
Figure 1. Experimental and data analysis workflow to study correlation between mRNA abundance and protein level degradation using DIA‐MS.
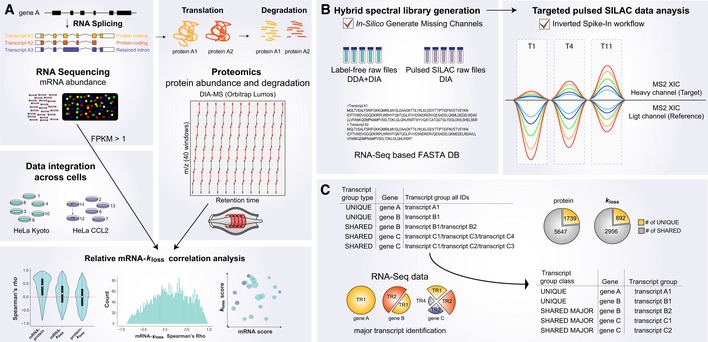
- Isoform‐resolved protein expression and degradation analysis using DIA‐MS. RNA splicing isoforms were analyzed using RNA‐Seq, and the total proteome and protein degradation were analyzed using DIA‐MS. A protein FASTA database was compiled using protein coding sequences expressing above a threshold of FPKM > 1. After data integration, the splicing isoform‐resolved matrix was used to study absolute and relative correlation between mRNA, protein, and protein degradation (k loss).
- pSILAC‐DIA workflow for determining protein degradation. A hybrid pSILAC library was created by combining both label‐free and labeled DIA‐ and DDA‐MS runs and enabling the In‐Silico Generate Missing Channels function (in, e.g., Spectronaut). To perform targeted pSILAC data analysis, the Inverted Spike‐In workflow (ISW) was used.
- mRNA abundance directed detection and quantification of protein isoforms and isoform groups. The non‐unique peptides were assigned a unique ID by using the average abundance on mRNA level for all proteins included in a protein group. The major (i.e., the most abundant) splicing isoform on mRNA level was selected as the best representative ID for the protein group (shared major).