of apoptotic cell compared to nifuroxazide treatment in MDA-MB231 from 6 ± 0.5 to 12 ± 2%, and in MCF-7 from 8 ± 1 to 46 ± 5 (Figure 4E).
The purpose of making a multi-layered nanoparticle system was to generate a stable nanoparticle system for drug delivery where drug is protected in a ‘prodrug’ form and core is also designed to provide systemic stability. By virtue of the design of the prodrug, the drug is retained stably within the shell since it is covalently linked to the oxidizable end of the lipid. Additionally, a polymeric core is also devised with a long carbon chain and melting point slightly above the physiological temperature. The presence of this core improves the stability of the colloidal system due to paired hydrophobic interactions with carbon chains of lipid and facilitates its intracellular delivery. It was found that nanoparticles with no core-shell-shell system as prepared by either nifuroxazide encapsulated in lipid assembly with PEGCE core (Nifuroxazide NP) or Pro-nifuroxazide assembly without PEGCE core (Pro-nifuroxazide NP (no-core)) showed considerably higher IC50 values in both MCF-7 and MDA-MB231 cells at both the experimental time points (Figure S13). Within these control formulations, Nifuroxazide NPs were found to be better than no-core assemblies. These observations support the benefit of using core-shell-shell arrangement of entities in Pro-nifuroxazide cored nanoparticles and were used for all the experimental procedures.
In vitro assays to delineate inhibition of STAT-3 expression at RNA and protein level.
The improved efficacy of Pro-nifuroxazide nanoparticles in cancer cell growth regression compared to nifuroxazide was successfully established by MTT assay, cell growth density analysis, and the determination of apoptotic cell population. Prompted by our initial findings, it was important to establish whether the modifications of nifuroxazide to Pro-nifuroxazide and Pro-nifuroxazide nanoparticles follow the same STAT-3 inhibition pathway or not. The chemical modification through the conversion of a Pro-nifuroxazide and its changes based on its assembly needed to be scrutinized for mechanistic pathway alternation during cancer cell growth inhibition including sub-population of stem-like cancer cells. PCR and Western blot studies were performed to verify the STAT-3 inhibition using Pro-nifuroxazide and Pro-nifuroxazide nanoparticle. Improvement in STAT-3 inhibition was also quantified using Quantitative PCR (StepOnePlus RT PCR system, Applied BioSystems). Analysis on fold inhibition of STAT-3 expression was compared with house-keeping gene β-actin.62 Fold STAT-3 inhibition for treatment with nifuroxazide was found to be 11 ± 5 which marginally improved to 13 ± 4 for Pro-nifuroxazide, and 26 ± 5 for Pro-nifuroxazide nanoparticles (Figure 6A). The trend in STAT-3 inhibition associated well with cancer cell growth regression efficiency of Pro-Nifuroxazide nanoparticles. Results confirmed that for all the agents of interest, the mechanism of cancer cell growth regression remained the same for inhibition of STAT-3. Furthermore, Western blots could reveal the decreased level of protein phospho-STAT-3 (Figure 6B) after treatment with nifuroxazide by ~20%. This improved to significant levels of approximately 50% and 90% in the cases of prodrug and prodrug nanoparticle treatments, respectively (Figure 6C). It signifies the role of nifuroxazide, Pro-nifuroxazide and Pro-nifuroxazide nanoparticles in averting phosphorylation of STAT-3. Thus, the pathway followed by Pro-nifuroxazide nanoparticles still remained the same as nifuroxazide with multiple folds improvement in the level of STAT-3 mRNA suppression and then the associated phosphorylated protein.
Figure 6.
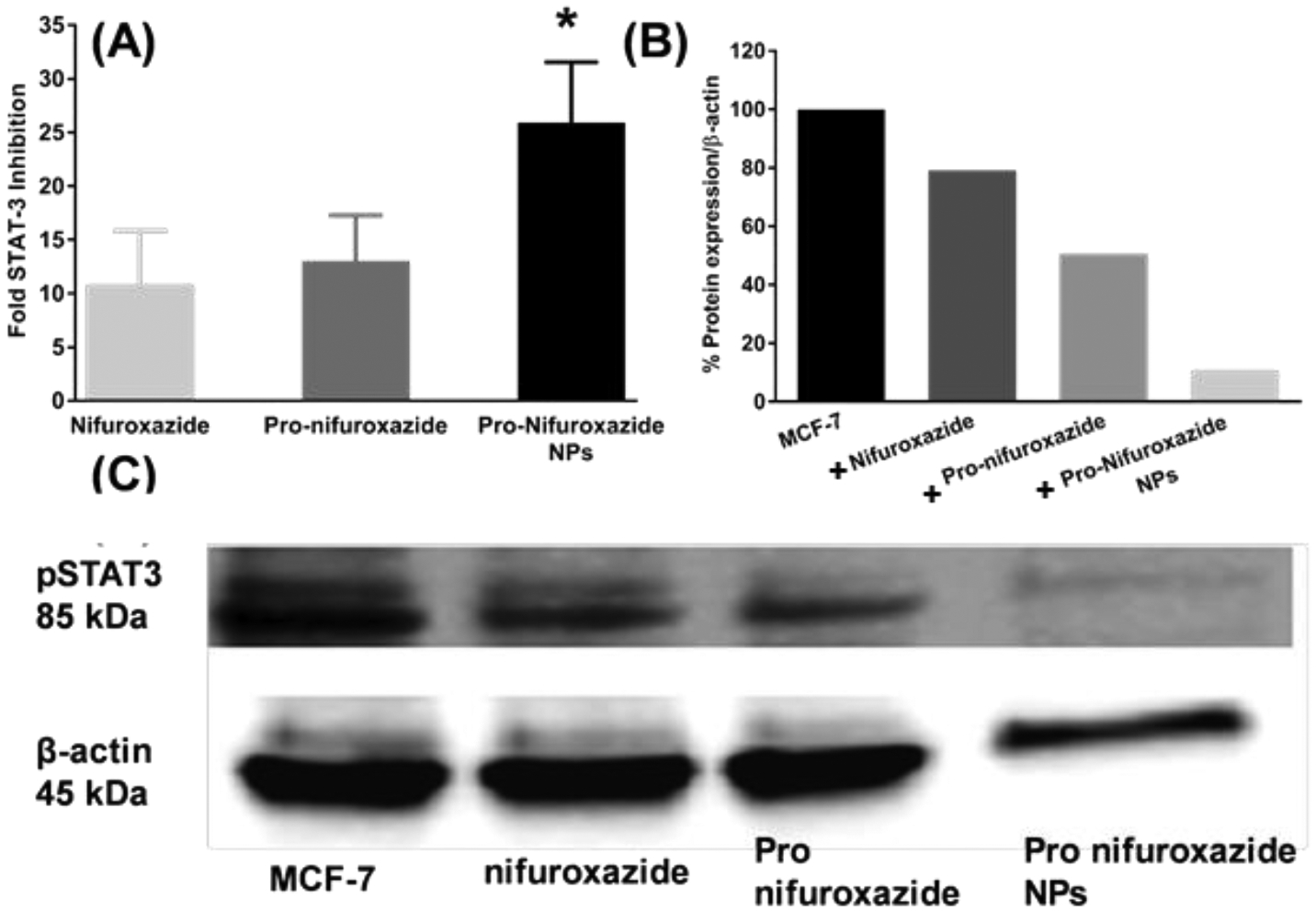
(A) STAT-3 inhibition study in MDA-MB231 cells at 48h incubation time point. Nifuroxazide was used in form of small molecule, Pro-nifuroxazide and prodrug nanoparticle at a concentration of 20 μM. Cells were incubated for 48h before collecting the RNA and performing PCR studies. Pro-nifuroxazide nanoparticle showed maximum inhibition in STAT-3 expression followed by Pro-nifuroxazide while nifuroxazide showed the minimum inhibition. (B) STAT-3 protein inhibition study in MDA-MB231 cells at 48h incubation time point. Nifuroxazide, Pro-nifuroxazide and Pro-nifuroxazide nanoparticle at final concentration of 20 μM were used. Cells were incubated for 48h before collecting the total protein and performing Western Blot studies (C). Pro-nifuroxazide nanoparticle showed maximum inhibition in STAT-3 expression followed by proPro-nifuroxazide while nifuroxazide alone showed the minimum inhibition. Biostatistical analysis was performed using ONE Way ANOVA with post Bonferroni test. Here * represents p values <0.05 and 0.001, respectively.
Functional and mechanistic activity of Pro-nifuroxazide nanoparticles in vivo.
The tumor regression ability of Pro-nifuroxazide nanoparticles was verified in vivo by treating xenografted tumors generated from MCF-7 cells grown in athymic nude mice (Figure 7A). Tumors grown to a minimum size of 0.5 × 0.5 cm2 were injected (40 μL) with control treatment (phosphate buffer) (Figure 7B, C) and Pro-nifuroxazide nanoparticles (Figure 7D, E) and on days of 0, 4, 8 and 12. The treatment was followed for 28 days before sacrificing the animal and collecting the tumor tissue (Figure 7C, E). The regression of tumor was found to be biologically significant for Pro-nifuroxazide nanoparticles (Figure 7D, F) compared to animals treated with buffer (Figure 7C, F). In the buffer-treated animals, the tumors continued to grow as high as 800% (Figure 7F) compared to the reduction in size for Pro-nifuroxazide nanoparticles treatment to as low as 400% (Figure 7G). Tumor reduction was corroborated with histopathologic analyses to detect tissue damage resulting from the treatment with Pro-nifuroxazide nanoparticles. On analyzing the tumor regression pattern in Figure 7F, a consistent inhibition in tumor volume was reported even after withdrawing the intra-tumoral Pro-Nifuroxazide NP injections on 12th day till 28th day of study. It indicates that plausibly these nanoparticles have intra-tumoral retention capabilities of around two weeks.
Figure 7.
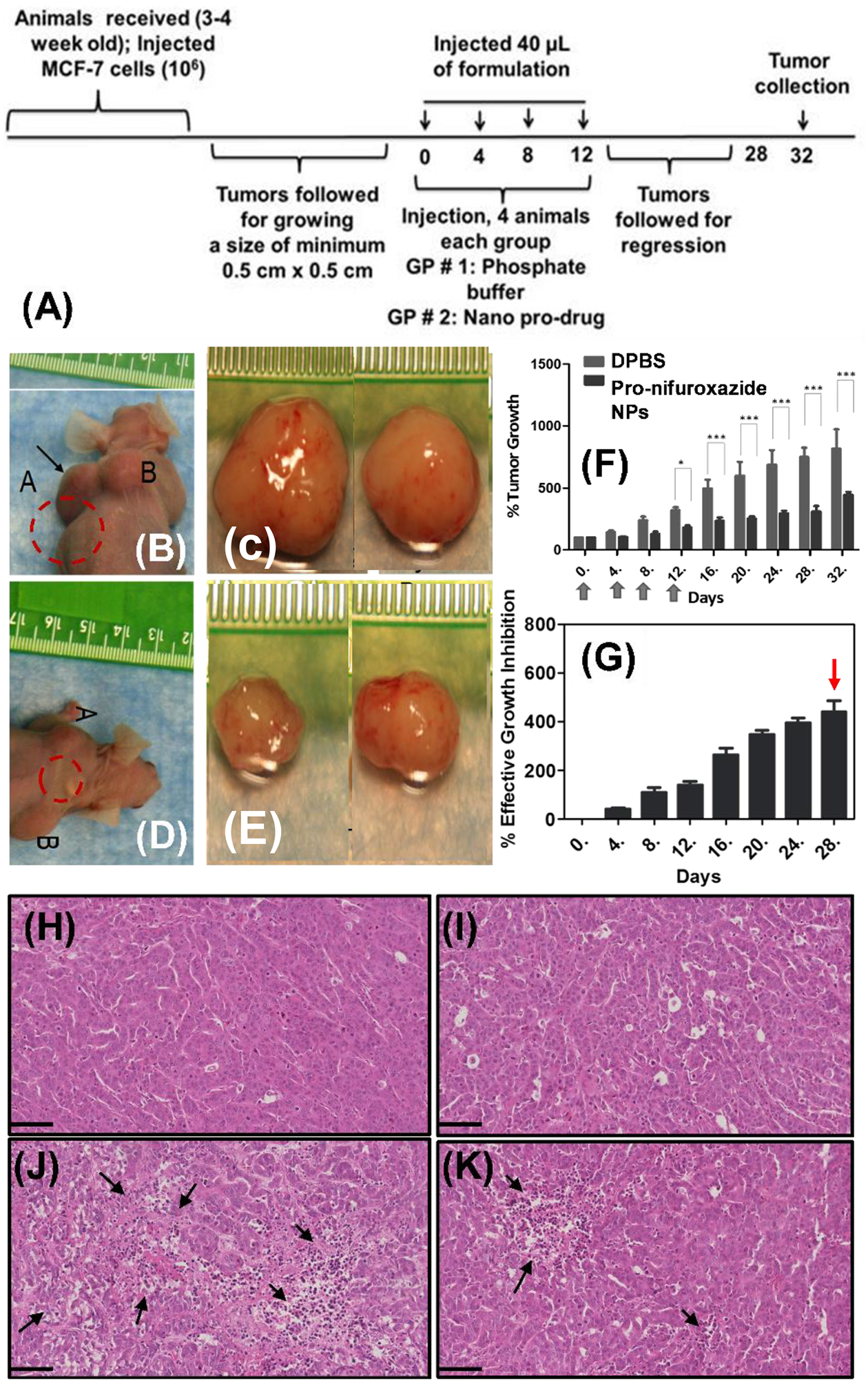
In vivo evaluation of tumor regression by treating xenograft tumors generated from MCF-7 cells grown in nude mice animal models. (A) Tumors were grown to a minimum size of 0.5×0.5 cm2 before injecting with phosphate buffer or Pro-nifuroxazide nanoparticles in 40 μL volume on day 0, 4, 8 and 12. Tumors were monitored for a total of 28 days (B, D) before sacrificing and (C, E) collected representative tumor tissue from animals treated with (B, C) phosphate buffer and (D, E) Pro-nifuroxazide nanoparticles on day 28. Analysis of (F) % tumor growth and (G) % effective growth inhibition across experimental time line for buffer and Pro-nifuroxazide NP treated animals. H&E staining on tumor sections from animals treated with (H, I) phosphate buffer and (J, K) Pro-nifuroxazide nanoparticles. Arrows represent areas of cellular degenerations in tumor sections from Pro-nifuroxazide nanoparticle treated animals. Biostatistical analysis was performed using ONE Way ANOVA with post Bonferroni test. Here * and *** represent p values <0.05 and 0.001, respectively.
These sub 50 nm sized nanoparticles would be passively accumulated in solid tumors due to the aberrant features of tumor vasculature and the poorly developed lymphatic system to pass through leaky vasculature. The concentration of PLA2 is only found in a significant amount in tumor cells capable of triggering pro-drug hydrolysis. Tumor cells are also known to have an abnormally acidic cytoplasmic pH due to Warburg effect,63,64 which have been found to be normalized in drug-resistant cells.65 Our results indicated that none of these external triggers (cytoplasmic pH or enzyme) are abundant in elevated level in MCF10A cells to cause liberation of the free drug and therefore avoids any significant detrimental side effect. Although some non-specific accumulation of nanoparticle is expected, the lack of trigger-able stimuli would prevent the pro-drug to undergo activation to do any considerable damage to non-cancerous tissue and cells.
Premature systemic drug loss is a major problem for drug delivery systems with encapsulated agents. Various strategies have been attempted to circumvent such leakages and improving assembly stability, however, a solution is still far-reaching. Here we show that a lipid-based prodrug-approach integrated with the introduction of a solid core may provide a novel alternative. In this approach, the drug is protected in a covalently stabilized ‘prodrug’ form. Following an intravenous administration of the nanoparticles (1 mg/ml) in mouse, a preliminary pharmacokinetic profile was studied. These particles follow a two-compartment bi-exponential model. The calculated half-lives were t1/2 (d) 30±1 and t1/2 (c) 285±10 min for the distribution and clearance phases, respectively.
Histopathological and Immuno-histochemical studies.
Representative tissue sections from animals treated with buffer (Figure 7H, I and S14) and Pro-nifuroxazide nanoparticles (Figure 7J, K and S15) were used to compare histological features generated after treatments. Hematoxylin/eosin (H&E) stained sections of tumors could show nuclear fragmentation and retracted cytoplasm if there is an occurrence of apoptotic features in tumor sections. Areas with nuclear fragmentation and retracted cytoplasm were clearly visible in representative sections of tumors from different animals treated with prodrug nanoparticles (Figure 7J, K and S15) with significant areas of interest shown with black arrows.
To image the expression of pSTAT-3 protein in Pro-nifuroxazide nanoparticle treated and buffer treated tumors, immuno-labeling was performed on paraffin-embedded tumor micro-sections. Immuno-labeling was performed by following standard protocol. β-actin was used as background protein for all the sections investigated. Representative immune-labelled cross sections of tumors treated with phosphate buffer saline (Figure 8A–C and S16) and Pro-nifuroxazide nanoparticles (Figure 8D–F and S17) revealed the down regulation activity of prodrug nanoparticles against pSTAT-3. Sections were incubated with pSTAT-3 antibody (red) and background protein β-actin (green) around cell nuclei stained with DAPI (blue) to show significantly high level of pSTAT-3 in phosphate buffer saline treated tumors compared to treated with prodrug nanoparticles.
Figure 8.
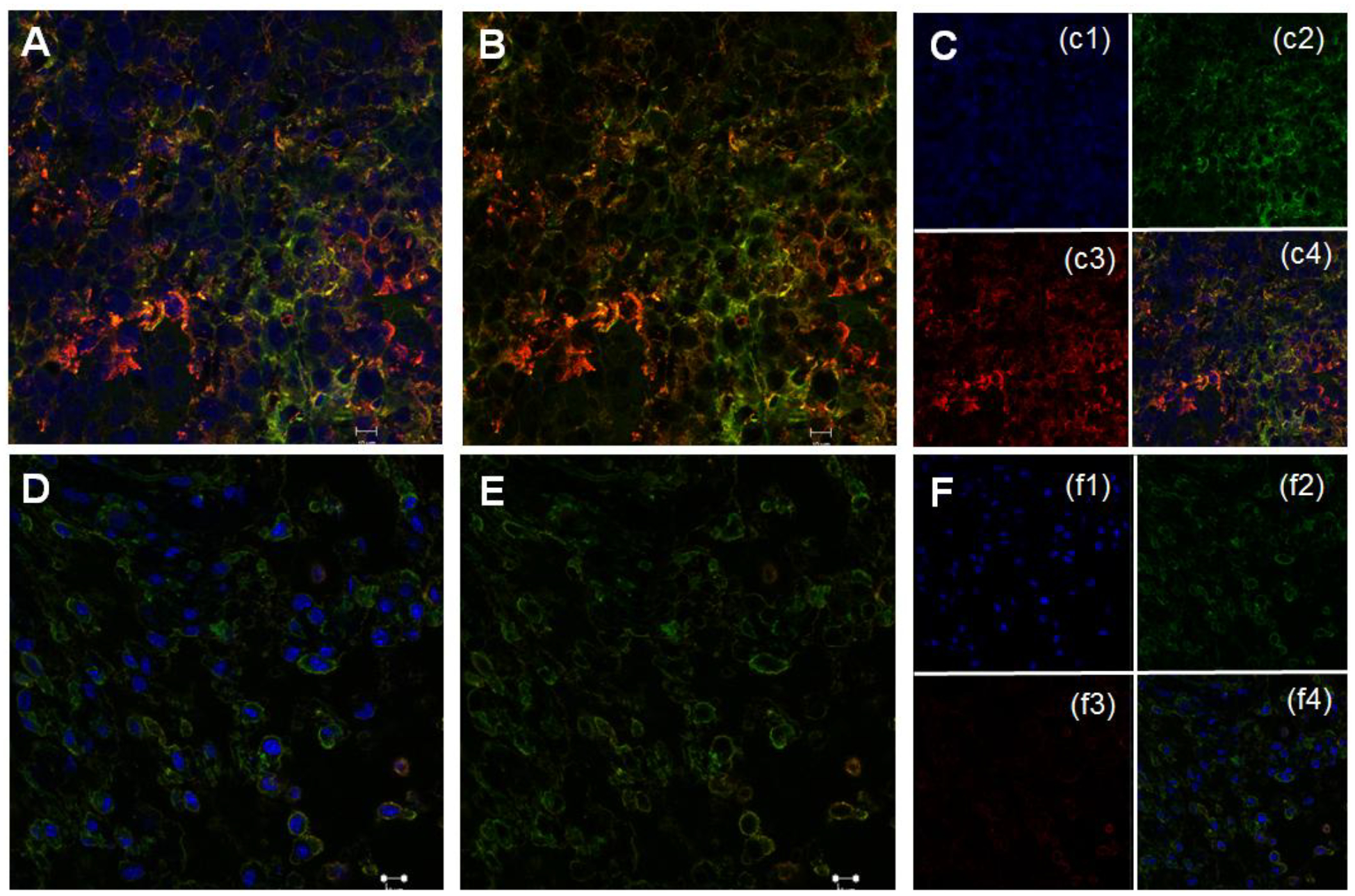
Representative immune-labeled cross sections of tumors treated with phosphate buffer saline (A-C) and prodrug nanoparticles (D-F). Sections were incubated with pSTAT-3 antibody (red) and background protein β-actin (green) around cell nuclei stained with DAPI (blue) to show significantly high level of pSTAT-3 in phosphate buffer saline treated tumors compared to treated with Pro-nifuroxazide nanoparticles.
Conclusions.
We have designed and synthesized a nanoscale system to improve the delivery and solubility of a poorly soluble drug resulting in enhanced effectivity against cancer. Our systematic development first analyzed interaction between the prodrug and cells through computational methods to predict the formation of ~10 nm Pro-nifuroxazide nanoparticles. A stable multi-layered nanoparticle with highly localized drug-regions was illustrated. The drug moiety is highly localized and easily accessible from the particle surface. Such particle structure assures an immensely high drug local concentration, and as a result greatly enhances drug effectivity. The design of prodrug enhances drug-membrane binding affinity as confirmed by the simulation studies. After establishing this, extensive physiochemical tests were conducted, which robustly indicated the success of synthesis. Subsequently, biological assays in an in vitro breast cancer model demonstrated the promoted inhibitory effect of the nanoparticles compared with nifuroxazide. In vivo studies also revealed the controlled growth of tumors treated with Pro-nifuroxazide nanoparticles compared to controls while a suppression of STAT-3 pathway was observed in the treatment group via immune-histochemical assessments. Overall, these results imply a high potential of our approach for the enhanced inhibition of STAT-3 pathway guided by molecular simulation. We envision that this methodology can be exploited in the future for the delivery of other poorly soluble agents.
Materials and Methods
Materials
Polyethylene glycol cetyl ether (PEGCE) was obtained from Sigma Life Sciences (St. Louis, MO, U.S.A). Tetrahydrofuran and Chloroform were obtained from Avantor Performance Materials (Center Valley, PA, U.S.A.). Nifuroxazide, 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), and N,N-dimethylaminopyridine (DMAP) solution were purchased from Sigma-Aldrich, USA. 1-palmitoyl-2-azelaoyl-sn-glycero-3-phosphocholine (PAzPC) was purchased from Avanti polar lipids. The hydrodynamic diameter was measured on Malvern Zetasizer machine equipped with 633 nm laser. UV-Vis spectra were recorded on Genesys 10S UV-Vis Spectrophotometer machine. Zeta potential measurement was performed on Malvern Zetasizer instrument. The TEM images were acquired on JEOL 2100 Cryo TEM machine and imaged by Gatan UltraScan 2kx2k CCD. Flow assisted cell sorting was performed on an iCyt Reflection machine from iCyt Mission Technology equipped with software Win List 3D.
Self-assembly simulation parameters for prodrug nanoparticle formation.
Coarse-grained (CG) dissipative particle dynamics (DPD) particle types were defined based on previous studies on membrane lipids.66–70 DPD provide an efficient model to study self-assembled structures where the driving force for structural formation is the balance of hydrophilic and hydrophobic interactions. Therefore, a minimum set of particle types could be used to represent different hydrophobicity in the system. Three particle types were defined: H, T, and W. H represented hydrophilic groups, such as the ether group in PEGCE, and the lipid head groups (Figure S3). T represented hydrophobic groups, such as the aliphatic carbon chains in PEGCE, cyclohexane solvent, and the lipid tails (Figure S3). W represented water. Such mapping is derived based on previous studies on membrane lipids.66 Non-bonded force parameters were taken directly from previous studies,66 where the balance of hydrophobic and hydrophilic interactions has been well justified. All interactions in DPD are defined in Equations (1)–(5). All self-assembly simulations were performed with Lammps.
The elementary units of DPD simulations are soft CG beads with mass m0 and diameter r0, whose dynamics are governed by Newton’s equations of motion supplemented by friction and random forces. When two CG beads are at positions ri and rj, their separation vector is rij=ri-rj and unit vector is . The set of conservative force parameters aij in Equation (1) are listed in Table S1. All the other parameters are identical to Ref. 1: friction parameters γij, where , , and . The bonding force constant is , and all reference bond lengths are l0=0.5r0. The angular force constant is .
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
Simulating prodrug self-assembly into prodrug nanoparticles.
To examine the molecular level structure of the self-assembled prodrug nanoparticles, DPD simulations were performed using a CG representation of the molecular system. See Supporting Information for detailed atom-to-CG particle mapping and parameters. All DPD simulations were performed with LAMMPS.
In the first stage, 20 PEGCE molecules were randomly placed in cyclohexane to simulate the formation of the core of the prodrug nanoparticles. The number of polyethylene moieties was selected based on its experimental critical micellar number. Due to the segregation of hydrophobic and hydrophilic segments, the PEGCE molecules self-assembled into a sphere with hydrophilic segments in the core and hydrophobic segments on the surface. Such a core-shell structure was observed in all 50 independent simulations within 1,000,000 simulation steps. These core particles were then used to prepare the prodrug nanoparticles in the next stage of the simulations.
In the second stage, self-assembly simulations were performed to allow the nifuroxazide prodrug molecules to aggregate around the PEGCE core particle and to form the final prodrug nanoparticles. To mimic the solvent evaporation method in experiments, the hydrophilic core of the PEGCE sphere was fixed and the organic solvent was replaced by water. 300 prodrug molecules were randomly placed in solution and 60 independent DPD simulations, each with 100,000,000 steps, were performed.
The number of PEGCE and prodrug molecules included in the system was estimated based on the molar concentrations of these components used experimentally. The resulting nanoparticle hydrodynamic radius was calculated to characterize the particle size.
Parameters and system setup in calculation of membrane-prodrug interaction.
Umbrella sampling MD simulations were carried out with NAMD 2.9 to calculate the potential of mean force (PMF) (http://www.ks.uiuc.edu/Research/namd/2.9/ug/). Different from the Self-assemble simulation, atomistic models were employed in these simulations. Parameters for the nifuroxazide group were generated using the CHARMM general force field (CGenFF) for the drug and prodrug molecules. The CHARMM (c36) force field was employed for the oxidized lipid and the membrane POPC lipids. A modified TIP3P water model in the CHARMM force field was used. Particle-mesh Ewald (PME) was used for long-range electrostatic interactions. r-RESPA multiple time-step integrator was employed with time steps of 2 and 4 fs for short-range non-bonded and long-range electrostatic interactions, respectively. The SETTLE algorithm maintained water rigid geometry while RATTLE constrained the length of covalent hydrogen bonds. All simulations were performed with NPT ensemble, where the temperature was set to 300 K for all systems by a Langevin thermostat and pressure was kept constant at 1 atm by the Langevin piston method.
The membrane insertion PMF of the drug moiety as a part of the prodrug molecule is compared with the parent drug molecule using umbrella sampling simulations. In each simulation, two copies of the drug (prodrug) molecules were placed in the system, one at the membrane and one away from the membrane, and thus two independent PMFs were obtained from a single set of umbrella sampling simulations. Each simulation was performed for 30 ns, with the umbrella potentials only acting on the center of mass of the drug moiety. In the case of the prodrug molecule, umbrella potentials were employed along the membrane normal at 1 Å intervals, thus covering prodrug-membrane interactions over a span stretching from −15 Å to 55 Å from the membrane-water interface. In the case of the parent drug molecule, 31 umbrella simulations along the membrane normal were set up at 1 Å intervals, covering drug-membrane interactions from −15 Å to 15 Å from the membrane-water interface. The resulting distributions of the drug (or prodrug) were obtained and the PMFs were calculated based on the distributions via the weighted histogram analysis method (WHAM). The PMFs from the two molecules, one in the membrane and one outside, were then averaged to obtain the final PMF, while their difference gave the error estimation.”
Calculation of the membrane-prodrug interaction.
Umbrella sampling MD simulations employing atomistic representations of the membrane, solvent, and drug molecules were carried out with NAMD 2.9 to calculate the potential of mean force (PMF) associated with membrane association of the molecules. See Supporting Information for detailed parameterization of nifuroxazide and system setup. Two drug (or prodrug) molecules were placed in each simulation system, one at the membrane and one away from the membrane, and thus two PMFs were obtained from a single simulation. umbrella simulations for nifuroxazide and 71 simulations for Pro-nifuroxazide, each 30 ns, along the membrane normal at 1 Å intervals were carried out. The resulting distribution of the drug (or the drug moiety in the case of the prodrug) was obtained, and the PMFs were calculated based on the distributions via the weighted histogram analysis method (WHAM).80 The PMFs from the two copies of the molecule of interest, one in the membrane and one outside, were then Boltzmann averaged to obtain the final PMF, while their difference was used for error estimation. Both the nifuroxazide (parent drug) and the Pro-nifuroxazide (prodrug) molecules are charge-neutral, as to give a fair comparison.
Preparation of prodrug nanoparticles.
In brief, PEGCE (0.5, 1 and 2 mg) was melted at 65°C for 5 minutes, followed by addition of 1 mL of autoclaved water drop-wise. The prepared micellar suspension was left to stir for next 20 minutes at 1,150 rpm. Pro-nifuroxazide/THF solution (250 μL; 2 mg/mL) was added drop-wise (approximately 1 drop per 10 s) to the stirring micellar suspension. Similarly, lipid/THF solution (250 μL; 2 mg/mL) was added drop-wise (approximately 1 drop per 10 seconds) to the stirring micellar suspension. The organic solvent was evaporated under stirring condition for >12 h. Total volume was made up to 1 mL with added autoclaved water. The suspension was further allowed to stir for 10 min. prepared prodrug nanoparticles were stored at 4°C overnight for curing the core of the particle and then the cured particles were characterized with various physicochemical techniques.
Preparation of control nanoparticles.
To study the functional controls for Pro-nifuroxazide NPs, two more batch of nanoparticles were synthesized using loading of nifuroxazide in lipid nanoparticles PEGCE core (Nifuroxazide-core-nanoparticles; Nifuroxazide NP) and by preparing Pro-nifuroxazide particles without PEGCE core (Pro-nifuroxazide-no-core-nanoparticles; Pro-nifuroxazide NP (no-core)). Nifuroxazide NP were prepared by melting PEGCE (0.5 mg) was at 65°C for 5 minutes, followed by drop-wise addition of 1 mL of autoclaved water. The prepared micellar suspension was left to stir for next 20 minutes at 1,150 rpm. A mixture of nifuroxazide and soy lecithin in THF solution (250 μL; 2 mg/mL of each) and was added drop-wise (approximately 1 drop per 10 s) to the stirring micellar suspension. The organic solvent was evaporated under stirring condition for >12 h. Total volume was made up to 1 mL with added autoclaved water. The suspension was further allowed to stir for 10 min. Prepared Nifuroxazide NP were stored at 4°C overnight for curing the core of the particle and then the cured particles were characterized with various physicochemical techniques.
Preparation of Rhodamine loaded Pro-Nifuroxazide NPs (Rh-Pro-nifuroxazide NPs).
To study the cell internalization efficiency of Pro-Nifuroxazide NPs and effect of endocytic inhibitors using confocal studies, rhodamine loaded Pro-Nifuroxazide NPs (Rh-Pro-nifuroxazide NPs) were prepared by method described above with minor modifications. In brief, prepared PEGCE micellar suspension was stirred with rhodamine B for 20 min before adding Pro-nifuroxazide in THF solution (250 μL; 2 mg/mL). Prepared particles were processed as discussed earlier.
Preparation of Iodine-labeled Pro-Nifuroxazide NPs (I-Pro-nifuroxazide NPs).
To study the cell internalization efficiency of Pro-Nifuroxazide NPs using electron microscopy studies, nanoparticles were labeled with a small molecule containing heavy element iodine. To achieve this, Pro-Nifuroxazide NPs (Ihx-Pro-nifuroxazide NPs) were prepared by method described above with minor modifications by encapsulated trace amount of 5-[N-(2,3-Dihydroxypropyl)acetamido]-2,4,6-triiodo-N,N’-bis(2,3-dihydroxypropyl)isophthalamide (iohexol). In brief, prepared PEGCE micellar suspension was stirred with iohexol for 20 min before adding Pro-nifuroxazide in THF solution (250 μL; 2 mg/mL). Particles were dialyzed in 10 KDa dialysis cassette against water. Purified samples were characterized with various physicochemical techniques. Dialyzed particles were processed as discussed earlier.
Dynamic light scattering.
Dialysis (MWCO 20 kDa dialysis tubing, Spectrum Laboratories, Rancho Dominguez, CA) was performed on nanoparticle suspensions into deionized water (0.2 μM) before performing characterization measurements and applications. A nano series Zetasizer instrument was used to measure the hydrodynamic diameter of prepared nanoparticles. The suspension was stored at 4°C and mildly vortexed before acquiring particle size again and/or other experiments. All determinations were made in multiples of 3 consecutive measurements with more than 15 runs each time.
Zeta potential measurements.
Zeta potential measurement was performed on samples of prodrug nanoparticles using a Malvern zetasizer of nano series. All the experiments were performed at 25°C. Data were acquired in the phase analysis light scattering (PALS) mode following solution equilibration at 25°C. Calculation of ζ from the measured nanoparticle electrophoretic mobility (μ) employed the Smoluchowski equation: μ = εζ/η, where ε and η are the dielectric constant and the absolute viscosity of the medium, respectively. Measurements of ζ were reproducible to within ±2 mV of the mean value given by 3 determinations of 10 data accumulations.
UV-Vis spectroscopy studies.
UV-Vis spectroscopy was performed on nifuroxazide, Pro-nifuroxazide and prodrug NP samples, using a Genesys 10S UV-Vis spectrophotometer. Aqueous samples were scanned for nifuroxazide concentration of 10 μM in free or nanoparticle loaded form in aqueous solution. Nifuroxazide was also scanned for absorbance spectra in ethanol, where solubility of nifuroxazide was much better compare to aqueous medium.
Transmission electron microscopy.
An aliquot of nano pro-drug was drop-casted onto a carbon coated copper grid (200 mesh size). Samples were stained with 10 μl of uranyl acetate (1%). TEM images were acquired on JEOL 2100 Cryo TEM machine and imaged by Gatan UltraScan 2kx2k CCD. For drop-casting, prodrug nanoparticles were used at the concentration of 300 nM.
Stability of prodrug nanoparticles.
Stability of formulations incubated with 10% fetal bovine serum was determined by evaluating the hydrodynamic diameters at different time points (0, 24, 48 and 72 h) using a Malvern Zetasizer ZS90 particle size analyzer. Formulations were incubated at 37°C for different time points before acquiring the dynamic light scattering data.
Stability of nanoparticles under different physiological conditions.
Stability of nanoparticles incubated in aqueous medium at pH 7.4, 4 and 12 were determined by evaluating the hydrodynamic diameters at different time points (0, 24, 48, 72, 96, 120 and 144h) using a Malvern Zetasizer ZS90 particle size analyzer. Formulations were incubated at 37°C for above time points before acquiring the dynamic light scattering data.
Effect of serum concentration on nanoparticle stability and protein corona formation on Pro-nifuroxazide NPs.
Zeta potential values of nanoparticles are generally found to be a good indicator of their colloidal stabilities. A change in extent of protein corona formation could determine the fate of these particles in systemic circulation and can be monitored by zeta potential measurements. Measurements were performed on samples after incubating them with or without 10% FBS solution at 37°C for 4h and 7 days using a Malvern zetasizer of nano series. All the experiments were performed at 25°C. Data were acquired in the phase analysis light scattering (PALS) mode following solution equilibration at 25°C. Calculation of ζ from the measured nanoparticle electrophoretic mobility (μ) employed the Smoluchowski equation: μ = εζ/η, where ε and η are the dielectric constant and the absolute viscosity of the medium, respectively. Measurements of ζ were reproducible to within ±2 mV of the mean value given by 3 determinations of 10 data accumulations.
Drug Release studies.
Release of Nifuroxazide from Pro-nifuroxazide NPs in water, blood serum and pH 4.5 mediums were investigated by incubating samples for different time points. Release of the drug was followed by UV absorption from Nifuroxazide at 372 nm after incubating at 37°C. Release of drug was also compared with other control nanoparticles of Nifuroxazide NP and Pro-nifuroxazide NP (no-core).
Cell internalization mechanism of Pro-nifuroxazide NPs.
Lipid fusion is a vital process in facilitating the efficient transport of lipid vesicles across biological membranes. Various interactions involved in lipid fusion process generally depend on head group chemistry of amphiphilic molecules. Changes at hydrophobic ends like conjugated drug molecules in Pro-drugs with drug molecules buried in hydrophobic reason are supposed to have less effect on membrane interaction properties. Thus, nanoparticles from Pro-nifuroxazide would follow similar entry mechanism as lipid vesicles. To prove cell entry mechanism of Pro-nifuroxazide NPs, different in vitro cell studies were performed. It was found that Pro-nifuroxazide NPs enter in cells by membrane fusion mechanisms as revealed by electron microscopy studies.
Electron microscopy of cell membrane fused Pro-nifuroxazide NPs.
An aliquot of I-Pro-nifuroxazide NPs was incubated with MCF-7 cells grown on coverslips for 24 h with ~80% confluence. Cells were incubated for 30 min, washed with fixing buffer twice before being fixed with 500 μL of fixing solution. Fixed cells were used for preparing slides for microscopic studies. The samples were embedded in epoxy resin, and allowed to harden overnight in an 80°C oven. The samples were then sectioned using an ultramicrotome (Ultracut UCT, Leica Micosystems, Germany). The images were obtained using a Philips CM200 transmission electron microscope Philips/FEI, Hillsboro, OR.) at 120 kV.
Effect of cell entry inhibitors on functional activity of Pro-nifuroxazide NPs.
Blocking of cellular entry pathways could be an alternative way to look at functional pathways of Pro-nifuroxazide NPs entry in cells. A series of cellular entry inhibitors were used for the study and cells were pre-incubated with specific concentration before being treated with different concentrations of Pro-nifuroxazide NPs (20, 10 and 5 μM) for next 72h before performing cell viability assay. 24h grown MCF-7 cells (10,000 cells, per well in 96 well plate) were incubated with NaN3/DOG (10/50 mM), Nystatin (28 μM), CPM (180 μM), Dynasore (80 μM) and sucrose 500 mM for 1h, before treating with Pro-nifuroxazide NPs (20, 10 and 5 μM) for 72h and control cells without any inhibitor treatment. At the end of incubation MTT assay was performed as described before.
Human transformed cancer cell culture.
MDA-MB231 cells (ER(−) Breast cancer cells) and MCF-7 cells (ER(+) Breast cancer cells) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Sigma) supplemented with 10% fetal bovine serum (FBS) in T25 culture flasks and were incubated at 37°C in a 99% humidified atmosphere containing 5% CO2. MCF-10A cells were grown in MEGM medium with supplements provided by ATCC with 10% fetal bovine serum (FBS) in T25 culture flasks and were incubated at 37°C in a 99% humidified atmosphere containing 5% CO2. Cells were regularly trypsinized with 0.1% trypsin (EDTA 0.02%, dextrose 0.05%, and trypsin 0.1%) in DPBS (pH 7.4). Non-synchronized cells were used for all the experiments.
In vitro cell studies.
The % cell viability of various cells treated with nifuroxazide, Pro-nifuroxazide and Pro-drug NP was investigated for 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay in presence of 10% FBS in antibiotic free media. Experiment was performed in 96 well plates growing 7,000 cells per well 24 h before treatments. Experiments were performed for various concentrations of Pro-nifuroxazide in pro-drug nanoparticles or free form ranging from 0.5 to 20 μM. Cells were incubated for 72h before performing the MTT assay. After incubation period, cells were treated with MTT as 20 ml (5 mg/mL) per well and further incubated for 4 h. At the end of the incubation the entire medium was removed from wells and 200 μL DMSO was added to dissolve blue colored formazan crystals. The %-cell viability was obtained from plate reader and was calculated using the formula:
Cells were similarly treated for apoptosis assay, CD44 presence assay, mRNA expression analysis and pSTAT3 protein expression analysis in vitro in nifuroxazide, Pro-nifuroxazide and prodrug nanoparticles treatment with suitable concentration and time as discussed thoroughly in Supporting Information.
Cell imaging and apoptosis assay.
Cells were treated with prodrug and prodrug nanoparticles followed by flow assisted cell sorting (FACS) analysis on propidium iodide (PI) stained treated and untreated cells to measure the apoptotic cell population. Cells (0.3 × 106 per well) were plated in 6-well plates and grown till it achieved ~80% confluence. After ~24 h of incubation, cells were treated with 10 μM of prodrug in free or nanoparticles form. At the end of 72 h time point, cells were imaged under bright field microscope for morphology determination. Cells were trypsinized and collected in 100 μL of reconstituted medium (DMEM containing 10% FBS). Cell pellets were fixed with chilled ethanol while vortexing. Fixed cells were stored at −20°C for >12 h. At the end of the incubation, cells were washed with DPBS at least two times and incubated with RNase A (1 μg/mL) at 37°C for >12 h. Cells were incubated with PI (2 μg/mL) for 30 min before scanning on FACS machine. Cells were treated in triplicated and pooled down before acquiring the FACS data.
Animal studies.
While advanced 3D cell culture techniques can mimic some of the aspects of the in vivo tumor environment, these techniques are still lacking in the complexity found in vivo. In addition to tumor architecture, the global setting of the tumor in the animal cannot yet be replicated with cell culture techniques. Thus, after optimizing treatment strategies in vitro, treatments have to be established in animal models. To evaluate the efficacy of adopted strategy using prodrug nanoparticles, animal experiments were performed. All experiments were designed to minimize the use of animals. In order to detect a least of 20% difference in tumor size, we decided to generate 4 tumors per animal as 4 mice per group. Athymic mice were bought from Charles River Laboratories International, Inc. USA. Upon arrival, athymic mice are allowed 1 week for acclimation. Animals were single-cage housed and had free access to food and water. Animals were housed in Institute of Genomic Biology (IGB), University of Illinois at Urbana-Champaign (UIUC).
Development of MCF-7 (ER (+) human breast cancer cells) xenografts in flanks of athymic mice.
Animals were anesthetized with isoflurane before injecting the MCF-7 cells suspended in Matrigel (50%, v/v). Using a Hamilton auto-injector or standard syringe tipped with a 26 gauge 1/2” long needle, we subcutaneously injected approximately 5×106 MCF-7 human breast cancer cells suspended in 40 μL of Matrigel into four sites in the flank of each mouse. Mice were monitored during recovery from the anesthesia in a clean cage. MCF-7 tumors were grown on the back of mice after cell injection. We did not find that in the time frame of completing the experiment grown tumors caused any significant discomfort to the mice. We monitored the mice daily for signs of discomfort and behavior change. Mice body weight was measured every week. The change in physiological function or abnormal behavior including shortness of breath, unsteady gait, abnormal eating behavior, physical abnormalities, rough hair coat due to lack of grooming, or lethargy were reported to division of animal research. Criteria for interventions were set up as animal body weight drops by 20% or tumor increase to 17mm × 17mm. Tumor size was determined by measuring the length and width of the tumor and then calculating the tumor volume via formulae
Animal Treatments.
Animals were followed till tumors grew to a minimum of 5 mm × 5 mm before starting the treatment protocol. Prodrug NP samples were prepared with Pro-nifuroxazide as described and injected to animals with grown tumors of at least 5 mm × 5 mm dimensions at a concentration (1.5 mg/kg) well below the LD50 for Nifuroxazide IP injections into mice reported as 1,000 mg/kg. Isoflurane-oxygen mixture was used to anesthetize the animals with 3–4% isoflurane gas from a vaporizer and constant anesthesia maintained with 1–2% isoflurane via an inlet tube. A second tube was used to remove carbon dioxide and excess anesthetic. All the personnel involved wore protective lab coats, face masks, sterile gloves during experimental procedures. A total of 40 μL particle suspensions were injected on every 4th day till 16th day while further followed the tumor size growth and regression till 28th day.
Tumor dissection, collection, processing, embedding and sectioning.
At the end of experiment, animals were euthanized with CO2 influx. Animals were dissected to collect tumors and stored in tissue cassettes dipped in 10% formalin before performing the tissue fixation protocol in Leica ASP300 tissue processor. The processing protocol was used including steps of tissue incubation in neutral buffered saline for 5h, twice with ethanol (70%) for 5h, ethanol (80%) for 5h, twice with ethanol (95%), twice with absolute ethanol for 5h, twice with xylene for 5h and finally thrice with paraffin wax for 5h. Processed tumors were embedded in paraffin wax melted at 65°C using metal cast. Embedded tumor blocks were clamped in microtome (Leica) and sectioned at 7 μm thickness.
RNA extraction and PCR studies.
RNA was collected from treated cells and controls using an RNeasy Mini Kit (Qiagen) following manufacturers protocols. cDNA was synthesized by incubating 125 ng of purified RNA with 4 μL of gene specific reverse primers (1 μM), and 2 μL of dNTPs (10 nM) in total volume of 26 μL at 65°C for 5 min followed by incubation at 4°C for 5 min. 5′GCTTCCTGCAAGAGTCGAAT3′ as forward and 5′ATTGGCTTCTCAA GATACCTG3′ as reverse primers were used against STAT-3 and 5’-GAGCGCGGCTACAGCTT-3’. Forward, 5’-TCCTTAATGTCACGCACGATTT-3’ Reverse for β-actin. At 4°C, 2 μL of Superscript III Reverse Transcriptase (200 U/μL; Invitrogen), 2 μL DTT (100 mM), 8 μL of 5x buffer, and 2 μL RNase Out (40 U/μL; Invitrogen) were mixed and the reaction incubated for 1 hour at 55°C followed by incubation for 15 minutes at 70°C. Quantitative PCR was performed using StepOnePlus RT PCR system (Applied Biosystems). Each qPCR reaction used 2 μL directly from the cDNA synthesis, 1 μL forward primer (8 μM), 1 μL reverse primer (8 μM), and 10 μL FastStart Universal SYBR Green Master (Thermo Fisher Scientific) in a total volume of 20 μL
Western blotting.
SDS-page Western blotting was used to examine the expression of pSTAT3 protein. Total protein content in the treated cells was extracted using Minute™ Total Protein Extraction Kit (Invent Biotechnologies, USA) as per manufacturer’s protocol. The concentration of protein samples was determined by a colorimetric assay using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, USA). A 25 μg of protein extract was separated by gradient (4–20%) sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane (Bio-Rad, USA) using a wet transfer unit (company name), blocked with 2.5% BSA in TBS-Tween buffer (0.12 M Tris-base, 1.5 M NaCl, 0.1% Tween 20) for an hour at room temperature, and incubated overnight at 4°C with the appropriate primary antibodies; β-actin (1:3000) and pSTAT3 (1:3000). This was followed by incubation with a HRP-conjugated goat anti-mouse secondary antibody (1:1000; Thermo Fisher Scientific, USA) for an hour and subsequent washing in TBST at RT. The blots were developed using Chemiluminescent kit (Thermo Fisher Scientific) to detect the target protein as per manufacturer’s protocol.
Histology and H&E staining.
Paraffin-embedded sections of 7 μm thickness were subjected to hematoxylin and eosin (H&E) staining. H&E staining was performed by following standard protocol supplied by core facility at IGB. The morphological changes of H&E-stained tissue with each fixation were analyzed at magnification ×100.
Immunolabeling of tissue sections for pSTAT-3 and β-actin.
To image the pSTAT-3 expression in treated and untreated tumors, immunolabeling was performed on paraffin-embedded sections (7 μm). Immunolabeling was performed by standard protocol supplied by core facility IGB. Prepared slides were imaged under confocal microscope.
Supplementary Material
Funding Sources
Funding support from the UIUC, National Science Foundation and Children’s Discovery Institute, NIH P41-GM104601 and computational support from XSEDE and NCSA Blue Waters are gratefully acknowledged. We thank Frederick Seitz Materials Research Laboratory for characterization techniques and IGB for animal studies.
Footnotes
Supporting Information.
Chemical characterization of Pro-nifuroxazide synthesized from nifuroxazide, Characterizations of drug and prodrug nanoparticles, atomistic and coarse-grained models of Pro-nifuroxazide, coarse-grained model parameters for Pro-nifuroxazide and PEGCE, Stability of nanoparticles, protein interaction properties, in vitro analysis of cancer cell growth regression as effect of prodrug, in vitro analysis of cancer cell growth regression as effect of prodrug nanoparticles, representative histogram of PI stained MCF-7 cells, MTT assay using control nanoparticles, representative H&E sections of tumors treated with buffer, representative H&E sections of tumors treated with prodrug nanoparticles, representative immune-labeled cross sections of tumors treated with buffer, representative immune-labeled cross sections of tumors treated with prodrug nanoparticles. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Lu Y; Park K Polymeric Micelles and Alternative Nanosized Delivery Vehicles for Poorly Soluble Drugs. Int. J. Pharm 2013, 453, 198–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Liechty WB; Kryscio DR; Slaughter BV; Peppas NA Polymers for Drug Delivery Systems. Annu. Rev. Chem. Biomol. Eng 2010, 1, 149–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ma D; Hettiarachchi G; Nguyen D; Zhang B; Wittenberg JB; Zavalij PY; Briken V; Isaac L Acyclic Cucurbit[n]uril Molecular Containers Enhance the Solubility and Bioactivity of Poorly Soluble Pharmaceuticals. Nat. Chem 2012, 4, 503–510. [DOI] [PubMed] [Google Scholar]
- (4).Gao Z; Lukyanov AN; Singhal A; Torchilin VP Diacyllipid-Polymer Micelles as Nanocarriers for Poorly Soluble Anticancer Drugs. Nano Lett. 2002, 2, 979–982. [Google Scholar]
- (5).Boztas AO; Karakuzu O; Galante G; Ugur Z; Kocabas F; Altuntas CZ; Yazaydin AO Synergistic Interaction of Paclitaxel and Curcumin with Cyclodextrin Polymer Complexation in Human Cancer Cells. Mol. Pharmaceutics 2013, 10, 2676–2683. [DOI] [PubMed] [Google Scholar]
- (6).Rautio J; Kumpulainen H; Heimbach T; Oliyai R; Oh D; Järvinen T; Savolainen J Prodrugs: Design and Clinical Applications. Nat. Rev. Drug Disc 2008, 7, 255–270. [DOI] [PubMed] [Google Scholar]
- (7).Müller CE Prodrug Approaches for Enhancing the Bioavailability of Drugs with Low Solubility. Chem. Biodivers 2009, 6, 2071–2083. [DOI] [PubMed] [Google Scholar]
- (8).Baudy RB; Butera JB; Abou-Gharbia MA; Chen H; Harrison B; Jain U; Magolda R; Sze JY; Brandt MR; Cummons TA; Kowal DA; Pangalos MN; Zupan B; Hoffmann M; May M; Mugford C; Kennedy J; Childers WE Jr Prodrugs of Perzinfotel with Improved Oral Bioavailability. J. Med. Chem 2009, 52, 771–778. [DOI] [PubMed] [Google Scholar]
- (9).Kleeb S; Jiang X; Frei P; Sigl A; Bezençon J; Bamberger K; Schwardt O; Ernst B FimH Antagonists: Phosphate Prodrugs Improve Oral Bioavailability. J. Med. Chem 2016, 59, 3163–3182. [DOI] [PubMed] [Google Scholar]
- (10).Misra SK; Ghoshal G; Ye M; Wu Z; Gartia M; Bromfield CR; Williams EM; Tangella KV; Schulten K; Ray PS; Burdette EC; Pan D Trimodal Therapy: Combining Hyperthermia with Repurposed Bexarotene and Ultrasound for Treating Liver Cancer. ACS Nano 2015, 9, 10695–10718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Misra SK; Ye M, Ostadhossein F, Pan D Pro-haloacetate Nanoparticles for Efficient Cancer Therapy via Pyruvate Dehydrogenase Kinase Modulation. Sci Rep. 2016, 6, 28196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Esser AK; Schmieder AH; Ross MH; Xiang J; Su X; Cui G; Zhang H; Yang X; Allen JS; Williams T; Wickline SA; Pan D; Lanza GM; Weilbaecher KN Dual-therapy with αvβ3-targeted SN2 Lipase-labile Fumagillin-prodrug Nanoparticles and Zoledronic acid in the Vx2 Rabbit Tumor Model. Nanomedicine. 2016, 12, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pan D; Pham CT; Weilbaecher KN; Tomasson MH; Wickline SA; Lanza GM Contact-Facilitated Drug Delivery with SN2 Lipase Labile Prodrugs Optimize Targeted Lipid Nanoparticle Drug Delivery. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2016, 8, 85–106, DOI: 10.1002/wnan.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Pan D; Pham CT; Weilbaecher KN; Tomasson MH; Wickline SA; Lanza GM Contact-facilitated Drug Delivery with SN2 Lipase Labile Prodrugs Optimize Targeted Lipid Nanoparticle Drug Delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016, 8, 85–106. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sun T; Zhang YS; Pang B; Hyun DC; Yang M; Xia Y Engineered Nanoparticles for Drug Delivery in Cancer Therapy. Angew. Chem. Int. Ed 2014, 53, 12320–12364. [DOI] [PubMed] [Google Scholar]
- (15).Kampert T; Misra SK; Srivastava I; Tripathi I; Pan D Phenotypically Screened Carbon Nanoparticles for Enhanced Combinatorial Therapy in Triple Negative Breast Cancer. Cellular and Molecular Bioengineering 2017, 10, 371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Liu J; Lu Y Preparation of Aptamer-linked Gold Nanoparticle Purple Aggregates for Colorimetric Sensing of Analytes. Nat. Protocols 2006, 1, 246–252. [DOI] [PubMed] [Google Scholar]
- (17).Puri A; Loomis K; Smith B; Lee J-H; Yavlovich A; Heldman E; Blumenthal R Lipid-based Nanoparticles as Pharmaceutical Drug Carriers: from Concepts to Clinic. Crit. Rev. Ther. Drug Carrier Syst 2009, 26, 523–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Warren PB Dissipative Particle Dynamics. Curr. Opin. Colloid Interface Sci 1998, 3, 620–624. [Google Scholar]
- (19).Shillcock JC Lipowsky R Equilibrium Structure and Lateral Stress Distribution of Amphiphilic Bilayers from Dissipative Particle Dynamics Simulations. J. Chem. Phy 2002, 117, 5048–5061. [Google Scholar]
- (20).Vermaas JV; Tajkhorshid E A Microscopic View of Phospholipid Insertion into Biological Membranes. J. Phys. Chem. B 2014, 118, 1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Plimpton J Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys 1995, 117, 1–19. [Google Scholar]
- (22).Koo OM; Rubinstein I; Onyuksel H Role of Nanotechnology in Targeted Drug Delivery and Imaging: a Concise Review. Nanomedicine 2005, 1, 193–212. [DOI] [PubMed] [Google Scholar]
- (23).Singh R; Lillard JW Jr. Nanoparticle-based Targeted Drug Delivery. Exp. Mol. Pathol 2009, 86, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Huynh NT; Passirani C; Saulnier P; Benoit JP Lipid Nanocapsules: a New Platform for Nanomedicine. Intern. J. Pharmaceu 2009, 379, 201–209. [DOI] [PubMed] [Google Scholar]
- (25).Ali MM; Yoo B; Pagel MD Tracking the Relative In Vivo Pharmacokinetics of Nanoparticles with PARACEST MRI. Mol. Pharm 2009, 6, 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Normandin MD; Yuan H; Wilks MQ; Chen HH; Kinsella JM; Cho H; Guehl NJ; Absi-Halabi N; Hosseini SM; El Fakhri G; Sosnovik DE; Josephson L Heat-Induced Radiolabeling of Nanoparticles for Monocyte Tracking by PET. Angew. Chemie, Inter. Ed 2015, 54, 13002–13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chang B; Chen D; Wang Y; Chen Y; Jiao Y; Sha X; Yang W Bioresponsive Controlled Drug Release Based on Mesoporous Silica Nanoparticles Coated with Reductively Sheddable Polymer Shell. Chem. Mat 2013, 25, 574–585. [Google Scholar]
- (28).Li Y; Lin J; Yang X; Li Y; Wu S; Huang Y; Ye S; Xie L; Dai L; Hou Z Self-Assembled Nanoparticles Based on Amphiphilic Anticancer Drug-Phospholipid Complex for Targeted Drug Delivery and Intracellular Dual-Controlled Release. ACS Appl. Mat. Inter 2015, 7, 17573–17581. [DOI] [PubMed] [Google Scholar]
- (29).Pan D; Schmieder AH; Wang K; Yang X; Senpan A; Cui G; Killgore K; Kim B; Allen JS; Zhang H; Caruthers SD; Shen B; Wickline SA; Lanza GM Anti-angiogenesis Therapy in the Vx2 Rabbit Cancer Model with a Lipase-cleavable SN-2 taxane Phospholipid Prodrug using α(v)β₃-targeted Theranostic Nanoparticles. Theranostics 2014, 4, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhou H-f.; Yan H; Senpan A; Wickline SA; Pan D; Lanza GM; Pham CTN Suppression of Inflammation in a Mouse Model of Rheumatoid Arthritis using Targeted Lipase-labile Fumagillin Prodrug Nanoparticles. Biomaterials 2012, 33, 8632–8640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Shen Y; Jin E; Zhang B; Murphy CJ; Sui M; Zhao J; Wang J; Tang J; Fan M; Van Kirk E; Murdoch WJ Prodrugs Forming High Drug Loading Multifunctional Nanocapsules for Intracellular Cancer Drug Delivery. J. Am. Chem. Soc 2010, 132, 4259–4265. [DOI] [PubMed] [Google Scholar]
- (32).Feng L; Gao M; Tao D; Chen Q; Wang H; Dong Z; Chen M; Liu Z Cisplatin-Prodrug-Constructed Liposomes as a Versatile Theranostic Nanoplatform for Bimodal Imaging Guided Combination Cancer Therapy. Adv. Func. Mater 2016, 26, 2207–2217. [Google Scholar]
- (33).Wang H; Liu X; Wang Y; Chen Y; Jin Q; Ji J Doxorubicin Conjugated Phospholipid Prodrugs as Smart Nanomedicine Platforms for Cancer Therapy. J. Mat. Chem. B: Mat. Bio. Med 2015, 3, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (34).Groot RD; Warren PB Dissipative Particle Dynamics: Bridging the Gap between Atomistic and Mesoscopic Simulation. J. Chem. Phy 1997, 107, 4423–4435. [Google Scholar]
- (35).Yamamoto S; Maruyama Y; Hyodo S Dissipative Particle Dynamics Study of Spontaneous Vesicle Formation of Amphiphilic Molecules. J. Chem. Phy 2002, 116, 5842–5849. [Google Scholar]
- (36).Kim CS; Duncan B; Creran B; Rotello VM Triggered Nanoparticles as Therapeutics. Nano Today 2013, 8, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wendum D; Svrcek M; Rigau V; Boëlle P-Y; Sebbagh N; Parc R; Masliah J; Trugnan G; Fléjou J-F COX-2, Inflammatory Secreted PLA2, and Cytoplasmic PLA2 Protein Expression in Small Bowel Adenocarcinomas Compared with Colorectal Adenocarcinomas. Mod. Pathol 2003, 16, 130–136. [DOI] [PubMed] [Google Scholar]
- (38).Awino JK; Gudipati S; Hartmann AK; Santiana JJ; Cairns-Gibson DF; Gomez N; Rouge JL Nucleic Acid Nanocapsules for Enzyme-Triggered Drug Release. J. Am. Chem. Soc 2017, 139, 6278–6281. [DOI] [PubMed] [Google Scholar]
- (39).Andresen TL; Thompson DH; Kaasgaard T Enzyme-triggered Nanomedicine: Drug Release Strategies in Cancer Therapy. Mol. Membr. Biol 2010, 27, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jørgensena K; Davidsena J; GMouritsen O Biophysical Mechanisms of Phospholipase A2 Activation and Their use in Liposome-based Drug Delivery. FEBS Lett. 2002, 531, 23–27. [DOI] [PubMed] [Google Scholar]
- (41).Ortiz V; Nielsen SO; Discher DE,; Klein, M. L.; Lipowsky, R.; Shillcock, J. Dissipative Particle Dynamics Simulations of Polymersomes. J. Phys. Chem. B 2005, 109, 17708–17714. [DOI] [PubMed] [Google Scholar]
- (42).Kranenburg M; Nicolas J; Smit B Comparison of Mesoscopic Phospholipid–water Models. Phys. Chem. Chem. Phys 2004, 6, 4142–4151. [Google Scholar]
- (43).Guo XD; Zhang LJ; Wu ZM; Qian Y Dissipative Particle Dynamics Studies on Microstructure of pH-Sensitive Micelles for Sustained Drug Delivery. Macromolecules 2010, 43, 7839–7844. [Google Scholar]
- (44).Bassolino D; Alper H; Stouch TR Drug-membrane Interactions Studied by Molecular Dynamics Simulation: Size Dependence of Diffusion. Drug Design Dis. 1996, 13, 135–141. [PubMed] [Google Scholar]
- (45).Sum AK Molecular Simulation Study of the Influence of Small Molecules on the Dynamic and Structural Properties of Phospholipid Bilayers. Chem. Biodiv 2005, 2, 1503–1516. [DOI] [PubMed] [Google Scholar]
- (46).Sykesa EA; Daia Q; Sarsonsb CD; Chenc J; Rocheleaua JV; Hwange DM; Zhengc G; Crambf DT; Rinkerb KD; Chan WCW Tailoring Nanoparticle Designs to Target Cancer Based on Tumor Pathophysiology. PNAS 2016, E1142–E1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Mozafari MR; Pardakhty A; Azarmi S; Jazayeri JA; Nokhodchi A; Omri A Role of Nanocarrier Systems in Cancer Nanotherapy. J. Lipos. Res 2009, 19, 310–321. [DOI] [PubMed] [Google Scholar]
- (48).Won C; Kim B-H; Yi EH; Choi K-J; Kim E-K; Jeong J-M; Lee J-H; Jang J-J; Yoon J-H; Jeong W-I; Park I-C; Kim TW; Bae SS; Factor VM; Ma S; Thorgeirsson SS; Lee Y-H; Ye S-K Signal Transducer and Activator of Transcription 3-Mediated CD133 Up-Regulation Contributes to Promotion of Hepatocellular Carcinoma. Hepatology, 2015, 62, 1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yang F; Hu M; Lei Q; Xia Y; Zhu Y; Song X; Li Y; Jie H; Liu C; Xiong Y; Zuo Z; Zeng A; Li Y; Yu L; Shen G; Wang D; Xie Y; Ye T; Wei Y Nifuroxazide Induces Apoptosis and Impairs Pulmonary Metastasis in Breast Cancer Model. Cell Death Dis. 2015, 6, e1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhao Z; Lu P; Zhang H; Xu H; Gao N; Li M; Liu C Nestin Positively Regulates the Wnt/ss-catenin Pathway and the Proliferation, Survival, and Invasiveness of Breast Cancer Stem Cells. Breast Cancer Res. 2014, 16, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Tannock IF; Hotins D Acid pH in Tumors and Its Potential for Therapeutic Exploitation. Cancer Research 1989, 49, 4173–4384. [PubMed] [Google Scholar]
- (52).Jong WHD; Borm PJA Drug Delivery and Nanoparticles: Applications and Hazards. Int. J. Nanomedicine 2008, 3, 133–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fröhlich E The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomedicine 2012, 7, 5577–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Oh N; Park J-H Endocytosis and Exocytosis of Nanoparticles in Mammalian Cells. Int. J. Nanomedicine 2014, 9(Suppl 1), 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Ahn S; Seo E; Kim K; Lee SJ Controlled cellular uptake and drug efficacy of nanotherapeutics. Sci. Rep 2013, 3, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Taylor RC; Cullen SP; Martin SJ Apoptosis: Controlled Demolition at the Cellular Level. Nat. Rev. Mol. Cell Biol 2008, 9, 231–241. [DOI] [PubMed] [Google Scholar]
- (57).Eguchi Y; Ewert DL; Tsujimoto Y Isolation and Characterization of the Chicken bcl-2 gene: Expression in a Variety of Tissues Including Lymphoid and Neuronal Organs in Adult and Embryo. Nucleic Acids Res. 1992, 20, 4187–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Dong B; Matsumura F Roles of Cytosolic Phospholipase A2 and Src Kinase in the Early Action of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin through a Nongenomic Pathway in MCF10A Cells. Mol. Pharmacol 2008, 74, 255–263. [DOI] [PubMed] [Google Scholar]
- (59).Zhang Y; Yazdanpanah V; Yang M; Ozkan M; Ozkan CS Normal and Cancer Breast Epithelial Cells Endocytosis Study of Nanoparticles by Combined AFM and NSOM Microscopy. In 2007 7th IEEE Conference on Nanotechnology (IEEE NANO); 2007; pp 1028–1032. [Google Scholar]
- (60).Visvader JE; Lindeman GJ Cancer Stem Cells in Solid Tumours: Accumulating Evidence and Unresolved Questions. Nat. Rev. Cancer 2008, 8, 755–768. [DOI] [PubMed] [Google Scholar]
- (61).Dragu DL; Necula LG; Bleotu C; Diaconu CC; Chivu-Economescu M Therapies Targeting Cancer Stem Cells: Current Trends and Future Challenges. World J. Stem Cells, 2015, 7, 1185–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable Molecular Dynamics with NAMD. J. Comput. Chem 2005, 26,,1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Warburg O On Respiratory Impairment in Cancer Cells. Science 1956, 124, 269–270. [PubMed] [Google Scholar]
- (64).Simon SM; Schindler M Cell Biological Mechanisms of Multidrug Resistance in Tumors. Proc. Natl. Acad. Sci. U.S.A 1994, 91, 3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Schindler M; Grabski S; Hoff E; Simon SM Defective pH Regulation of Acidic Compartments in Human Breast Cancer Cells (MCF-7) is Normalized in Adriamycin-Resistant Cells (MCF-7adr). Biochemistry. 1996, 35, 2811–2817. [DOI] [PubMed] [Google Scholar]
- (66).Shillcock JC; Lipowsky R Equilibrium Structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J. Chem. Phys 2002, 117, 5048. [Google Scholar]
- (67).Groot RD; Rabone KL Mesoscopic Simulation of Cell Membrane Damage, Morphology Change and Rupture by Nonionic Surfactants. Biophys. J 2001, 81, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Yamamoto S; Maruyama Y; Hyodo S Coarse Gaining and Scaling in Dissipative Particle Dynamics. J. Chem. Phys 2002, 116, 5842. [Google Scholar]
- (69).Ortiz V; Nielsen SO; Discher DE; Klein ML; Lipowsky R; Shillcock J Dissipative particle dynamics simulations of polymersomes. J. Phys. Chem. B 2005, 109, 17708. [DOI] [PubMed] [Google Scholar]
- (70).Kranenburg M; Nicolas J; Smit B Comparison of Mesoscopic Phospholipid–water Models. Phys. Chem. Chem. Phys 2004, 6, 4142. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.