

Abstract
Species occupying habitats subjected to frequent natural and/or anthropogenic changes are a challenge for conservation management. We studied one such species, Viola uliginosa, an endangered perennial wetland species typically inhabiting sporadically flooded meadows alongside rivers/lakes. In order to estimate genomic diversity, population structure, and history, we sampled five sites in Finland, three in Estonia, and one each in Slovenia, Belarus, and Poland using genomic SNP data with double‐digest restriction site‐associated DNA sequencing (ddRAD‐seq). We found monophyletic populations, high levels of inbreeding (mean population F SNP = 0.407–0.945), low effective population sizes (N e = 0.8–50.9), indications of past demographic expansion, and rare long‐distance dispersal. Our results are important in implementing conservation strategies for V. uliginosa, which should include founding of seed banks, ex situ cultivations, and reintroductions with individuals of proper origin, combined with continuous population monitoring and habitat management.
Keywords: conservation, demography, genomic diversity, population genomics, RAD sequencing, Viola
We investigated endangered and nonendangered populations of a violet, Viola uliginosa, to estimate genomic diversity, population structure, and history from genomic data. We found monophyletic populations, high levels of inbreeding, and low effective population sizes and indications of past demographic expansion and rare long‐distance dispersal. These are proposed to include the founding of seed banks, ex situ cultivations, and reintroductions with individuals of proper origin, combined with continuous population monitoring and habitat management.

1. INTRODUCTION
Endangered species inhabiting patchy, periodically changing habitats are especially challenging for conservation efforts. Populations of such species have a high risk of extinction simply due to environmental stochasticity, and if populations are sparse, as is usually the case with endangered species, recolonizations of locally extinct populations will be unlikely. The temporal extinction–recolonization dynamics, that is, metapopulation dynamics, has been shown to depend on the number of available habitats, extinction rate, and colonization rate. However, the classic metapopulation model (e.g., Hanski, 1998; Levins, 1969) assumes that the dynamics of a species is greater than stability of the habitat patches. Thus, species occupying patchy and rapidly changing habitats must use habitat tracking for successful colonization (Thomas, 1994).
Colonization of a new habitat exposes the population to a founder effect, or a population bottleneck due to a small number of colonizing and often related individuals. As the founding population represents only a part of the source population, it most likely will be genetically differentiated from the source at emergence. For example, riparian populations of oregano (Origanum vulgare; Van Looy, Jacquemyn, Breyne, & Honnay, 2009) and roadside populations of white campion (Silene latifolia; Fields & Taylor, 2014) display strong genetic differentiation between populations due to founder effects. A small founding population also has a small effective population size, which combined with genetic drift can lead to further genetic differentiation between newly founded populations and their source population. This was illustrated in the annual jewelweed (Impatiens capensis), which showed strong genetic structuring along bodies of water, due mainly to genetic drift (Toczydlowski & Waller, 2019). Additionally, differential selection pressures can increase divergence between the populations at the newly occupied and the source sites, if the newly occupied site stays occupied long enough for natural selection to operate. For instance, the endangered Spanish catchfly (Silene otites), in situ, and their respective ex situ populations became highly genetically differentiated within a few decades, partly attributed not only to the effect of genetic drift, but also to unintended selection during cultivation (Lauterbach, Burkart, & Gemeinholzer, 2012).
Genetic diversity, therefore, tends to be lost during founding of new populations. Such cases have been reported in an isolated population of alder buckthorn (Frangula alnus) in Ireland (Finlay, Bradley, Preston, & Provan, 2017) and in invasive populations of Carolina geranium (Geranium carolinianum) in China (Shirk, Hamrick, Zhang, & Qiang, 2014). Moreover, small populations commonly have lower genetic diversity than large populations, clearly demonstrated in a survey of 247 plant species, where rare and common species were compared at a generic level using allozymes and the rare species had significantly lower genetic diversity than their common counterparts (Cole, 2003). Further, small populations are prone to increased homozygosity, inbreeding, and inbreeding depression, leading to reduced fitness of highly related individuals (Charlesworth & Willis, 2009), which can threaten their viability (Frankham, 2005).
The above features pose challenges for conservation management. If a habitat for a population is lost for a period of time, a population could be reintroduced to the site provided that the habitat once again becomes favorable and a proper source population exists. When an imminent threat to an occupied habitat exists, the population could be relocated to another suitable site. Identification of proper source populations or introduction sites requires understanding of genetic structure and diversity of the species at the range in question, in addition to understanding the evolutionary processes underlying the observed genetic structure and diversity.
Here, we studied endangered populations of Viola uliginosa, which are at the northern edge of the species’ range in Finland, to clarify the amount of genetic diversity, inbreeding, and population structure. Our goal was to define whether and which populations could be used as source populations for reintroductions to sites from which the species became locally extinct, but are still suitable for V. uliginosa. In addition, we sampled nearby populations from Estonia and distant populations at the center and western edge of the distribution range to infer the origin and phylogenetic position of the Finnish populations.
2. MATERIALS AND METHODS
2.1. Study species and sampling
Viola uliginosa Besser is a perennial wetland species that inhabits typically rich, flooded meadows along rivers/lakes. The stronghold for the species distribution is in Eastern Europe, with sporadic sites at the western and northern edges of the main distribution (Figure 1). Viola uliginosa is listed as endangered in Finland (Ryttäri et al., 2019), existing at present in only six sites. It has been monitored for 30 years in an attempt to reduce further loss (Siitonen, 1990). As it occupies rare eutrophic swamp forests and flooded meadows, it can be considered as an indicator species for these habitats (Kuris & Ruskule, 2006). The species also has been classified as near‐threatened, vulnerable, endangered, or critically endangered in many other parts of its range, primarily because of the habitat type (e.g., temporal flooding mostly at springtime), habitat loss, and human disturbance (Ranta, Jokinen, & Laaka‐Lindberg, 2016).
Figure 1.
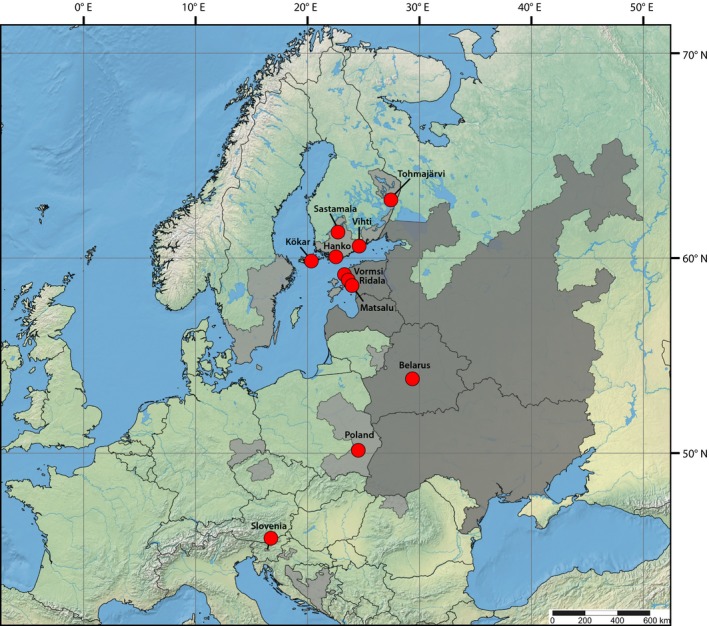
The updated distribution map of Viola uliginosa redrawn from Ranta et al. (2016) with slight modifications according to Matulevičiūté (2015) and the Swedish Species Information Centre (2019). The core areas are colored dark gray, and the peripheral areas, light gray. Red circles indicate sampling sites of this study
Viola uliginosa reproduces primarily by clonal propagation using subterranean stems (Cieślak, Paul, & Ronikier, 2006; Paul et al., 2016), but it also forms seeds that are able to exist in a seed bank and can be viable for years (Ranta & Siitonen, 2011). Individual plants can develop both chasmogamous (CH, open flowers, enabling cross‐pollination) and cleistogamous (CL, closed flowers, resulting in obligatory self‐pollination) flowers, as well as intermediate forms (semi‐CL) (Małobęcki et al., 2016). Pollination in this species has not been studied in detail, but cross‐pollination occurs by several species of bees, hoverflies, and flies, as in other Viola species, in addition to self‐pollination (Beattie, 1971). Even though reproduction is thought to be mostly clonal, seeds have a high germination capacity, putatively allowing rapid adaptation to changes in the habitat (Ranta et al., 2016). Local‐level seed dispersal by snails and ants has been observed to occur, and long‐distance dispersal can occur by floating in water currents (P. Ranta, personal communication).
We focused our sampling on extant Finnish populations of V. uliginosa, which constitute the northern edge of its global distribution. For inferring the history of the Finnish populations, we also collected samples from Estonia (isolated by the Gulf of Finland) and Belarus, both of which form the species’ center of distribution and from Poland and Slovenia, which form the southern and western edge populations, respectively. Thus, our sampling provides a latitudinal perspective. We collected one or two leaves per individual from each sampling site during summer 2016 and 2017 (with all relevant permits), with a special effort to sample individuals as distantly from each other as possible from each site. We sampled five sites in Finland (Kökar, Åland N = 30, Hanko N = 30, Vihti N = 30, Sastamala N = 20, Tohmajärvi N = 30), three in Estonia (Maatsalu N = 21, Vormsi, two populations N = 10 + 20, Ridala N = 21), one in Slovenia (Ljubljansko Barje N = 30), one in Belarus (Iljinka, consisting of two adjacent sites, N = 10 + 30), and one in Poland (Lipa, consisting of three adjacent sites, N = 10 in all) (Figure 1). The sixth known Finnish site in Mäntsälä was rapidly declining at the time of sampling, thus was not sampled (see Table S1 for more detailed sampling information). Leaf samples were stored dry in plastic bags containing silica gel. DNA was extracted with DNeasy Plant Kit (Qiagen) or PowerPlant DNA Isolation Kit (Mobio) according to manufacturer's protocols. Based on the quality and quantity of obtained DNA, we chose 96 individuals (eight individuals per population except 16 for Vormsi) including one outgroup species (V. mirabilis) for further analyses (see Table S1).
2.2. ddRAD library preparation and sequencing
The quantity of genomic DNA (gDNA) was determined with the PicoGreen Kit (Molecular Probes, Eugene, OR, USA). The ddRAD‐seq library was implemented following protocols described in Peterson, Weber, Kay, Fisher, and Hoekstra (2012) and Lee et al. (2018) with the following modifications. Briefly, gDNA was digested at 37°C for 3 hr using the restriction enzymes PstI and MseI (NEB) followed by a ligation step, whereby each sample was assigned to one of 48 adaptors. Pools of 48 individuals were combined and run on 1.5% agarose cartridge in the automated size‐selection technology, BluePippin (Sage Science), where 300‐bp fragments were excised. Each pool was amplified using 16 PCR cycles in 50 µl reactions containing 30 µl Phusion High‐Fidelity PCR Master Mix (Thermo Fisher Scientific), 15 µl of library DNA, and a unique indexing primer for each pool that corresponds to the standard Illumina multiplexed sequencing protocol. PCRs were performed in a Veriti 96‐Well Thermal Cycler (Life Technologies) using the following protocol: initial step heating at 98°C for 30 s, 16 cycles (98°C for 10 s, 60°C for 30 s, and 72°C for 40 s), followed by a final step at 72°C for 5 min. DNA libraries were quantified using the High‐Sensitivity DNA Analysis Kit in a 2100 Bioanalyzer (Agilent Technologies). Pools were combined in equimolar concentration to form a single genomic library and sequenced in one lane of a HiSeq 2500 Illumina sequencer (paired‐end, v4 reagents). The demultiplexed V. uliginosa fastq data are archived in the NCBI SRA: PRJNA540749.
2.3. Bioinformatics
Raw paired‐end reads were demultiplexed with no mismatches tolerated using their unique barcode and adapter sequences with ipyrad v.0.7.23 (Eaton & Overcast, 2016). The quality of raw demultiplexed reads was checked with FastQC software (available at http://www.bionformatics.babraham.ac.uk/projects/fastq/). The demultiplexed paired‐end reads were run through PEAR (Zhang, Kobert, Flouri, & Stamatakis, 2014) using default setting to merge overlapping reads, and input into the ipyrad pipeline. All ipyrad defaults were used, with the following exceptions: The minimum depth at which majority rule base calls are made was set to 3, the cluster threshold was set to 0.90, the minimum number of samples that must have data at a given locus for it to be retained was set to 48, 70, and 95, and the assembly method was set to “denovo” and “reference” for independent testing. Consensus sequences that had low coverage (<6 reads), excessively undetermined, or characterized with many heterozygous sites (>8), potentially resulting from paralogs or highly repetitive genomic regions, were discarded. Additionally, we excluded all loci with excessive (>50% of samples) shared polymorphic sites as they likely represented clustering of paralogs. Up to four shared polymorphic sites per called locus were allowed to accommodate polyploid genomes. This was done, because tetraploid marker data can be indistinguishable from diploid data (Gompert & Mock, 2017) and the species has been reported to produce polyploid individuals in a micropropagation experiment (Slazak et al., 2015); thus to our knowledge, polyploidy has not been observed in the wild. As there was no evidence of polyploidy, all samples were treated as diploids in the following analyses, thus allowing two haplotypes per polymorphic site. In the “denovo” assembly, sequences were assembled without any reference genome with homology inferred during alignment clustering by sequence similarity using the program vsearch (http://github.com/torognes/vsearch). In the “reference” assembly, the sequences were mapped to the whole genome of Viola pubescens (GenBank, GCA_002752925) using BWA with default bwa‐mem setting (Li, 2013) based on 90% of sequence similarity.
Phylogenetic trees were generated for “ddrad_m48” data matrices (Table 1) using a maximum‐likelihood method in RAxML v.8.2.0 (Stamatakis, 2014) with node supports estimated by a 1,000 rapid bootstrap replicates based on aligned concatenated sequences (with the following commands: ‐f v ‐m GTRCAT). One individual sampled from Tohmajärvi, identified as V. mirabilis based on BLASTN results of the ddRAD sequences, was used as an outgroup. Viola mirabilis belongs to the same subgroup Rostratae than V. uliginosa and these two species have even been suggested as sister species, making it a good species to be used as an outgroup (van den Hof, Berg, & Gravendeel, 2008; Małobęcki et al., 2016). The resulting phylogeny was visualized using FigTree v.1.4.2 (Rambaut, 2015).
Table 1.
Summary statistics of ddRAD datasets
| Data matrix | ddrad_m48 | ddrad_m95 | ref_ddrad |
|---|---|---|---|
| Assembly method | De novo | De novo | Referencea |
| Loci | 11,398 | 2,273 | 14,495 |
| SNPs | 31,724 | 4,143 | 51,435 |
| Parsimony informative | 8,770 | 1,713 | 21,405 |
| Alignment length (bp) | 2,040,210 | 432,991 | 2,512,418 |
| Missing (%) | 15.0 | 2.0 | 71.8 |
Viola pubescens genome (GCA_002752925) was used as reference.
2.4. Genetic differences among populations
The ddrad_m95 dataset (Table 1) was used for population analysis. Patterns of genetic diversity among populations were examined using Arlequin v.3.5.1 (Excoffier & Lischer, 2010). The analyses were calculated on a per‐locus basis, using the consensus SNP set. To avoid possible biases due to low coverage in estimating nucleotide diversity, one haplotype for each individual was randomly sampled, and calculations were performed using this haploid subset. Pairwise F ST values and nucleotide diversity (π) for all populations were calculated. AMOVA (analysis of molecular variance) quantified the proportion of variation at each organizational level based on unlinked SNP data with 1,000 permutations. All five Finnish populations were grouped into one group, the three Estonian populations into another group, and Belarusian, Polish, and Slovenian populations were retained as separate groups. Reasoning for the grouping was that Finnish and Estonian populations are separated by the Gulf of Finland, likely a strong dispersal barrier, and the Central European populations locate far apart. The grouping also reflects latitudinal locations of the study populations. Isolation by distance was tested using the Mantel test implemented in Arlequin, with Slatkin's linearized F ST and natural logarithms of geographic distances.
SNP‐based inbreeding coefficients were calculated using vcftools (Danecek et al., 2011). Vcftools calculates the inbreeding coefficient F SNP per individual using the equation F SNP = (O – E) / (N – E), where O is the observed number of homozygotes, E is the expected number of homozygotes (given population allele frequency), and N is the total number of genotyped loci. Boxplots between populations and F SNP values were executed with R v.3.5.2 (R Core Team, 2015) and graphically represented using the packages corrplot (Wei, 2013) and ggplot2 (Wickham, 2009).
2.5. Clustering analysis
Population clustering with admixture from SNP frequency data was inferred to better visualize genomic variation between individuals with STRUCTURE v.2.3.1 (Pritchard, Stephens, & Donnelly, 2000). In this analysis, 1,958 putatively unlinked SNPs were identified by selecting a single SNP from each locus in the “ddrad_m95” data matrix (Table S1). Ten replicates were run with K = 1–11. Each run had a burn‐in of 50K generations followed by 500K generations of sampling. Replicates were permuted using CLUMPP (Jakobsson & Rosenberg, 2007) and the optimal K value was inferred using StructureHarvester (Earl & VonHoldt, 2012) according to the ad hoc ΔK statistics (Evanno, Regnaut, & Goudet, 2005), which is the second‐order rate of change in the likelihood function. Structure results were visualized using DISTRUCT (Rosenberg, 2004).
The package FineRADstructure was also used to investigate the genetic structure in V. uliginosa (Malinsky, Trucchi, Lawson, & Falush, 2018). The package includes RADpainter, a program designed to infer the co‐ancestry matrix and estimate the number of populations within the dataset. The input file used was an allele.loci matrix (“ddrad_m48” = 15% of missing data) generated with ipyrad program. The allele data were converted using a python script (available at http://github.com/edgardomortiz/fineRADstructure-tools; last accessed 21 June 2019). Samples were assigned to populations using 100,000 iterations as burn‐in prior to sampling with 100,000 iterations. The trees were constructed using 10,000 iterations and the output visualized using the fineradstructureplot.r and finestructurelibrary.r R scripts (http://cichlid.gurdon.cam.ac.uk/fineRADstructure.html).
To generate an unrooted genetic network, Neighbor‐Net analysis (Bryant & Moulton, 2003) was implemented in SplitsTree v.4.14.2 (Huson & Bryant, 2006) using uncorrected p‐distances with heterozygous ambiguities averaged and normalized with 1,000 bootstrap replicates (>75% shown). This method uses aspects of Neighbor‐Joining (Saitou & Nei, 1987) and SplitsTree to create a network that visualizes multiple hypotheses simultaneously.
2.6. Population history
Population demographic changes and deviation from neutrality were estimated based on mismatch distribution (Excoffier, 2004), the raggedness index (Harpending, 1994), and Tajima's D (Tajima, 1989) using Arlequin v.3.5.1 (Excoffier & Lischer, 2010). Mismatch distributions were constructed from the full dataset and based on combined Finnish and Estonian population to attain a sufficient sample size.
Effective population sizes (N e) for the contemporary and historical samples were calculated using the bias‐corrected measure of linkage disequilibrium (Waples & Do, 2010), as implemented in NeEstimator v.2.1 (Do et al., 2014) based on allele frequencies of all 4,143 loci. We estimated N e for each population using minor allele frequency cutoff of 0.05.
History was further evaluated using the program DIYABC v.2.0.3 (Cornuet et al., 2014), based on approximate Bayesian computation (ABC). Two different historical scenarios were compared, based on (a) branching order of the phylogenetic tree and (b) an otherwise similar branching order, but a simultaneous divergence of Estonian populations followed by a simultaneous divergence of the Finnish populations (Figure 2). After preliminary runs, the following priors were set: present effective population size (N e) for Poland, Slovenia, and Belarus of 10–1,000, and for other populations of 10–100, ancient population sizes of 10–10,000 for all and divergence times (t) of 10–1,000 for all. We ran 200,000 simulations with this scenario and used the 2,000 sets closest to the observed data for parameter estimation.
Figure 2.
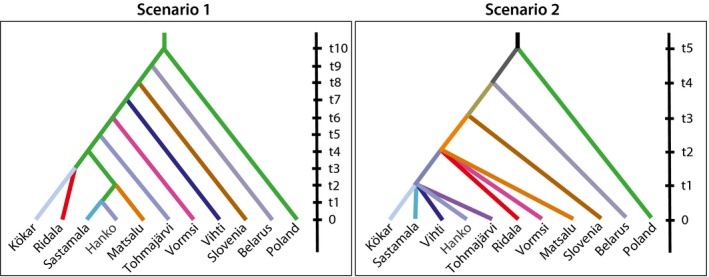
Scenarios used for DIYABC analysis
3. RESULTS
We obtained over 2,000,000‐bp sequence and 31,000 SNPs for the ddrad_m48 dataset and over 430,000‐bp sequence and over 4,100 SNPs for the ddrad_m95 dataset with only 15% and 2.0% of missing data, respectively. There were 8,770 parsimony‐informative sites in ddrad_m48 data and 1,713 in the ddrad_m95 data. The alignment with the reference sequence exceeded 2,500,000 bp and 51,400 SNPs (Table 1).
Nucleotide diversities ranged from 0.013 (Vihti) to 0.023 (Ridala). The three sites with lowest diversity values were all from Finland (Vihti, Kökar, and Hanko) (Table 2). Individual inbreeding coefficients were all positive and mostly very high. Out of the 39 Finnish individuals, 36 had inbreeding coefficients > 0.500, with 20 individuals above 0.900. One exceptionally low value of 0.084 was found for one individual from Tohmajärvi (range: 0.084–0.982). Of the 32 individuals from Estonia, 21 had values > 0.500, with two individuals having a value below 0.200 (range: 0.100–0.836). Inbreeding coefficients in Belarus were 0.386–0.741, in Poland 0.263–0.777, and in Slovenia 0.226–0.626. The average inbreeding coefficients of the studied populations were highest in the Finnish populations, with the most extreme value (0.945) obtained from the island population of Kökar (Figure S1 and Table 3). All populations had small effective population sizes based on NeEstimator, some even extremely small (Tohmajärvi 0.8, Poland 1.5). The largest effective population sizes were in two Estonian populations (Matsalu 50.9, Ridala 38.9) (Table 3).
Table 2.
Diversity measures, Tajima's D, and mismatch distribution parameter estimates
| Population | n | S | π | Tajima's D | HRag | τ | θ 0 | θ 1 |
|---|---|---|---|---|---|---|---|---|
| Belarus | 8 | 685 | 0.02042 | −1.64723** | 0.0714 | 76.000 | 9.429 | 1,000 |
| Poland | 8 | 430 | 0.01906 | −1.44254* | 0.0816 | 54.992 | 5.972 | 1,000 |
| Slovenia | 8 | 406 | 0.02113 | −1.99967** | 0.0587 | 50.410 | 8.268 | 1,000 |
| Matsalu | 8 | 695 | 0.01842 | −2.15492** | 0.0561 | 68.103 | 6.861 | 1,000 |
| Ridala | 8 | 756 | 0.02339 | −1.13695 | 0.1327 | 86.079 | 5.635 | 1,000 |
| Vormsi | 16 | 840 | 0.01877 | −2.13757** | 0.0114 | 58.170 | 7.905 | 1,000 |
| Hanko | 8 | 624 | 0.01586 | −1.21846 | 0.0740 | 66.192 | 4.558 | 1,000 |
| Kökar | 8 | 660 | 0.01494 | −1.43031* | 0.0485 | 79.612 | 2.996 | 1,000 |
| Sastamala | 8 | 779 | 0.02253 | −1.67537** | 0.0893 | 78.904 | 11.132 | 1,000 |
| Tohmajärvi | 7 | 659 | 0.02068 | −1.78349** | 0.0680 | 73.870 | 11.225 | 1,000 |
| Vihti | 8 | 589 | 0.01301 | −1.64673** | 0.0842 | 54.742 | 9.579 | 1,000 |
| All | 95 | 1,579 | 0.04395 | −1.66120* | 0.0004 | 58.951 | 10.677 | 1,000 |
n = number of individuals, S = number of polymorphic sites, π = nucleotide diversity, HRag = raggedness statistic r, τ = 2 µt, where µ is the mutation rate and t is time in generations, θ 0 = theta before population size change, θ 1 = theta after population size change.
Significant p‐values are indicated by *p < .05; **p < .02.
Table 3.
Average linkage disequilibrium (r 2), effective population sizes (N e) with 95% confidence intervals, and average inbreeding coefficients (F SNP) with standard deviations in brackets of Viola uliginosa populations. Note that infinite estimates of the effective population size are likely due to a correction for the sample size that is larger than r2 rather than a truly infinite population size (Macbeth, Broderick, Buckworth, & Ovenden, 2013)
| Population | r 2 | N e | 95% confidence interval | F SNP |
|---|---|---|---|---|
| Hanko | 0.208 | 23.9 | 21.1–27.4 | 0.749 (0.184) |
| Kökar | 0.187 | Infinite | Infinite – infinite | 0.945 (0.064) |
| Sastamala | 0.229 | 7.6 | 7.2–8.0 | 0.779 (0.228) |
| Tohmajärvi | 0.440 | 0.8 | 0.8–0.8 | 0.835 (0.332) |
| Vihti | 0.184 | Infinite | Infinite ‐ infinite | 0.872 (0.051) |
| Matsalu | 0.199 | 50.9 | 42.9–62.2 | 0.510 (0.167) |
| Ridala | 0.202 | 38.9 | 34.7–44.1 | 0.442 (0.161) |
| Vormsi | 0.102 | 13.2 | 12.8–13.5 | 0.593 (0.149) |
| Belarus | 0.213 | 14.4 | 13.6–15.4 | 0.526 (0.123) |
| Poland | 0.306 | 1.5 | 1.5–1.5 | 0.505 (0.201) |
| Slovenia | 0.218 | 12.1 | 11.6–12.7 | 0.407 (0.123) |
Infinite N e estimates occur when genetic variation is high and the sampling error among individuals is stronger than the signal of genetic drift from a finite number of parents (Do et al., 2014), thus infinite ≠ infinitely large population.
The maximum‐likelihood tree (Figure 3 and Figure S2) showed that each population is monophyletic with high support and that Estonian and Finnish populations were mixed among each other. The Polish population was the closest to the root, with the Belarusian and Slovenian populations forming the next level of branching. Adjacent subpopulations in Poland and in Belarus were monophyletic and formed sister clades to subpopulations in the same locality. The subpopulations from Vormsi, Estonia, showed paraphyly. The similar topology with all 11 major clades was also recovered in the phylogenetic network (Figure S3), where the Central European populations (Poland, Slovenia, and Belarus) were separated from North European populations by a long branch and the Finnish and Estonian populations were close to each other. However, the reticulation observed in the phylogenetic network between Finnish and Estonian populations, especially in adjacent Vormsi and Ridala populations, is indicative of hybridization and/or incomplete lineage sorting.
Figure 3.
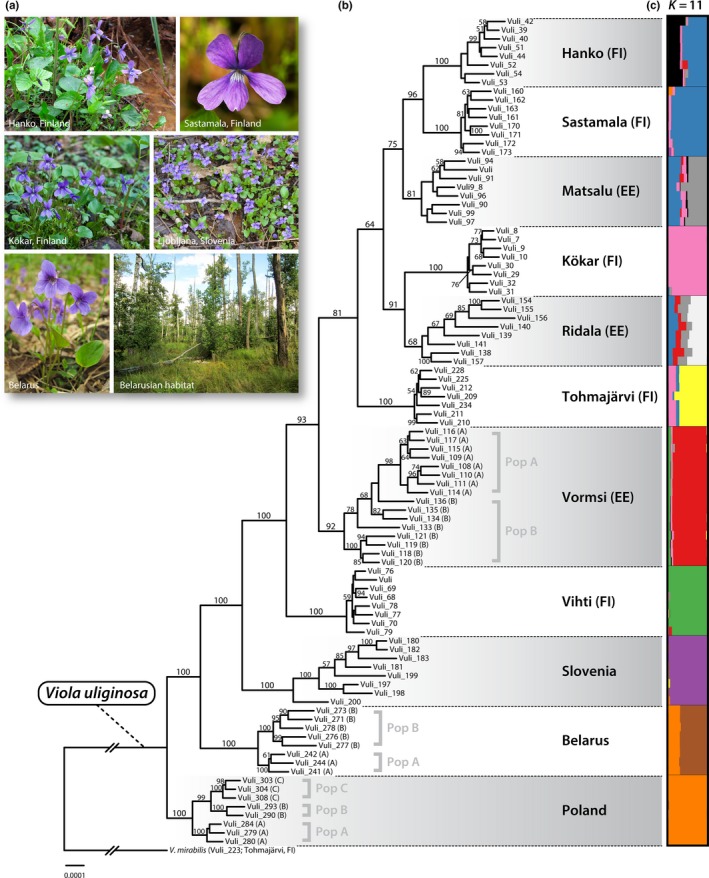
(a) Representative photographs of Viola uliginosa. (b) Phylogenetic relationships of V. uliginosa including one outgroup species, V. mirabilis (Vuli_223). Maximum‐likelihood tree inferred from RAxML analysis based on de novo assembly method. The data matrix consisted of 31,724 SNPs in 2,040,210 bp. The bootstrap values shown near the branches are from 1,000 rapid bootstrap resamplings. (c) The barplot, generated through Structure software, shows the assignments of individuals into 11 genetic clusters. Each bar represents one individual, and colors represent the proportion of the individuals that belong to each of the genetic cluster
Structure analysis (Figure 3) suggested the optimal number of estimated genetic clusters was 11, corresponding to the total number of populations studied. However, some indications of admixture were present, and specifically, individuals from Hanko (Finland, Q values ranged between 0.39 and 0.60), Matsalu (Estonia, Q values: 0.17–0.29), and Ridala (Estonia, Q values: 0.08–0.19) had parts of their genomes assigned to the same cluster with individuals from Sastamala (Finland). Similarly, individuals from Belarus were partially assigned to the same cluster as individuals from Poland (Q values: 0.26–0.32). The FineRADstructure analysis showed a clear separation of the southern (Slovenia, Poland, and Belarus) from northern (Finland and Estonia) populations and also weak structuring within the northern populations (Figure S4).
AMOVA and pairwise F ST results further supported strong genetic differentiation of the populations. AMOVA analysis suggested that 37.29% of variation arose from differentiation among populations within groups (F SC = 0.423) and 11.85% from differentiation among groups (F CT = 0.119), with a total F ST of 0.492 (Table S2). Pairwise F ST values were high, ranging from 0.154 between Ridala and Matsalu to 0.628 between Kökar and Poland, and all had p‐values < .02 (Table S3). Mantel test revealed the correlation coefficient r = 0.471 (p = .023), with 22.2% of the geographic distance determined by the genetic distance.
All populations, except Ridala and Hanko, showed negative Tajima's D values and small raggedness with p‐values < .05, suggesting past population expansion (Table 2). This was further supported by the mismatch distributions constructed from all the data and from the Finnish and Estonian populations (Figure S5). The best scenario according to the DIYABC was scenario 2 in Figure 2 (simultaneous divergence of first Estonian and then Finnish populations) that received 100% support against the one constructed using the branching order of the phylogenetic tree. Based on the principal component analysis, the observed data were located among the data simulated according to scenario 2 and distant from data simulated according to scenario 1 (see Figure S6). Divergence times were given in generations, and as there were no estimates for a generation time in V. uliginosa, we used a mean of estimates obtained from V. elatior, V. pumila, and V. stagnina from Eckstein, Danihelka, and Otte (2009), 9.44 years, to transfer the generations into years (Table S4). Using this estimate, divergence from the common ancestor occurred 9,140 years BP (95% CI: 4,630–9,360; 824 generations), divergence of Estonian populations 550 years BP (95% CI: 280–1,650; 85.1 generations), and divergence of Finnish populations just 100 years BP (95% CI: 94.4–140; 11.6 generations).
4. DISCUSSION
4.1. Genetic diversity, inbreeding, and effective population size
Estimates of nucleotide diversities from our study populations (0.013–0.023) were at an intermediate level compared with previous studies on other plant species using ddRAD sequencing. For example, much lower levels were found in endangered Hawaiian lobelioid Clermontia (0.0014) and Cyanea (0.0012) populations (Jennings et al., 2016) and much higher estimates were detected in endangered Indian orchids Geodorum densiflorum (0.036), Dendrobium densiflorum (0.106), and Rhynchostylis retusa (0.113). Levels similar to our study were found in Cymbidium aloifolium (0.014) (Roy, Moitra, & Sarker, 2017). We are not aware of other studies of violets using ddRAD sequencing; however, other methods have been used for population analyses. For example, RAPD markers and population demographic methods revealed high genetic polymorphism and both clonal and sexual reproduction in Viola riviniana (Auge, Neuffer, Erlinghagen, Grupe, & Brandl, 2001). Gene diversity estimates from AFLP data from three violet species in Central Europe, Viola elatior, V. pumila, and V. stagnina, were relatively low (0.113–0.174) and varied between populations located in the central or peripheral parts of the species ranges (Eckstein, Hölzel, & Danihelka, 2006). Asian violets Viola grayi, V. kusanoana, and V. grypoceras showed expected microsatellite heterozygosity of 0.078–0.773, depending on the species and population, with the endangered coastal violet V. grayi having the lowest genetic diversity (Hirai, Kubo, Ohsako, & Utsumi, 2012). Thus, the genus Viola in general has wide variation in genetic diversity, likely stemming from different levels of sexual reproduction, varying population sizes, generation lengths, and ages of species and populations, and in addition to the strength and type of selection, the species and/or populations are facing. Genetic diversity of V. uliginosa in Poland suggested high genetic uniformity over the study populations, when AFLP markers were applied. Similar to our results, each individual possessed an own genotype profile although individuals from a given subpopulation were very alike, with similarity coefficients typically of 0.94–0.99 (Cieślak et al., 2006).
Individual inbreeding coefficients in our study populations were high, with 68 individuals from the 95 studied having values above 0.500. Similar high inbreeding coefficients have been observed in the endangered seaside violet V. grayi (Hirai et al., 2012). In addition, all effective population sizes for V. uliginosa were < 51, supporting a reproductive strategy favoring clonal propagation. The northernmost population in Finland had the highest inbreeding coefficient and the N e estimate for one population was < 1, possibly due to a higher proportion of clonality. It has been commonly suggested that ecologically marginal populations suffer from small effective population sizes, because of fragmentation and lack of gene flow due to lack of suitable habitat (e.g., Caughley, Grice, Barker, & Brown, 1988; Young, Boyle, & Brown, 1996). The small N e is manifested by an increase in self‐pollination, inbreeding, and clonality in plants at the edge populations when compared to central populations (Arnaud‐haond et al., 2006; Beatty, McEvoy, Sweeney, & Provan, 2008; Eckert, 2002; Silvertown, 2008). Our data support the abundant center hypothesis, which predicts that genetic diversity is lowest at the species margin because biological tolerances of a species reach their limit and/or due to repeated founder events during postglacial colonization (Brown, 1984; Petit et al., 2003; Sagarin & Gaines, 2002). However, the hypothesis is subject to further evaluation, as a meta‐analysis reported a decline in genetic diversity toward range margins in only 64.2% of the reviewed 134 studies (Eckert et al., 2010).
Another factor explaining low genetic variation, small effective population sizes, and high inbreeding coefficients, characteristic of some V. uliginosa populations, may be that their habitats are affected by temporal stochastic changes. In Finland, the occupied habitats are separated by 60–520 km compared with the Estonian populations at 35–63 km. Thus, if a strong environmental disturbance exposes the population to a genetic bottleneck, there is no gene flow to rescue the population. This is supported further by the high pairwise F ST values among the Finnish populations (average 0.520) compared with Estonian populations (average 0.201) and among the eastern and southern European populations from Slovenia, Belarus, and Poland (average 0.454). The latter are separated by 530–1,890 km, yet their F ST values were smaller than between Finnish populations. This can be explained by that occasional gene flow among the southern populations may be more likely than among the Finnish populations. Most likely V. uliginosa has never been common in Finland, as it requires a rare, eutrophic, and flooded habitat and thus gene flow seldom, if ever occurs. Only 17 sites have been recorded since 1851, and all but six are now extinct. The six extant populations have existed for a range of time with the oldest being Vihti (1851) and the youngest at Tohmajärvi (discovered in 1999). The present sizes of the populations vary between tens and thousands of rosettes (Ranta & Siitonen, 2011). Thus, if the populations have been founded via long‐distance dispersal by just a few seeds and/or have experienced repeated bottlenecks, they have lost genetic diversity several times in their history, simultaneously resulting in low effective population sizes and high inbreeding coefficients.
Nevertheless, some individuals displayed strikingly smaller inbreeding coefficients than others, (e.g., Tohmajärvi, Finland, 0.084; Ridala, Estonia, 0.100), strongly suggesting that occasional outbreeding reduced homozygosity of otherwise very homogeneous populations. In vitro experiments revealed that a high percentage of seeds actually germinate (Ranta et al., 2016) supporting the potential importance of sexual reproduction. Indeed, the species was reintroduced in 2014 at one site after 40 years of absence and was still flowering in 2017 (Hyvärinen, 2018; Ranta, 2014). The reintroduction used seeds collected from a private garden with plants originating from almost the same site, which had been destroyed due to construction work in 1975 (this population was not sampled for this study, Kulmala, Ryttäri, & Laaka‐Lindberg, 2016; Ranta, 2014). Thus, although sexual reproduction may not be common, it can play an important role in genetic diversity of V. uliginosa populations, especially at the northern edge of the species’ range.
4.2. Population history and the origin of the Finnish populations
The phylogenetic tree constructed using the maximum‐likelihood method shows mixing of Estonian and Finnish populations with the more southern populations located closer to the outgroup (V. mirabilis). However, DIYABC results strongly supported divergence of Estonian populations more or less simultaneously about 550 years BP and the Finnish populations more recently, about 100 years BP (95% CI: 94.4–140). The generation time used for these time estimates is adopted from other Viola species (9.44 years, calculated from Eckstein et al., 2009), which possibly is not valid for our study species. On the other hand, the estimate for the branching from the most common ancestor (9 140 years ago) fits nicely to the onset of a warmer and moister period after the Younger Dryas in Europe (e.g., Mauri, Davis, Collins, & Kaplan, 2015). The mismatch distributions, Tajima's D and raggedness values suggest past expansion for most populations and for the whole data. The time of the expansion based on τ values does not seem to vary much between populations, suggesting that all populations still carry the signs of the same past expansion. Based on the estimates of ancestral effective population sizes from DIYABC, the population sizes have indeed been thousands of individuals and at the time of divergence of Slovenian and Belarusian populations, about three times larger than in the common ancestral population.
Finnish populations are phylogenetically very close to the Estonian populations, which are also geographically close. The pattern is supported by mixing of these populations in the phylogenetic tree (Figure 3), branching of the Finnish populations from the Estonian ancestor in the DIYABC analysis (Figure 2, scenario 2), and existence of the same genetic clusters in several Estonian and Finnish populations as detected with Structure analysis (“blue,” “pink,” and “gray” clusters in Figure 3). It is likely that at the time of divergence of populations in present Estonia, some individuals dispersed to Finland, which then founded the ancestral Finnish population(s) from which the present remaining populations originated. By now, population sizes have declined in most populations, probably due to founder effects as new populations have been founded by very few individuals and due to repeated bottlenecks caused by temporal changes in the habitat. The estimates of present effective sizes vary by about 10‐fold between the two different methods (linkage disequilibrium, NeEstimator and coalescence, DIYABC), but still show basically the same trend, and Finnish populations have smaller effective population sizes than Estonian populations.
4.3. Conservation and reintroductions
IUCN guidelines for reintroductions state that “Founders should show characteristics based on genetic provenance, and on morphology, physiology and behaviour that are assessed as appropriate through comparison with the original or any remaining wild populations” (IUCN/SSC, 2013). Maschinski and Albrecht (2017) recently presented additional guidelines for reintroduction of rare plants stating that measuring of genetic structures should be performed before reintroductions, especially if, for example, the populations are highly fragmented and isolated or if populations have less than 50 individuals setting fruit. This number likely stems from a previously recommended minimum effective population size of 50 for the prevention of loss of fitness, which has since been amended to a value of 100 (Frankham, Bradshaw, & Brook, 2014). Here, we focused on examining the few remaining natural populations of V. uliginosa in Finland. Three of the existing populations can be considered abundant, each with thousands of individuals (Hanko, Kökar and Tohmajärvi, Ranta et al., 2016); however, the remaining two populations are much smaller: Sastamala with < 100 individuals and Vihti with 100s individuals (unsampled population in Mäntsälä has < 10 individuals). Irrespective of the census size, the effective population sizes are much smaller than the presently recommended 100 for the prevention of loss of fitness and particularly smaller than the recommended size of 1,000 for retaining evolutionary potential (Frankham et al., 2014). Furthermore, all Finnish populations are clearly genetically differentiated and are located as isolated patches separated by 10s and 100s of km. Even though there is evidence of past common ancestry in Finnish and Estonian populations, the observed genetic distinctiveness of all populations opposes mixing of individuals between populations. Thus, for conserving these remaining populations and considering previous success in reintroduction, we propose that seed banks from the remaining populations should be founded, cultivated ex situ, and introduced to suitable, but other sites close to the original locations. This will minimize the chance that stochastic habitat changes will eliminate whole populations. Using sites that are closely located to the source population for introduction also fulfills the IUCN guideline for genetic provenance and acts as a precaution in avoiding source populations that possibly are maladapted to the site. Usage of distant source populations may even result in outbreeding depression within original populations if they crossbreed. Founding of multiple close sites can also help in increasing or at least maintaining the effective population sizes, especially if different founder individuals are used for different sites and if there is fast diversifying selection at the sites. Furthermore, the remaining populations, as well as the newly founded sites, should be monitored annually to (a) found a population ecological study to estimate vital rates and the importance of different life stages to population growth, (b) perform population viability analyses, and (c) address any stochastic changes in the population size or in the habitat as soon as possible.
CONFLICT OF INTEREST
None declared.
AUTHOR CONTRIBUTIONS
L. Kvist conceived and designed the study, performed some data analysis, and drafted the manuscript; K.M. Lee carried out the molecular laboratory work, performed the bioinformatics and data analysis, and drafted the manuscript; P. Ranta, J. Saarikivi, L. Kutnar, B. Vreš, and M. Dzhus collected field samples and drafted the manuscript; and M. Mutanen conceived the study and drafted the manuscript. All authors gave final approval for publication.
Supporting information
ACKNOWLEDGMENTS
We are grateful for the invaluable help from Laura Törmälä for assistance in the laboratory. The authors also wish to acknowledge CSC‐IT Centre for Science, Finland, for providing computational resources and Dr. James A. Coyer for revising English grammar in this manuscript. This work was financed by University of Oulu to L. Kvist, Academy of Finland (grant number: 277984) to M. Mutanen, and the Kvantum Institute to K.M. Lee.
Lee KM, Ranta P, Saarikivi J, et al. Using genomic information for management planning of an endangered perennial, Viola uliginosa . Ecol Evol. 2020;10:2638–2649. 10.1002/ece3.6093
DATA AVAILABILITY STATEMENT
The demultiplexed fastq data are archived in the NCBI SRA (BioProject ID: PRJNA540749). All Supporting Information material accompanying the article is posted on Dryad Digital Repository, http://doi.org/10.5061/dryad.6v78j4c.
REFERENCES
- Arnaud‐haond, S. , Teixeira, S. , Massa, S. I. , Billot, C. , Saenger, P. , Coupland, G. , … Serrão, E. A. (2006). Genetic structure at range edge: Low diversity and high inbreeding in Southeast Asian mangrove (Avicennia marina) populations. Molecular Ecology, 15(12), 3515–3525. 10.1111/j.1365-294X.2006.02997.x [DOI] [PubMed] [Google Scholar]
- Auge, H. , Neuffer, B. , Erlinghagen, F. , Grupe, R. , & Brandl, R. (2001). Demographic and random amplified polymorphic DNA analyses reveal high levels of genetic diversity in a clonal violet. Molecular Ecology, 10(7), 1811–1819. 10.1046/j.0962-1083.2001.01311.x [DOI] [PubMed] [Google Scholar]
- Beattie, A. (1971). Pollination mechanisms in Viola . New Phytologist, 70, 343–360. 10.1111/j.1469-8137.1971.tb02533.x [DOI] [Google Scholar]
- Beatty, G. E. , McEvoy, P. M. , Sweeney, O. , & Provan, J. (2008). Range‐edge effects promote clonal growth in peripheral populations of the one‐sided wintergreen Orthilia secunda . Diversity and Distributions, 14(3), 546–555. 10.1111/j.1472-4642.2008.00469.x [DOI] [Google Scholar]
- Brown, J. H. (1984). On the relationship between abundance and distribution of species. The American Naturalist, 124(2), 255–279. 10.1086/284267 [DOI] [Google Scholar]
- Bryant, D. , & Moulton, V. (2003). Neighbor‐Net: An agglomerative method for the construction of phylogenetic networks. Molecular Biology and Evolution, 21(2), 255–265. 10.1093/molbev/msh018 [DOI] [PubMed] [Google Scholar]
- Caughley, G. , Grice, D. , Barker, R. , & Brown, B. (1988). The edge of the range. The Journal of Animal Ecology, 57(3), 771 10.2307/5092 [DOI] [Google Scholar]
- Charlesworth, D. , & Willis, J. H. (2009). The genetics of inbreeding depression. Nature Reviews Genetics, 10(11), 783–796. 10.1038/nrg2664 [DOI] [PubMed] [Google Scholar]
- Cieślak, E. , Paul, W. , & Ronikier, M. (2006). Low genetic diversity in the endangered population of Viola uliginosa in its locus classicus at Rząska near Cracow (Southern Poland) as revealed by AFLP markers. Acta Societatis Botanicorum Poloniae, 75(3), 245–251. 10.5586/asbp.2006.029 [DOI] [Google Scholar]
- Cole, C. T. (2003). Genetic variation in rare and common plants. Annual Review of Ecology, Evolution, and Systematics, 34(1), 213–237. 10.1146/annurev.ecolsys.34.030102.151717 [DOI] [Google Scholar]
- Cornuet, J.‐M. , Pudlo, P. , Veyssier, J. , Dehne‐Garcia, A. , Gautier, M. , Leblois, R. , … Estoup, A. (2014). DIYABC v2. 0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics, 30(8), 1187–1189. 10.1093/bioinformatics/btt763 [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , … Durbin, R. (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do, C. , Waples, R. S. , Peel, D. , Macbeth, G. M. , Tillett, B. J. , & Ovenden, J. R. (2014). NeEstimator v2: Re‐implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Molecular Ecology Resources, 14(1), 209–214. 10.1111/1755-0998.12157 [DOI] [PubMed] [Google Scholar]
- Earl, D. A. , & VonHoldt, B. M. (2012). STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- Eaton, D. A. R. , & Overcast, I. (2016). ipyrad: Interactive assembly and analysis of RADseq data sets. Retrieved from http://ipyrad.readthedocs.io/. [DOI] [PubMed]
- Eckert, C. G. (2002). The loss of sex in clonal plants. Evolutionary Ecology, 15(4–6), 501–520. 10.1007/978-94-017-1345-0_15 [DOI] [Google Scholar]
- Eckert, C. G. , Kalisz, S. , Geber, M. A. , Sargent, R. , Elle, E. , Cheptou, P.‐O. , … Winn, A. A. (2010). Plant mating systems in a changing world. Trends in Ecology & Evolution, 25(1), 35–43. 10.1016/J.TREE.2009.06.013 [DOI] [PubMed] [Google Scholar]
- Eckstein, R. L. , Danihelka, J. , & Otte, A. (2009). Variation in life‐cycle between three rare and endangered floodplain violets in two regions: Implications for population viability and conservation. Biologia, 64(1), 69 10.2478/s11756-009-0002-1 [DOI] [Google Scholar]
- Eckstein, R. L. , Hölzel, N. , & Danihelka, J. (2006). Biological flora of central europe: Viola elatior, V. pumila and V. stagnina . Perspectives in Plant Ecology, Evolution and Systematics, 8(1), 45–66. 10.1016/J.PPEES.2006.01.001 [DOI] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: A simulation study. Molecular Ecology, 14(8), 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Excoffier, L. (2004). Patterns of DNA sequence diversity and genetic structure after a range expansion: Lessons from the infinite‐island model. Molecular Ecology, 13(4), 853–864. 10.1046/j.1365-294X.2003.02004.x [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Fields, P. D. , & Taylor, D. R. (2014). Determinants of genetic structure in a nonequilibrium metapopulation of the plant Silene latifolia . PLoS ONE, 9(9), e104575 10.1371/journal.pone.0104575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay, C. M. V. , Bradley, C. R. , Preston, S. J. , & Provan, J. (2017). Low genetic diversity and potential inbreeding in an isolated population of alder buckthorn (Frangula alnus) following a founder effect. Scientific Reports, 7(1), 3010 10.1038/s41598-017-03166-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankham, R. (2005). Genetics and extinction. Biological Conservation, 126(2), 131–140. 10.1016/J.BIOCON.2005.05.002 [DOI] [Google Scholar]
- Frankham, R. , Bradshaw, C. J. A. , & Brook, B. W. (2014). Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biological Conservation, 170, 56–63. 10.1016/J.BIOCON.2013.12.036 [DOI] [Google Scholar]
- Gompert, Z. , & Mock, K. E. (2017). Detection of individual ploidy levels with genotyping‐by‐sequencing (GBS) analysis. Molecular Ecology Resources, 17(6), 1156–1167. 10.111/1755-0998.12657 [DOI] [PubMed] [Google Scholar]
- Hanski, I. (1998). Metapopulation dynamics. Nature, 396(6706), 41–49. 10.1038/23876 [DOI] [Google Scholar]
- Harpending, H. C. (1994). Signature of ancient population growth in a low‐resolution mitochondrial DNA mismatch distribution. Human Biology, 66(4), 591–600. 10.2307/41465371 [DOI] [PubMed] [Google Scholar]
- Hirai, M. , Kubo, N. , Ohsako, T. , & Utsumi, T. (2012). Genetic diversity of the endangered coastal violet Viola grayi Franchet et Savatier (Violaceae) and its genetic relationship to the species in subsection Rostratae . Conservation Genetics, 13(3), 837–848. 10.1007/s10592-012-0333-2 [DOI] [Google Scholar]
- Huson, D. H. , & Bryant, D. (2006). Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution, 23(2), 254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Hyvärinen, M. (2018). LIFE+2011 BIO/FI/917 FINAL Report. Retrieved from https://www.luomus.fi/en/ex-situ-conservation-finnish-native-plant-species. Accessed 16 July 2019. [Google Scholar]
- IUCN, SSC., (2013). Guidelines for reintroductions and other conservation translocations. Version 1.0. Gland, Switzerland: IUCN Species Survival Commission. [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. A. (2007). CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 23(14), 1801–1806. 10.1093/bioinformatics/btm233 [DOI] [PubMed] [Google Scholar]
- Jennings, H. , Wallin, K. , Brennan, J. , Valle, A. D. , Guzman, A. , Hein, D. , … Givnish, T. J. (2016). Inbreeding, low genetic diversity, and spatial genetic structure in the endemic Hawaiian lobeliads Clermontia fauriei and Cyanea pilosa ssp. longipedunculata . Conservation Genetics, 17(2), 497–502. 10.1007/s10592-015-0785-2 [DOI] [Google Scholar]
- Kulmala, P. , Ryttäri, T. , & Laaka‐Lindberg, S. (2016). LIFE+2011 BIO/FI/917 ESCAPE Action D.2 deliverable report. Retrieved from https://www.luomus.fi/en/ex-situ-conservation-finnish-native-plant-species. Accessed 16 July 2019. In Finnish with an English summary. [Google Scholar]
- Kuris, M. , & Ruskule, A. (2006). Favourable conservation status of boreal forests: Monitoring, assessment, management. Tallinn, Estonia: Baltic Environmental Forum. [Google Scholar]
- Lauterbach, D. , Burkart, M. , & Gemeinholzer, B. (2012). Rapid genetic differentiation between ex situ and their in situ source populations: An example of the endangered Silene otites (Caryophyllaceae). Botanical Journal of the Linnean Society, 168(1), 64–75. 10.1111/j.1095-8339.2011.01185.x [DOI] [Google Scholar]
- Lee, K. M. , Kivelä, S. M. , Ivanov, V. , Hausmann, A. , Kaila, L. , Wahlberg, N. , & Mutanen, M. (2018). Information dropout patterns in restriction site associated DNA phylogenomics and a comparison with multilocus Sanger data in a species‐rich moth genus. Systematic Biology, 67(6), 925–939. 10.1093/sysbio/syy029 [DOI] [PubMed] [Google Scholar]
- Levins, R. (1969). Some demographic and genetic consequences of environmental heterogeneity for biological control. Bulletin of the Entomological Society of America, 15(3), 237–240. 10.1093/besa/15.3.237 [DOI] [Google Scholar]
- Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv, 1303.3997. Retrieved from http://arxiv.org/abs/1303.3997
- Macbeth, G. M. , Broderick, D. , Buckworth, R. C. , & Ovenden, J. R. (2013). Linkage disequilibrium estimation of effective population size with immigrants from divergent populations: A case study on spanish mackerel (Scomberomorus commerson). G3: Genes, Genomes. Genetics, 3(4), 709–717. 10.1534/g3.112.005124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinsky, M. , Trucchi, E. , Lawson, D. J. , & Falush, D. (2018). RADpainter and fineRADstructure: Population inference from RADseq data. Molecular Biology and Evolution, 35(5), 1284–1290. 10.1093/molbev/msy023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Małobęcki, A. , Marcussen, T. , Bohdanowicz, J. , Migdałek, G. , Słomka, A. , & Kuta, E. (2016). Cleistogamy and phylogenetic position of Viola uliginosa (Violaceae) re‐examined. Botanical Journal of the Linnean Society, 182, 180–194. 10.1111/boj.12460 [DOI] [Google Scholar]
- Maschinski, J. , & Albrecht, M. A. (2017). Center for plant conservation’s best practice guidelines for the reintroduction of rare plants. Plant Diversity, 39(6), 390–395. 10.1016/J.PLD.2017.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulevičiūté, D. (2015). Notes on the status of Viola uliginosa in Lithuania. Botanica Lithuanica, 21(1), 64–67. 10.1515/botlit-2015-0008 [DOI] [Google Scholar]
- Mauri, A. , Davis, B. A. S. , Collins, P. M. , & Kaplan, J. O. (2015). The climate of Europe during the Holocene: A gridded pollen‐based reconstruction and its multi‐proxy evaluation. Quaternary Science Reviews, 112, 109–127. 10.1016/J.QUASCIREV.2015.01.013 [DOI] [Google Scholar]
- Paul, W. , Cieślak, E. , Ronikier, M. , Migdałek, G. , Słomka, A. , & Żabicka, J. (2016). Low Genetic Diversity of Declining Viola uliginosa (Violaceae) at its Southern Range Limits in Poland. Acta Biologica Cracoviensia s. Botanica, 58(2), 71–82. 101010/abcsb-2016-0015 [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS ONE, 7(5), e37135 10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit, R. J. , Aguinagalde, I. , de Beaulieu, J.‐L. , Bittkau, C. , Brewer, S. , Cheddadi, R. , … Vendramin, G. G. (2003). Glacial refugia: Hotspots but not melting pots of genetic diversity. Science (New York, N.Y.), 300(5625), 1563–1565. 10.1126/science.1083264 [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. 10.1111/j.1471-8286.2007.01758.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2015). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Retrieved from http://www.r-project.org/. [Google Scholar]
- Rambaut, A. (2015). FigTree, v1.4.2: Tree figure drawing tool. Molecular evolution, phylogenetics and epidemiology. Retrieved from http://tree.bio.ed.ac.uk/software/figtree/. [Google Scholar]
- Ranta, P. (2014). Villit vihreät kaupungit. Suomen kaupunkikasvio. Tampere, Finland: Vastapaino. [Google Scholar]
- Ranta, P. , Jokinen, A. , & Laaka‐Lindberg, S. (2016). Surviving in Europe: Geopolitics of biodiversity conservation illustrated by a proxy species Viola uliginosa . Ecosphere, 7(9), e01401 10.1002/ecs2.1401 [DOI] [Google Scholar]
- Ranta, P. , & Siitonen, M. (2011). Kuinka käy luhtaorvokin? Lutukka, 3, 74–87. In Finnish with an English summary. [Google Scholar]
- Rosenberg, N. (2004). DISTRUCT: A program for the graphical display of population structure. Molecular Ecology Notes, 4(1), 137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- Roy, S. C. , Moitra, K. , & De Sarker, D. (2017). Assessment of genetic diversity among four orchids based on ddRAD sequencing data for conservation purposes. Physiology and Molecular Biology of Plants, 23(1), 169–183. 10.1007/s12298-016-0401-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryttäri, T. , Reinikainen, M. , Haeggström, C. A. , Hakalisto, S. , Hallman, J. , Kanerva, T. , … Vainio, O. (2019).Putkilokasvit. Vascular plants. Trachaeophyta In Hyvärinen E., Juslén A., Kemppainen E., Uddström A., & Liukko U.‐M. (Eds.), Suomen lajien uhanalaisuus 2019: The 2019 Red List of Finnish Species (pp. 182–202). Helsinki, Finland: Ministry of the Environment & Finnish Environment Institute. [Google Scholar]
- Sagarin, R. D. , & Gaines, S. D. (2002). The “abundant centre” distribution: To what extent is it a biogeographical rule? Ecology Letters, 5(1), 137–147. 10.1046/j.1461-0248.2002.00297.x [DOI] [Google Scholar]
- Saitou, N. , & Nei, M. (1987). The neighbor‐joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4(4), 406–425. 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- Shirk, R. Y. , Hamrick, J. L. , Zhang, C. , & Qiang, S. (2014). Patterns of genetic diversity reveal multiple introductions and recurrent founder effects during range expansion in invasive populations of Geranium carolinianum (Geraniaceae). Heredity, 112(5), 497–507. 10.1038/hdy.2013.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siitonen, M. (1990). Luhtaorvokin (Viola uliginosa) kasvupaikat ja kasvupaikkojen historia Suomessa. Master’s thesis, University of Helsinki. [Google Scholar]
- Silvertown, J. (2008). The evolutionary maintenance of sexual reproduction: Evidence from the ecological distribution of asexual reproduction in clonal plants. International Journal of Plant Sciences, 169(1), 157–168. 10.1086/523357 [DOI] [Google Scholar]
- Slazak, B. , Sliwinska, E. , Saługa, M. , Ronikier, M. , Bujak, J. , Słomka, A. , … Kuta, E. (2015). Micropropagation of Viola uliginosa (Violaceae) for endangered species conservation and for somaclonal variation‐enhanced cyclotide biosynthesis. Plant Cell, Tissue and Organ Culture, 120(1), 179–190. 10.1007/s11240-014-0592-3 [DOI] [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30(9), 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123(3), 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Swedish Species Information Centre . (2019). https://artportalen.se/. Accessed 24 July 2019.
- Thomas, C. D. (1994). Extinction, colonization, and metapopulations: Environmental tracking by rare species. Conservation Biology, 8(2), 373–378. 10.2307/2386461 [DOI] [Google Scholar]
- Toczydlowski, R. H. , & Waller, D. M. (2019). Drift happens: Molecular genetic diversity and differentiation among populations of jewelweed (Impatiens capensis Meerb.) reflect fragmentation of floodplain forests. Molecular Ecology, 28(10), 2459–2475. 10.1111/mec.15072 [DOI] [PubMed] [Google Scholar]
- van den Hof, K. , van den Berg, R. G. , & Gravendeel, B. (2008). Chalcone synthase gene lineage diversification confirms allopolyploid evolutionary relationships of European Rostrate Violets. Molecular Biology and Evolution, 25(10), 2099–2108. 10.1093/molbev/msn157 [DOI] [PubMed] [Google Scholar]
- Van Looy, K. , Jacquemyn, H. , Breyne, P. , & Honnay, O. (2009). Effects of flood events on the genetic structure of riparian populations of the grassland plant Origanum vulgare . Biological Conservation, 142(4), 870–878. 10.1016/J.BIOCON.2008.12.006 [DOI] [Google Scholar]
- Waples, R. S. , & Do, C. (2010). Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evolutionary Applications, 3(3), 244–262. 10.1111/j.1752-4571.2009.00104.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, T. Y. (2013). corrplot: Visualization of a correlation matrix. Retrieved from R package version 0.73. http://cran.r-project.org/package=corrplot.
- Wickham, H. (2009). ggplot2: Elegant graphics for data analysis. New York, NY: Springer. [Google Scholar]
- Young, A. , Boyle, T. , & Brown, T. (1996). The population genetic consequences of habitat fragmentation for plants. Trends in Ecology & Evolution, 11(10), 413–418. 10.1016/0169-5347(96)10045-8 [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Kobert, K. , Flouri, T. , & Stamatakis, A. (2014). PEAR: A fast and accurate Illumina Paired‐End reAd mergeR. Bioinformatics, 30(5), 614–620. 10.1093/bioinformatics/btt593 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The demultiplexed fastq data are archived in the NCBI SRA (BioProject ID: PRJNA540749). All Supporting Information material accompanying the article is posted on Dryad Digital Repository, http://doi.org/10.5061/dryad.6v78j4c.