

Abstract
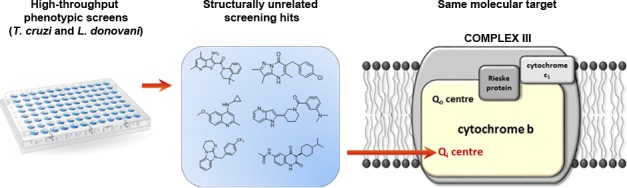
Available treatments for Chagas’ disease and visceral leishmaniasis are inadequate, and there is a pressing need for new therapeutics. Drug discovery efforts for both diseases principally rely upon phenotypic screening. However, the optimization of phenotypically active compounds is hindered by a lack of information regarding their molecular target(s). To combat this issue we initiate target deconvolution studies at an early stage. Here, we describe comprehensive genetic and biochemical studies to determine the targets of three unrelated phenotypically active compounds. All three structurally diverse compounds target the Qi active-site of cytochrome b, part of the cytochrome bc1 complex of the electron transport chain. Our studies go on to identify the Qi site as a promiscuous drug target in Leishmania donovani and Trypanosoma cruzi with a propensity to rapidly mutate. Strategies to rapidly identify compounds acting via this mechanism are discussed to ensure that drug discovery portfolios are not overwhelmed with inhibitors of a single target.
Keywords: cytochrome b, Trypanosoma cruzi, Leishmania donovani, drug target, mechanism of action
Neglected tropical diseases (NTDs) are prevalent infectious diseases afflicting the world’s poorest people, often living in extreme poverty earning less than $2 per day. Consequently, it is hardly surprising that the economic drivers for the development of novel diagnostics, drugs, vaccines, and other interventions are sadly lacking. Among the “most neglected” NTDs are Chagas’ disease (CD) infecting 7–8 million in Central and South America and visceral leishmaniasis (VL), with up to 100 000 new cases arising each year predominantly in rural India, Sudan, South Sudan, Kenya, Somalia, Ethiopia, and Brazil. Combined, these diseases are responsible for more than 40 000 fatalities annually and the loss of over 1.2 million disease adjusted life years.1,2 The accompanying economic burden of these vector-borne diseases provides a major obstacle to improving human health.3 Current treatments for both VL and CD suffer from a range of issues including severe toxic side effects4,5 and acquired drug resistance.6 To compound these difficulties, many of these chemotherapeutics also require prolonged treatment regimens7 and are prohibitively expensive. Thus, there remains a definite and urgent need to develop low-cost, safe, effective, oral, and short-course drugs to strengthen the range of treatment options. Unfortunately, there are currently no new therapeutics in advanced clinical development for either disease and relatively few in preclinical development.8
Drug discovery efforts for CD (caused by infection with the protozoan parasite Trypanosoma cruzi) and VL (caused by Leishmania donovani or Leishmania infantum) are hampered by a dearth of well-validated molecular drug targets. This has severely curtailed target-focused drug discovery against these parasites, leaving drug discovery programs reliant upon whole cell (phenotypic) screening to identify effective start points.9 This approach has proven somewhat effective. However, the downstream development and optimization of phenotypically active compounds is often hindered by a lack of information regarding their mechanism(s) of action and/or molecular target(s). Specifically, issues related to compound toxicity and poor pharmacokinetic properties can be considerably more difficult to overcome without knowledge of the target and the molecular context in which the compound is binding. As a consequence, compounds identified via phenotypic screening can suffer from disproportionately high attrition rates.10
One strategy to combat the high failure rates associated with the development of phenotypically active compounds is to initiate target deconvolution studies at a relatively early stage. The association of active compounds with defined molecular targets during the development process can be extremely powerful. Toxic liabilities associated with the target can be assessed, and in cases where the target is structure-enabled, more selective and potent versions of compound series can be evolved. A more complete understanding of the compound mechanism of action (MoA) can also prevent enrichment of drug candidates against the same molecular target or the development of inhibitors with an unattractive or invalidated target, such as sterol 14α-demethylase (CYP51) in T. cruzi.11,12 Furthermore, this knowledge can inform future drug combination strategies, and may lead to the identification of novel drug targets that can be exploited by de novo target-based drug discovery.
Here, we describe our comprehensive genetic and biochemical studies to determine the MoA of 3 unrelated compounds that demonstrated promising in vitro activity against L. donovani and T. cruzi. Despite the structural diversity of these compounds, identified as a result of independent high-throughput screening initiatives, all 3 specifically target the Qi active site of cytochrome b, part of the cytochrome bc1 complex of the electron transport chain (ETC). Our studies identify the Qi site of cytochrome b as a promiscuous drug target in L. donovani and T. cruzi. Strategies to rapidly identify compounds acting via this MoA to prevent drug discovery portfolios from becoming overwhelmed with cytochrome b inhibitors are discussed.
Results
Pyrazolopyrimidinone Compound Demonstrating Promising Activity against L. donovani and T. cruzi
High-throughput screening of GSK’s 1.8 M diverse compound library against L. donovani, T. cruzi, and Trypanosoma brucei resulted in the identification of a significant number of compounds active against these parasites.13 Among these hits, TCMDC-143087 was moderately active against T. cruzi intracellular amastigotes with an EC50 value of 250 nM. Using TCMDC-143087 as a start point, a hit-to-lead drug discovery program was initiated and resulted in the development of a compound series exemplified by DDD01542111 (compound 1, Figure 1). Compound 1 demonstrated promising potency against both the mammalian (intracellular amastigote) and insect (promastigote) stages of L. donovani with EC50 values of 1500 ± 400 and 19 ± 1 nM, respectively (Table 1). In addition, compound 1 was active against both developmental stages of T. cruzi (EC50 values of 210 ± 7 and 20 ± 3 nM for epimastigotes and intracellular amastigotes, respectively). For T. brucei, this pyrazolopyrimidinone compound was also extremely potent against the procyclic stage of the parasite (EC50 = 97 ± 3 nM); however, there was a pronounced drop in activity of more than 3 orders of magnitude against the bloodstream form (EC50 = 6600 ± 470 nM) (Table 1).
Figure 1.

Chemical structures.
Table 1. Compound Efficacy: Potencies of Compounds Were Determined against L. donovani Promastigotes (Pro), L. donovani Intramacrophage Amastigotes (Intra-MAC), T. cruzi Epimastigotes (Epi), T. cruzi Intra-Vero Cells (Intra-Vero), T. brucei Bloodstream Forms (BSF), T. brucei Procyclics (Pro), and HepG2 Cellsa.
| EC50 values, nM |
|||||||
|---|---|---|---|---|---|---|---|
|
T. cruzi |
L. donovani |
T. brucei |
|||||
| compound ID | Epi | Intra-Vero | Pro | Intra-MAC | BSF | Pro | HepG2 |
| DDD01542111 (compound 1) | 210 ± 7 | 20 ± 3 | 19 ± 1 | 1500 ± 400 | 6700 ± 470 | 97 ± 3 | >50 000 |
| DDD01716002 (compound 2) | 71 ± 2 | 30 ± 10 | 24 ± 2 | 1100 ± 600 | 8000 ± 970 | 140 ± 14 | >50 000 |
| DDD00808408 (compound 3) | 320 ± 12 | 150 ± 46 | 1640 ± 54 | >50 000 | 8700 ± 470 | 88 ± 2 | 7300 ± 60 |
| GNF7686 | 710 ± 17 | 150 ± 30b | 570 ± 51 | >50 000 | >50 000 | 520 ± 13 | >50 000 |
| antimycin A | 28 ± 1 | NDc | 2 ± 0.1 | ND | >50 000 | 18 ± 0.5 | <1000 |
All data are the mean ± standard deviation of at least two biological replicates (n ≥ 2) with each biological replicate composed of three technical replicates. In all cases, Hill slope values ranged between 1.0 and 6.
Reported previously.14
ND: not determined.
Resistance Generation and Whole Genome Sequencing
To investigate the MoA of compound 1, populations of L. donovani and T. cruzi parasites resistant to this pyrazolopyrimidinone were selected. Clonal lines of drug-susceptible L. donovani promastigotes and T. cruzi epimastigotes were cultured in vitro in the continuous presence of compound 1 until significant levels of drug resistance emerged. L. donovani promastigotes were exposed for a total of 140 days until they were capable of growing in 2 μM compound 1 (equivalent to 100× the established EC50 value, Table 1). Resistance emerged more quickly in our T. cruzi cultures with epimastigotes exposed to drug for just 70 days capable of growing in 10 μM compound 1 (equivalent to >80× the established EC50 value, Table 1). Following drug selection, resistant parasites were cloned by limiting dilution; the susceptibility of each cloned cell line to compound 1 was determined and compared to that of wild-type parasites (Figure 2). All cloned cell lines demonstrated considerable levels of resistance to compound 1 with L. donovani clones between 38- and 62-fold and T. cruzi between 12- and 32-fold less sensitive than wild-type parental cell lines (Figure 2). In each case the resistance demonstrated by these clones was stable over 20 passages in culture in the absence of drug.
Figure 2.

Compound 1 resistance in vitro. Schematic representation of the generation of compound 1-resistant cell lines in Leishmania donovani (A) and Trypanosoma cruzi (B). Each passage of cells in culture (circles) is indicated with clones I, II, and III indicated in black, white, and gray, respectively. (C, D) EC50 values for compound 1 were determined for WT (closed circles) and RES I, II, and III-resistant cell lines (black, white, and gray squares, respectively). The curves are the nonlinear fits of data using a two-parameter EC50 equation provided by GraFit. EC50 values of 21.5 ± 0.5 and 120 ± 6 nM were determined for compound 1 against WT L. donovani and T. cruzi, respectively. EC50 values for L. donovani RES I, II, and III were 1300 ± 14, 1300 ± 96, and 790 ± 27 nM. For T. cruzi, RES I, II, and III returned values of 1400 ± 78, 2500 ± 62, and 3800 ± 160 nM, respectively. EC50 values are the weighted mean ± standard deviation of at least two biological replicates (n ≥ 2) with each biological replicate composed of three technical replicates.
Whole genome sequencing of all 6 L. donovani and T. cruzi clones resistant to compound 1 revealed mutations within the gene encoding Cytochrome b, part of the cytochrome bc1 complex (complex III) of the ETC (Figure 3A,B). All 3 T. cruzi clones analyzed shared the same L197F mutation in cytochrome b (Figure 3B). Interestingly, this specific mutation has previously been identified in T. cruzi epimastigotes that are resistant to GNF7686 (Figure 1), an established cytochrome b inhibitor.14 Compound 1-resistant L. donovani promastigotes maintained two separate mutations within cytochrome b, G37A in clones 1 and 2 and C222F in clone 3. Cytochrome b contains 2 discrete reaction sites involved in the Q cycle: a ubiquinone reduction center (Qi site) and a ubiquinol oxidation center (Qo site) (Figure 3C). All of the mutations identified in our compound 1-resistant parasites map to the Qi center of cytochrome b (Figure 3C).
Figure 3.

Mutations in cytochrome b confer resistance to compounds 1–3. The tables illustrate the collated mutations within L. donovani (A) and T. cruzi (B) cytochrome b identified by WGS of clones resistant to compounds 1–3. (C) Secondary structure model of the L. donovani cytochrome b based on the Saccharomyces cerevisiae enzyme.60 Amino acids in cytochrome b that were mutated in cell lines resistant to compounds 1–3 are indicated by light blue circles (L. donovani only), dark blue circles (T. cruzi only), and light blue/dark blue circles (found in both parasites). Please note that the numbering of amino acids in this model is representative of L. donovani cytochrome b. Please note that the numbering of amino acids in T. cruzi cytochrome b is equivalent to L. donovani residues −1.
Our sequencing analysis, at a genome coverage of between 46- and 88-fold, indicates that in all cases these mutations are homozygous. It should be noted that no other consistent single nucleotide polymorphisms (SNPs) or copy number variations (CNVs) that could indicate an alternative mechanism of action and/or resistance were identified in these resistant clones (Tables S1 and S2 and Figure S2). In the kinetoplastids, Cytochrome b is encoded solely by kinetoplast DNA and specifically by maxi-circle DNA.15 Mitochondrial networks can maintain up to 50 copies of maxi-circle DNA meaning that a single network encodes up to 50 copies of Cytochrome b. With this in mind, we hypothesize that these mutations are likely to have arisen from a point mutation on a single copy of maxi-circle DNA before spreading to all copies during the process of compound selection. Collectively, these data identify cytochrome b as the putative target of compound 1 in both L. donovani and T. cruzi.
Compound 1 Inhibits Complex III Activity and Respiration
Located in the mitochondrial inner membrane, complex III of the ETC is composed of cytochrome b associated with a Rieske iron–sulfur protein and cytochrome c. Complex III accepts ubiquinol from complex II of the ETC; the Qo and Qi sites of cytochrome b then act in tandem to reduce cytochrome c by quinone-based electron bifurcation, sequentially oxidizing two ubiquinol molecules to ubiquinone, and then reducing one ubiquinone to ubiquinol.16 Since Cytochrome b is encoded by kinetoplast DNA there is currently no technical way of editing these genes (∼50 copies) to validate the role of resistance-associated mutations, either by CRISPR-cas9 or by other more traditional methods of gene editing. In addition, episomal expression of Cytochrome b would be unlikely to confer resistance as the overexpressed protein would be localized to the cytoplasm rather than in the mitochondrion where it is functionally required. Thus, to establish if compound 1 specifically inhibits complex III activity in L. donovani and T. cruzi, clarified cell lysates of both parasites that were enriched for mitochondria were prepared. Using decylubiquinol as a pseudosubstrate, the activity of complex III in the presence and absence of test compounds was determined by monitoring the reduction of cytochrome c at 550 nm. In the first instance, the assay was validated using established inhibitors of the Qi site of cytochrome b, antimycin A17 and GNF768614 (Figure 1). As expected, both compounds were potent inhibitors of complex III activity in lysates of L. donovani and T. cruzi (Table 2, Figure S3). Antimycin A demonstrated relatively equal potency against complex III from both parasites with IC50 values of 25 ± 2 and 14 ± 3 nM in L. donovani and T. cruzi lysates, respectively. In keeping with its development as a small molecule inhibitor of T. cruzi, GNF7686 was a more potent inhibitor of complex III derived from T. cruzi epimastigotes than L. donovani promastigotes (IC50 values of 550 ± 97 nM versus 1610 ± 300 nM, respectively). Compound 1 also proved to be a potent inhibitor of complex III activity in both parasite lysates, returning IC50 values of 44 ± 7 and 72 ± 16 nM for L. donovani and T. cruzi, respectively. Importantly, these IC50 values correlate well with the established EC50 values for compound 1 against promastigote and epimastigote growth in vitro (Table 1) supporting our initial hypothesis that cytochrome b is the principal target of this pyrazolopyrimidinone compound. In further support of this hypothesis, complex III activity derived from a compound 1-resistant L. donovani clone demonstrated marked resistance to inhibition by compound 1 compared to the equivalent activity in lysates of WT parasites (Table S3).
Table 2. Assessment of Compounds in Assays Measuring Complex III Activity and Respirationa.
| complex III, IC50 values, nM |
O2 consumption, IC50 values, nM | ||
|---|---|---|---|
| compound ID | L. donovani | T. cruzi | T. cruzi |
| DDD01542111 (compound 1) | 44 ± 7 | 72 ± 16 | 150 ± 36 |
| DDD01716002 (compound 2) | 99 ± 13 | 100 ± 24 | 510 ± 119 |
| DDD00808408 (compound 3) | 13 000 ± 4800 | 86 ± 39 | 190 ± 38 |
| GNF7686 | 1600 ± 300 | 550 ± 97 | 210b |
| antimycin A | 25 ± 2 | 14 ± 3 | 200 ± 36 |
Inhibition of complex III (cytochrome c reduction) activity was determined in lysates enriched with mitochondria isolated from L. donovani promastigotes and T. cruzi epimastigotes. For all complex III assays, Hill slope values ranged from 0.6 to 1.3 with the majority of values close to 1.0. Therefore, we do not believe that these Hill slope values are indicative of cooperativity. The respiration of T. cruzi epimastigotes (3 × 106) at a range of compound concentrations was determined using MitoXpress-Xtra probe (see Materials and Methods for details). All complex III assay data are the weighted mean ± standard deviation of at least two biological replicates (n ≥ 2) with each biological replicate composed of three technical replicates. O2 consumption data are the weighted mean ± standard deviation of two biological replicates (n = 2). Representative data from both assays are shown in Figure S3.
Reported previously.14
During aerobic metabolism, the ETC mediates the transport of electrons, from electron donors such as NADH or FADH2, to the terminal electron acceptor, molecular oxygen. Thus, inhibition of cytochrome b should result in a measurable decrease in the consumption of O2 by parasites. To further confirm a role in inhibition of ETC function, the rate of O2 consumption of T. cruzi epimastigotes was determined using the MitoXpress Xtra reagent in the presence of varying concentrations of compound 1 (Table 2). Antimycin A was once again utilized as a positive control in these assays and returned an IC50 value of 200 ± 36 nM. Compound 1 was a similarly potent inhibitor of T. cruzi respiration with an IC50 value of 150 ± 36 nM.
Multiple Cytochrome b Inhibitors Identified through High-Throughput Screening
At the same time the MoA of compound 1 was established, two unrelated compounds demonstrating potent antikinetoplastid activity were identified as a result of various high-throughput screening initiatives. Compound 2 was developed from an original hit identified from screening of an in-house 15 659 compound diversity library against L. donovani (Figure 1). Following optimization, this thienopyridine compound maintained its activity against L. donovani and also proved to be a low nM inhibitor of T. cruzi (epimastigotes and intracellular amastigotes, Table 1). Compound 3 originated from a hit identified from screening of a Medivir library of compounds. An iterative process of optimization then led to the development of compound 3 (Figure 1). While compound 3 demonstrated promising activity against T. cruzi (EC50 values of 320 ± 12 and 150 ± 46 nM for epimastigotes and intracellular amastigotes, respectively), it was largely inactive against both developmental stages of L. donovani. Interestingly, both compounds demonstrated the same pronounced drop-off in activity against bloodstream form compared to procyclic T. brucei as seen with compound 1. This pattern of compound potency is particularly striking since procyclics are considered the more robust form of T. brucei and are generally more resilient to drug treatment. In the case of compound 1, this pattern of potency is entirely consistent with our hypothesis that compound 1 is a cytochrome b inhibitor. Procyclic parasites are dependent upon a classical ETC, including cytochrome b, for ATP production; while bloodstream forms rely almost entirely on glycolysis and do not express cytochrome b.18 Indeed, the same differential activity against procyclic and bloodstream T. brucei was observed with the established cytochrome b inhibitors GNF7686 (EC50 values of 521 versus >50 000 nM) and antimycin A (EC50 values of 18 versus >50 000 nM) (Table 1). Collectively, these observations reinforce cytochrome b as the likely molecular target of compound 1 and raise the possibility that compounds 2 and 3 may also specifically target this key enzyme of the ETC.
L. donovani and/or T. cruzi cell lines resistant to compounds 2 and 3 were generated in vitro, as described above (Figure S1). Whole genome and in-house Sanger sequencing revealed several homozygous point mutations within the Cytochrome b genes of these resistant clones (summarized in Figure 3A–C; see also Figure S2 and Table S4). Both compounds were confirmed as potent inhibitors of complex III activity in parasite cell lysates (Table 2 and Figure S3). Compound 2 inhibited complex III isolated from L. donovani and T. cruzi parasites (IC50 values of 99 ± 13 and 100 ± 24 nM, respectively), while compound 3 inhibited complex III from T. cruzi epimastigotes (IC50 value of 86 ± 39 nM) but showed little significant inhibition of complex III from L. donovani promastigotes (IC50 > 13 000 nM). As expected, both compounds also inhibited the respiration (O2 consumption) of wild-type T. cruzi epimastigotes with IC50 values of 510 ± 120 and 190 ± 38 nM for compounds 2 and 3, respectively (Table 2). This confirms that these apparently structurally unrelated compounds (2 and 3), derived from independent high-throughput screens, target the same molecular target as compound 1.
Complex Cross-Resistance Relationships
We next determined the relative sensitivity of each resistant line to compounds 1–3 and GNF7686 (Figure 4). This analysis revealed a complex pattern of susceptibility that appears to be entirely driven by the specific cytochrome b mutations represented in the respective clones. In L. donovani (Figure 4A), our resistant clones generated through selection with compound 1 maintained either a G37A or C222F mutation. Promastigotes bearing the G37A mutation demonstrated hypersensitivity to compound 2, relative to wild-type parasites, while remaining sensitive to GNF7686. In contrast, parasites with the C222F mutation were markedly cross-resistant to GNF7686 and hypersensitive to compound 2. Three separate point mutations (G31A, C207P, and F227I) were identified in the three L. donovani-resistant clones generated with compound 2. These individual cell lines responded very differently to treatment with compound 1 and GNF7686, ranging from hypersensitivity to decreased susceptibility depending on the mutations encoded in cytochrome b. Importantly, these observations were replicated when the L. donovani-resistant lines were assessed against compounds 1 and 2 as intracellular amastigotes within macrophages (Table S5). Our T. cruzi-resistant clones also exhibited stark, mutation-driven variability in their response to these various cytochrome b inhibitors (Figure 4B).
Figure 4.

Cross-resistance relationships. Resistant clones bearing specific cytochrome b mutations were assessed for their response to GNF7686 (light gray), compound 1 (white), compound 2 (dark gray), and compound 3 (black). EC50 values were determined and compared to the EC50 values established with these compounds against wild-type parasites. (A) Data generated with L. donovani-resistant promastigotes. (B) Data generated with T. cruzi epimastigotes. All data are the mean ± standard deviation of at least two biological replicates (n ≥ 2) with each biological replicate composed of three technical replicates. (C, D) Data from a single biological replicate.
Molecular Modeling
In order to rationalize the role of specific mutations in decreased susceptibility and/or hypersensitivity to compounds 1–3, homology models of the L. donovani and T. cruzi cytochrome b were generated using the crystallographic structure of the avian (Gallus gallus) homologue as a template.19 The Qi active sites of L. donovani and T. cruzi cytochrome exhibit 38% and 45% sequence identity compared to the avian sequence, respectively (Figure 5A). Each model was built with ubiquinone bound in the active site. Ramachandran plots indicated the robustness of the L. donovani and T. cruzi models with 89% and 87% of the residues in the most favored orientations, respectively. Modeling results obtained from docking calculations and refined using molecular dynamic (MD) simulations predict that compounds 1–3 share a similar mode of binding in the Qi active site (Figure 5B). Indeed, all 3 ligands exploit the same hydrophobic pocket as the well-established Qi site inhibitor antimycin A (Figure 5B,C, Figures S4 and S5). With regard to our model with compound 1, the planar pyrazolo-pyrimidinone moiety binds between haem (bH) and Phe223. The carbonyl group and the pyrazole N form two water-bridged hydrogen bonds with the conserved D231 while hydrophobic interactions are established between the bicycle ring and residues L20, L201, and F223. In addition, the chloro-phenyl ring extends toward G37, establishing hydrophobic contacts with L198 and a stacking interaction with F34. This predicted mode of binding goes some way to explaining the molecular role of the G37A mutation identified in L. donovani clones resistant to compound 1. The methyl group of the alanine side-chain could cause a steric clash with the chloro-phenyl moiety resulting in a drop in potency. Similarly, the L197F mutation identified in compound 1-resistant T. cruzi would likely hinder the binding of the ligand due to a possible steric clash with the phenyl ring.
Figure 5.

Docking and druggability of the Qi active sites of L. donovani and T. cruzi cytochrome b. (A) Sequence alignments of the Qi site of cytochrome b from L. donovani, T. cruzi, human, and Gallus gallus, generated using Jalview version.61 (B) Binding mode of antimycin A (green), compound 1 (pink), compound 2 (violet), and compound 3 (cyan) in the Qi active site of cytochrome b (light-yellow cartoon). The structures shown correspond to the L. donovani cytochrome b homology model except for the compound 3 complex where the T. cruzi model is displayed. Residues establishing the more relevant interaction with the ligands are displayed as sticks. Haem (bH) is shown in red at the back of the cavity. Please note that the numbering of amino acids in T. cruzi cytochrome b are equivalent to L. donovani residues −1. (C) Physicochemical properties of the cytochrome b Qi active site. Hydrophobic (yellow), hydrogen-bond donor (blue), and hydrogen-bond acceptor (red) maps of the Qi active site are annotated. Antimycin A (green sticks) is docked into the Qi site of cytochrome b.
In our model of compound 2 (Figure 1), the planar thienopyridine bicycle also binds close to the haem (bH), with the tetrahydroquinoline moiety extending out of the binding pocket toward the interface of transmembrane helices TM-A and TM-D (Figure 5B). A water-mediated hydrogen bond is established between D231 and the thienopyridine acceptor N, and a stacking interaction is established with F223. In addition, the dimethyl group fits neatly into a hydrophobic pocket formed by residues I194, F195, and L198. Interestingly, and in direct contrast to compound 1, a G37A mutation in the L. donovani enzyme (equivalent to G36C in T. cruzi) results in hypersensitivity to compound 2. Based on our current model, this can be rationalized by the mutated alanine causing a general increase in the overall hydrophobicity of the binding pocket that accommodates the dimethyl group of compound 2. Mutations in L. donovani cytochrome b directly associated with resistance to compound 2 include G31A, S207P, and F227I. G31 is in close proximity to the conserved water molecule that bridges D231 with the ligand. Bulkier residues in this position would disrupt this interaction notably affecting the binding of the compounds. S207 is located within loop 2 at the back of the hydrophobic pocket where the planar moiety of compound 2 binds. Mutation of S207 to proline is likely to impact the loop conformation and the shape of the binding pocket. F227 within the hydrophobic pocket defines the conformation of F223 responsible for forming a stacking interaction with the ligand. A F227I mutation in L. donovani cytochrome b alters the conformation of the F223 thus limiting the binding of the ligand.
In MD simulations, the binding of compounds 1 and 2 to the Qi site of both L. donovani and T. cruzi cytochrome b demonstrates a high degree of stability (Figures S6 and S7). However, simulations with compound 3 illustrate a significant divergence in behavior between the two parasite enzymes that may explain the comparatively poor antileishmanial activity of this compound. In complex with the T. cruzi enzyme, compound 3 demonstrates notable stability forming numerous ligand–protein interactions (Figure 5B). In contrast, the binding of compound 3 to L. donovani cytochrome b is considerably less stable due to the absence of key interactions (Figure S8). Of particular note, in the T. cruzi complex, amino acid M21 stabilizes the acetanilide moiety of compound 3 through van der Waals interactions. These contacts are not maintained with the L. donovani enzyme where M21 corresponds to T22. In binding to T. cruzi cytochrome b, the substituted cyclohexyl ring of compound 3 extends out of the binding cavity in close proximity to residue G36, establishing further hydrophobic contacts with residues F33, L37, and L197. These interactions play a key role in resistance to compound 3 in T. cruzi with mutations G36C, L197I, and L197F identified in resistant clones. Mutation of G36 or L197 to bulky residues, such as C or I, will induce a steric clash with the isopropyl group of compound 3 that accounts for the reduced potency observed against clonal lines bearing these mutations.
Assessing the Druggability of the L. donovani and T. cruzi Qi Site
In an attempt to explain the apparent promiscuity of the Qi site as a drug target in L. donovani and T. cruzi, the binding site recognition software SiteMap was used to assess “druggability” of the pocket.20 In both parasites, the Qi site is predominantly hydrophobic in nature and is of considerable size (volume = 394 Å3) (Figure 5C). The SiteMap analysis characterizes the Qi site as highly druggable. The computed value of the SiteScore and Druggability Score for the Qi site was 1.1, well above the cutoff value of 0.8 that is used to discriminate between drug-binding and non-drug-binding cavities. Calculation of the exposure and enclosure of putative binding sites can provide an estimate of how open any given pocket is to solvent. Highly druggable sites tend to display low values of exposure (<0.49) and high values of enclosure (>0.78). The calculated exposure and enclosure scores for the Qi site of L. donovani and T. cruzi were 0.35 and 0.87, respectively, and confirm the highly druggable properties of the pocket. In conclusion, the SiteMap calculations showed that the Qi site of cytochrome b from T. cruzi and L. donovani displays a combination of features that are optimal for ligand binding: a large pocket volume, an extended hydrophobic pocket, high druggability scores, a low cavity exposure, and a high cavity enclosure. This combination of physicochemical features confirms the highly druggable nature of this pocket and may go some way to explaining the significant numbers of Qi inhibitors identified in our phenotypic screens.
Strategy to Identify Cytochrome b Inhibitors at an Early Stage of the Drug Discovery Pipeline
Our studies indicate that a diverse range of chemotypes are capable of targeting the Qi site of cytochrome b in both L. donovani and T. cruzi. This structural diversity effectively precludes the use of structure-based methods to predict cytochrome b inhibitors at present. In order to prevent our drug discovery portfolios from becoming enriched with compounds that inhibit this single molecular target, strategies to rapidly and efficiently identify cytochrome b inhibitors at an early stage of development are required. With this mind, resistant clones bearing a range of representative cytochrome b mutations were selected to form a cytochrome b-resistant cell line panel. Hits identified in high-throughput screens (single-point and cidal potency assays) against T. cruzi intracellular amastigotes were then screened against our panel of resistant parasites. Inhibitors specifically targeting the Qi site of cytochrome b were identified by marked shifts in their potency against panel cell lines compared to wild-type parasites. To date, 54 primary screening hits have been assessed in this assay with 11 cytochrome b inhibitors positively identified representing a remarkable hit rate of almost 20%.
Three representative cytochrome b inhibitors identified via this screen are shown in Figure 6A and once again illustrate the structural diversity of compounds capable of targeting the Qi active site of this enzyme. Indeed, all 11 compounds identified by this secondary screen are chemically diverse heterocycles that do not share an obvious pharmacophore and are also structurally distinct from compounds 1–3. The three representative compounds chosen for further study elicited a range of responses from the 5 representative resistant cell lines in the panel, from hypo- to hypersensitivity (Figure 6A, Figure S9). All 3 compounds were subsequently confirmed as inhibitors of complex III activity in T. cruzi epimastigote lysates (Figure S10). The cytochrome b-resistant panel now forms a fundamental part of our screening cascade for T. cruzi(21) (Figure 6B) and sits alongside a high-throughput biochemical assay designed to identify specific inhibitors of CYP51,22 another promiscuous drug target in T. cruzi. We also intend to introduce an equivalent cytochrome b resistance panel into our L. donovani screening cascade.
Figure 6.
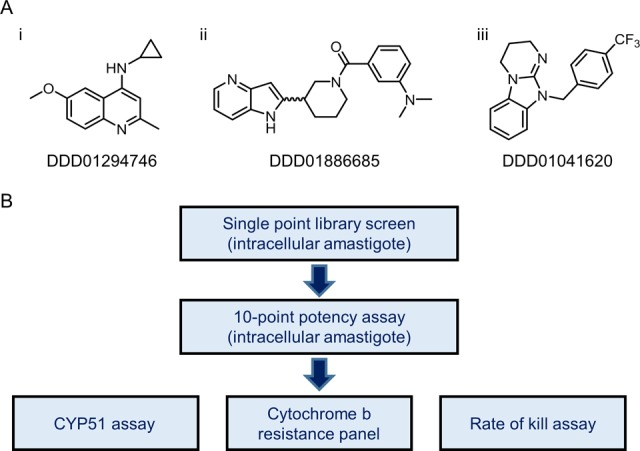
Cytochrome b-resistant cell line panel. (A) Three representative compounds identified as inhibitors of T. cruzi cytochrome b as a result of screening against our resistant cell line panel (also see Figures S3 and S4). (B) Early stages of our adapted in vitro screening cascade for T. cruzi with addition of the cytochrome b-resistant cell line panel.
Discussion
The past decade has seen a renewed focus on the discovery of novel therapeutics for the treatment of kinetoplastid diseases. In the absence of robustly validated molecular targets to support target-based studies, large-scale phenotypic screens have been used to identify start points for drug discovery. This approach not only allows the efficient identification of parasite growth inhibitors but also has the potential to identify new and exploitable molecular targets when coupled to drug target deconvolution studies. Regrettably, the actual number of new molecular targets identified via this approach for the kinetoplastids has been somewhat limited to date.14,23−26 There are likely to be several reasons why primary hits from phenotypic screens are failing to interact with a diverse range of molecular targets. However, our current study suggests that the presence of promiscuous and readily druggable targets within these parasites may well be masking the identification of novel targets.
The Qi site of cytochrome b is certainly not the only highly druggable target to be identified as a result of the growing number of high-throughput screens now being carried out against kinetoplastids. Recent screening initiatives against T. cruzi have identified a preponderance of sterol 14α-demethylase (CYP51) inhibitors, thus earmarking this enzyme as another promiscuous target in T. cruzi.13,27 Indeed, the proportion of “hits” from our in-house primary phenotypic screens demonstrating some level of CYP51 inhibition has been placed as high as 80% (Dr. Manu De Rycker, personal communication). These findings required steps to be taken to prevent an enrichment of candidates against this single molecular target, particularly in light of the recent failure of posaconazole, an established inhibitor of CYP51, in phase II clinical trials for CD.11,12 Rapid identification of CYP51 inhibitors is now achieved by triaging primary screening hits in a high-throughput fluorescence-based CYP51 inhibition assay22 (Figure 6B). Our studies indicate that similar action is now required to prevent the overenrichment of VL and CD drug development pipelines with compounds targeting the Qi site of cytochrome b. The diversity of chemotypes found to inhibit the Qi site of cytochrome b has so far prevented the development of computational solutions to identify the over-representation of such compounds in our screening libraries. It is hoped that the cumulative data sets generated by the triaging assays now in place for both promiscuous targets can be used to develop predictive algorithms. Undoubtedly, such computational tools would assist in the design of more diverse compound libraries.
Micro-organisms are often heavily dependent upon oxidative phosphorylation for cell maintenance and replication, such that inhibition of respiratory enzymes, including cytochrome b, can have dire consequences. Drugs specifically targeting these enzymes are seen as having significant potential for pathogen control. With this in mind, the past few years have seen the development of numerous cytochrome b inhibitors as antimicrobials.28 Overwhelmingly, these compounds target the Qo active site of cytochrome b, with the antimalarial atovaquone perhaps the best known example. Indeed, the Qo active site of cytochrome b could rightly be considered as something of a promiscuous drug target in Plasmodium spp and Mycobacterium tuberculosis.29−32 To our knowledge, there is currently only one Qi-targeting drug on the market: cyazofamid is an antifungal agent that is used to treat potato blight (Phytophthora infestans).33 Thus, it is highly unusual for the Qi active site of the kinetoplastids to be targeted in this way.
The range of cytochrome b mutations identified in the course of this study is noteworthy. As previously discussed, in kinetoplastids the Cytochrome b gene is encoded by the maxi-circle DNA of the kinetoplast, equivalent to the mitochondrial DNA of other eukaryotes. In vertebrates, mutation rates for mitochondrial-encoded genes have been reported as between 5 and 50 times higher than for genes encoded by nuclear DNA.34 This higher rate has been attributed to DNA damage resulting from exposure to reactive oxygen species released as byproducts of oxidative phosphorylation as well as the low efficiency of DNA repair pathways. The mutation rates for kinetoplastid-encoded genes have yet to be established but could be similarly high. Developing inhibitors against a molecular target with an intrinsically high rate of mutation clearly has the potential to deliver drugs with a particularly high resistance potential. However, it should be noted that malaria parasites bearing mutations in cytochrome b and resistant to atovaquone are not transmissible by mosquitoes.35 The apparent loss of fitness of atovaquone-resistant parasites in the mosquito has been associated with the higher respiratory rate required at this stage of the lifecycle. The failure of these mutated parasites to be transmitted effectively limits the spread of atovaquone resistance in the field. It remains to be seen if a similar fitness cost will be associated with T. cruzi and L. donovani parasites bearing Qi site mutations. Regardless, future treatment strategies for VL and CD are likely to focus on the development of combination therapies that should limit the propensity for resistance to emerge.
Our current study once again illustrates the vital importance of MoA studies at an early stage of the drug discovery process. Such studies have the potential to positively impact drug discovery in numerous ways. Here, the identification of multiple compounds targeting cytochrome b enabled the most promising of these series to be prioritized, while the development of others was efficiently terminated. Target identification allowed these series to be ranked primarily on the basis of selective toxicity versus human cytochrome b. In conclusion, MoA studies have the potential to guide key aspects of drug discovery and ultimately play an important role in the delivery of more effective drugs for NTDs to the clinic.
Materials and Methods
Test Compounds
Test compounds 1 and 2 were synthesized by medicinal chemists within the GlaxoSmithKline and the University of Dundee collaboration funded by the Wellcome Trust. Compound 3 was developed as part of the Trypobase Consortium funded under the European Union’s seventh Framework Programme.36 The details of the chemical syntheses of all 3 compounds will be provided in subsequent publications.
Purchased Compounds
Antimycin A was purchased from Sigma. GNF7686 was purchased from Vitas. On receipt, the purity of each purchased compound was verified by LCMS and NMR and confirmed to be >85%.
Cell Lines and Culture Conditions
The clonal Leishmania donovani cell line LdBOB (derived from MHOM/SD/62/1S-CL2D) was grown as promastigotes at 28 °C in modified M199 media.37T. cruzi epimastigotes from the Silvio strain (MHOM/BR/78/Silvio; clone X10/7A38) were grown at 28 °C in RTH/FBS [RPMI 1640 medium supplemented with trypticase (0.4%), 25 μM hemin, 17 mM Hepes (pH7.4), and 10% (v/v) heat-inactivated fetal bovine serum (FBS, PAA Laboratories; now GE Healthcare)].39 Bloodstream T. brucei bloodstream form “single marker” S427 (T7RPOL TETR NEO) were cultured in the presence of G418 (15 μg mL–1) at 37 °C in HMI9-T media in the presence of 5% CO2.40 Procyclic form T. brucei 427 strain containing T7 RNA polymerase and Tet repressor protein, under control of G418 (15 μg mL–1) and hygromycin (50 μg mL–1), respectively, were grown at 28 °C without CO2 in SDM-79 medium41 containing 15% (v/v) heat-inactivated FBS, 2 g L–1 sodium bicarbonate, 2 mM glutamax (Invitrogen), and 22.5 μg mL–1 hemin.
Drug Sensitivity Assays (Extracellular Parasites)
T. cruzi Epimastigotes
To examine the effects of test compounds on growth, mid log epimastigotes were seeded into 96-well plates at a cell density of 5 × 105 cells mL–1. Cells were exposed to test compounds over a range of concentrations (2-fold serial dilutions). Cells were incubated for 4 days, after which 5 mM resazurin was added to each well, before measuring fluorescence (excitation of 528 nm and emission of 590 nm), after a further 24 h incubation. Data were processed using GRAFIT (Erithacus software) and fitted to a 2-parameter equation, where the data are corrected for background fluorescence, to obtain the effective concentration inhibiting growth by 50% (EC50):
In this equation, [I] represents inhibitor concentration, and m is the slope factor. Experiments were repeated at least two times, and the data are presented as the mean plus standard deviation.
L. donovani Promastigotes
To examine the effects of test compounds on growth, triplicate promastigote cultures were seeded with 5 × 104 parasites mL–1. Parasites were grown in 10 mL cultures in the presence of drug for 72 h, after which 200 μL aliquots of each culture were added to 96-well plates; 500 μM resazurin was added to each well and fluorescence (excitation of 528 nm and emission of 590 nm) measured after a further 3 h incubation. Data were processed using GRAFIT as described above.
T. brucei Bloodstream Form
EC50 values for test compounds were determined as previously described.40
T. brucei Procyclics
To examine the effects of test compounds on growth, mid log procyclics were seeded into 96-well plates at a cell density of 5 × 105 cells mL–1. Cells were exposed to test compounds over a range of concentrations (2-fold serial dilutions). Following 72 h of incubation in the presence of drug, 500 μM resazurin was added to each well and fluorescence measured after a further 2 h incubation.
Drug Sensitivity Assays (Intracellular Parasites)
T. cruzi and L. donovani intracellular amastigote drug sensitivity assays were conducted as previously described.42,43
Generation of Drug-Resistant Parasites
Compound-resistant cell lines were generated by subculturing clones of wild-type L. donovani or T. cruzi in the continuous presence of test compounds. Starting at sublethal concentrations, drug concentrations in 3 independent cultures were increased in a stepwise manner, usually by 2-fold. When parasites were able to survive and grow in concentrations of drug equivalent to 20 times the established EC50 value, the resulting cell lines were cloned by limiting dilution in the presence of compound. Three clones (RES I–III) were selected for further biological study.
Sequencing
Genomic DNA was isolated from resistant parasites (∼5 × 108) using a standard alkaline lysis protocol. Whole genome sequencing was performed using a HiSeq4000 next-generation sequencing platform (Beijing Genomics Institute, Hong Kong). Sequencing reads were aligned to the Leishmania donovani BPK282A1 genome (v39, tritrypDB) with maxi-circle (CP022652.1, NCBI) or Trypanosoma cruzi Silvio X10-1 genome (v39, tritrypDB) with maxi-circle (FJ203996.1, NCBI) using Bowtie244 and Samtools45 software. Single nucleotide polymorphisms (SNPs) and indels were called using Samtools and Bcftools46 where overall quality score (QUAL) was >100 when compared with the wild-type starter clone. Chromosome and gene copy number variation (CNV) analysis, as well as manual confirmation of putative SNPs, was performed using Artemis.47 All associated data sets have been deposited with the European Nucleotide Archive under the following accession numbers: L. donovani compound 1-resistant clones (PRJEB32040), T. cruzi compound 1-resistant clones (PRJEB32041), and L. donovani compound 2-resistant clones (PRJEB32039). The Cytochrome b genes from T. cruzi-resistant clones generated with compounds 2 and 3 were sequenced directly in-house using the following primers: 5′-GAGAGAGAGTTTCGAGAGGGA- 3′ (forward) and 5′-TCTAAATTCGCCCAAATTCCTCTTA-3′ (reverse). Cytochrome b sequences required RNA editing within the coding region prior to translation, as previously described.48
Intramacrophage Drug Sensitivity Assays (Resistant Cell Lines)
Human peripheral blood mononuclear cells (PBMCs) were isolated from buffy coat supplied by the Scottish National Blood Transfusion Service (SNBTS) from anonymized donors under agreement with the SNBTS Committee for Governance of Blood and Tissue Samples for Nontherapeutic Use. Human PBMCs were isolated from 2 donors/assay using Leucosep tubes filled with Ficoll-Paque plus, according to the manufacturer’s instructions (Greiner Bio-One). CD14+ monocytes were subsequently purified using CD14 microbeads (Miltenyi Biotec) and differentiated for 7 days using human M-CSF (Bio-Techne), as previously described.23 Macrophages were then incubated with metacyclic promastigotes (either wild-type or resistant cell lines) at a multiplicity of infection of 5 for 16–24 h at 37 °C 5% CO2. Extracellular parasites were removed by washing three times with fresh culture media. Adherent PBMCs were detached from Petri dishes using a cell scraper following incubation with Versene, plated at 5 × 104 cells per well into 96-well plates prestamped with 500 nL test compounds, and incubated at 37 °C in 5% CO2 for 96 h. Plates were processed as previously described.23 Images were analyzed using Columbus image analysis software (PerkinElmer) and compound potencies calculated in XLFit. The data were fitted to the following sigmoidal model:
Here, y = total number of amastigotes per well, D = minimum value of y, R = range of y, x = drug concentration, E = EC50, and n = slope factor.
Complex III Assay
Measurement of complex III activity was performed as described in Khare et al., 2015,14 with minor changes. Briefly, L. donovani promastigotes and T. cruzi epimastigotes were harvested by centrifugation (1600g, 10 min, RT) and washed in Buffer A [(10 mM Tris-HCl, pH 7.4, 0.23 M mannitol, 0.07 M sucrose, 0.2 mM EDTA, 0.2% BSA, 0.5 nM phenylmethanesulfonyl fluoride (PMSF)]. The pellet was resuspended at 10 mg protein mL–1 (equivalent to ∼1 × 109 cells mL–1) in Buffer A containing 0.1 mg digitonin mg–1 protein and incubated at 26 °C, for 10 min. Lysates were centrifuged (13 000g, 5 min, RT); the resulting pellets were washed and finally resuspended in Reaction Buffer (25 mM potassium phosphate, 5 mM MgCl2, 2.5 mg mL–1 BSA, pH 7.2). Pellet suspensions (3.6 μL, equivalent to 36 μg) were added to 491.4 μL of Reaction Buffer plus 1 mM KCN, 0.6 mM maltoside, and 0.1 mM yeast cytochrome c and incubated with a specific concentration of test compound for 8 min. Following incubation, absorbance at 550 nm was monitored for a further 2 min (UV-2401 PC, Shimadzu). Reactions were initiated by the addition of decylubiquinol (80 μM final concentration), and the change in absorbance at 550 nM was monitored for 2 min. For IC50 determinations, the decylubiquinol-dependent rate of cytochrome c reduction was calculated by subtracting the rate achieved prior to decylubiquinol addition. DMSO (0.2%) was used as a negative control. IC50 values were obtained using the 2-parameter fit (GraFit 7, Erithacus software).
Measurement of T. cruzi Epimastigote Respiration
Compounds were predispensed (150 nL well–1) into 384-well assay plates using an Echo liquid handler (Labcyte Inc.). For potency determinations, 11-point dilution curves were generated with a top concentration of 25 μM. The negative control for this assay was 0.2% DMSO, equivalent to the final concentration of solvent used in drug dilutions. The positive control in this assay was antimycin A at 25 μM.
Our O2 consumption assay was an adaptation of the assay described by Khare and colleagues.14 Briefly, assay buffer (250 mM sucrose, 15 mM KCl, 5 mM MgCl2, 1 mM EGTA, 30 mM K2HPO4, pH 7.4; 25 μL) was added to 384-well assay plates and incubated at 37 °C for 10 min. Mid log T. cruzi epimastigotes were harvested (1600g for 10 min at 4 °C), washed three times with buffer A (10 mM Tris-HCl, pH 7.4, 230 mM mannitol, 70 mM sucrose, 0.2 mM EDTA, 0.2% bovine serum albumin, 0.5 mM PMSF), and finally resuspended at 1.5 × 108 epimastigotes mL–1. Following resuspension, 3 × 106 epimastigotes (20 μL) were dispensed into each well followed by MitoXpress-Xtra probe (15 μL, Agilent) resulting in a final assay volume of 60 μL. Plates were gently shaken, and 30 μL of HS mineral oil (Agilent) was then added to each well to prevent oxygen exchange between the assay buffer and air. Plates were incubated for 90 min at 37 °C, and fluorescence was then measured using an EnVision MultiMode plate reader (PerkinElmer) using excitation and emission wavelengths of 380 and 650 nm, respectively.
Data were normalized to percent of biological response by using positive (i.e., highest response achieved using a chemical tool compound, RactinomycinA) or negative (i.e., lowest response achieved in the absence of any testing compound, RDMSO) controls by using the following equation:
where Rx is the assay response measured in the presence of test compounds.
Homology Modeling
The homology models of L. donovani and T. cruzi cytochrome b in complex with ubiquinone bound to the Qi site were built using Modeler (Version 9.20).49,50 The X-ray structure of avian cytochrome bc1 ubiquinone complex was used as a template for both models (PDB code 1BCC, chain C).19 The Qi sites of L. donovani and T. cruzi cytochrome exhibit 38% and 45% sequence identity compared to the template sequence. The sequence alignments were performed using the T-Coffee algorithm51 (see sequence alignments shown in Figure 5). The geometries of the homology models were verified using PROCHECK.52 The resulting Ramachandran plots indicate a good model quality with 89% and 87% of the residues in the most favored regions.
In Silico Docking Studies
Molecular docking was performed using the homology models of cytochrome b from L. donovani and T. cruzi built with ubiquinone bound in the Qi site. Compounds 1–3 were docked into the Qi site of both structures using GLIDE,53 in the standard precision (SP) mode. The center of the grid was defined using ubiquinone in the Qi site as a reference. Crystal structures of cytochrome b bound to ubiquinone or Qi site inhibitors revealed a conserved water molecule bridging the ligands with residues on transmembrane helices TM-A and TM-E (Figure S5). This water molecule was maintained in the docking calculations and binding mode studies performed in this work. This approach was validated by predicting the binding pose of antimycin A, a well-known inhibitor of cytochrome b binding to the Qi site: docking of antimycin A to L. donovani and T. cruzi cytochrome b reproduced the binding poses observed in the crystal structures from different species (avian and bovine, PDB codes 3H1I and 1PPJ, respectively)19,54 (RMSD = 1.6 and 1.5 Å) (Figure S6). The resulting complexes of cytochrome b bound to compounds 1–3 were further refined by means of molecular dynamics (MD) simulations (see details below).
Molecular Dynamics (MD) Simulations
The stability of each complex was examined by performing three independent 100 ns molecular dynamics (MD) simulations using Desmond.55 Periodic boundary conditions were used. OPLS3e force field56 was used for proteins and ligands, and the simple point charge (SPC) method water model was applied.57 The systems were neutralized by adding the appropriate number of counterions. The cutoff distance for the nonbonded interactions was 9 Å. The SHAKE algorithm was applied to all bonds involving hydrogens, and an integration step of 2.0 fs was used throughout.58 The systems were simulated at constant temperature (300 K) and pressure (1 atm) for 100 ns. Cytochrome b is a component of the cytochrome bc1 complex formed by multiple subunits embedded in the inner mitochondrial membrane. However, the simulated systems display a unique cytochrome b subunit. Hence, to prevent any major conformational changes within the cytochrome b structure as a result of the absence of the neighboring subunits, positional restraints (5 kcal mol–1 Å2) were applied on the backbone atoms of cytochrome b during simulations.
Qi Site Physicochemical Properties
The site recognition software SiteMap was used to interrogate the Qi sites of cytochrome b from L. donovani and T. cruzi in terms of physicochemical properties (hydrophobic/hydrophilic nature), volume, exposure, and enclosure.20 Based on those properties, an overall “SiteScore” and “Druggability” score (Dscore) were generated providing an estimate value of the druggability of the pocket. Using the default settings, scores >1.0 indicate a very promising druggable area while scores of 0.8 define the limit between drug-binding and non-drug-binding cavities.
Cytochrome b-Resistant Cell Line Panel
T. cruzi X10/7 epimastigotes (wild-type and representative cytochrome b-resistant lines) were maintained at 28 °C in RTH media. Test compounds were dispensed using a LabCyte ECHO into Greiner 384-well assay plates (Greiner 781080), including four standard control curves (nifurtimox, posaconazole, DDD0077085459 and DDD0101224824), a 0% effect vehicle control (DMSO in columns 11 and 23), and a 100% effect control (50 μM nifurtimox in columns 12 and 24). Cell suspension (25 μL at a density of 5 × 105 cell mL–1) was dispensed into each well of the assay plates and left to settle at room temperature for 40 min, followed by incubation at 28 °C for 96 h. Parasite growth was assessed with the BacTiter-Glo luminescence reagent (25 μL well–1) using a PerkinElmer Victor 3 or BMG Labtech PHERAstar plate reader. All raw data were normalized to percent inhibition based on the raw data values for the 0% and 100% effect controls. For potency determinations curve fitting was carried out using the following 4-parameter equation:
with A = minimum y value, B = maximum y value, C = log EC50, D = slope factor, y = % inhibition, and x = concentration [M].
Acknowledgments
We would like to thank the Wellcome Trust (Grants: 092340 and 105021) for funding. We would like to acknowledge Dr. Al Ivens, University of Edinburgh, for his advice regarding the analysis of our whole genome sequencing data. We would also like to thank the members of the Trypobase Consortium for providing access to compound 3.
Glossary
Abbreviations
- Chagas’ disease
CD
- CNV
copy number variation
- CYP51
sterol 14α-demethylase
- ETC
electron transport chain
- GSK
GlaxoSmithKline
- MD
molecular dynamics
- MoA
mode of action
- NTDs
neglected tropical diseases
- Qi
ubiquinone reduction center
- Qo
ubiquinol oxidation center
- SNP
single nucleotide polymorphism
- VL
visceral leishmaniasis
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.9b00426.
Additional data and figures including resistance generation in vitro; analysis for L. donovani and T. cruzi clones resistant to compounds 1 and compound 2; representative complex III and O2 consumption assay data; X-ray structures of cytochrome b; binding mode of antimycin A in the cytochrome b Qi site; structural analyses; cytochrome b-resistant panel screen; complex III assays; whole genome sequencings for L. donovani and T. cruzi clones resistant to compound 1; compounds 1 and 2 in complex III assays with WT and resistant cell lysates; whole genome sequencing for L. donovani clones resistant to compound 2; and compound potencies against wild-type and resistant L. donovani cell lines in intramacrophage assays (PDF)
Author Present Address
⊥ R. Milne: Astex Pharmaceuticals Inc., 436 Cambridge Science Park, Milton Road, Cambridge CB4 0QA, UK.
Author Contributions
∥ R. J. Wall and S. Carvalho contributed equally. Author contributions were as follows: R. J. Wall, S. Carvalho, J. A. Bueren-Calabuig, I. Cotillo, L. MacLean, S. Thompson, F. Zuccotto, M. G. Thomas, A. H. Fairlamb, I. H. Gilbert, and S. Wyllie designed the research. R. J. Wall, S. Carvalho, R. Milne, J. A. Bueren-Calabuig, S. Moniz, J. Cantizani-Perez, L. MacLean, A. Kessler, I. Cotillo, L. Sastry, S. Manthri, and S. Wyllie performed the research. F. Zuccotto, S. Thompson, J. Martin, M. Marco, T. J. Miles, M. De Rycker, M. G. Thomas, I. H. Gilbert, and S. Wyllie supervised the research. R. J. Wall, S. Carvalho, J. A. Bueren-Calabuig, I. Cotillo, F. Zuccotto, J. Martin, M. De Rycker, A. H. Fairlamb, and S. Wyllie analyzed data. A. Kessler, S. Patterson, S. Thompson, and M. G. Thomas provided reagents. S. Wyllie wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Control of Neglected Tropical Diseases (NTD) World Health Organization (2015) Status of endemicity of visceral leishmaniasis worldwide. www.who.int/leishmaniasis/burden/Status_of_endemicity_of_VL_worldwide_2015_with_imported_cases.pdf?ua=1.
- Hay S. I. (2017) Global, regional, and national disability-adjusted life-years (DALYs) for 333 diseases and injuries and healthy life expectancy (HALE) for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 390 (10100), 1260–1344. 10.1016/S0140-6736(17)32130-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K.; Brun R.; Croft S.; Fairlamb A.; Gurtler R. E.; McKerrow J.; Reed S.; Tarleton R. (2008) Kinetoplastids: related protozoan pathogens. J. Clin. Invest. 118 (4), 1301–10. 10.1172/JCI33945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldasoro E.; Posada E.; Requena-Mendez A.; Calvo-Cano A.; Serret N.; Casellas A.; Sanz S.; Soy D.; Pinazo M. J.; Gascon J. (2018) What to expect and when: benznidazole toxicity in chronic Chagas’ disease treatment. J. Antimicrob. Chemother. 73 (4), 1060–1067. 10.1093/jac/dkx516. [DOI] [PubMed] [Google Scholar]
- Soto J.; Soto P. (2006) Miltefosine: oral treatment of leishmaniasis. Expert Rev. Anti-Infect. Ther. 4 (2), 177–85. 10.1586/14787210.4.2.177. [DOI] [PubMed] [Google Scholar]
- Croft S. L.; Sundar S.; Fairlamb A. H. (2006) Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19 (1), 111–26. 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijal S.; Ostyn B.; Uranw S.; Rai K.; Bhattarai N. R.; Dorlo T. P.; Beijnen J. H.; Vanaerschot M.; Decuypere S.; Dhakal S. S.; Das M. L.; Karki P.; Singh R.; Boelaert M.; Dujardin J. C. (2013) Increasing failure of miltefosine in the treatment of Kala-azar in Nepal and the potential role of parasite drug resistance, reinfection, or noncompliance. Clin. Infect. Dis. 56 (11), 1530–8. 10.1093/cid/cit102. [DOI] [PubMed] [Google Scholar]
- DNDi Diseases and Projects. https://www.dndi.org/diseases-projects/.
- Don R.; Ioset J. R. (2014) Screening strategies to identify new chemical diversity for drug development to treat kinetoplastid infections. Parasitology 141 (1), 140–6. 10.1017/S003118201300142X. [DOI] [PubMed] [Google Scholar]
- Gilbert I. H. (2013) Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J. Med. Chem. 56 (20), 7719–26. 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina I.; Salvador F.; Sanchez-Montalva A. (2014) Posaconazole versus benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 371 (10), 965. 10.1056/NEJMc1407914. [DOI] [PubMed] [Google Scholar]
- Molina I.; Gomez i Prat J.; Salvador F.; Trevino B.; Sulleiro E.; Serre N.; Pou D.; Roure S.; Cabezos J.; Valerio L.; Blanco-Grau A.; Sanchez-Montalva A.; Vidal X.; Pahissa A. (2014) Randomized trial of posaconazole and benznidazole for chronic Chagas’ disease. N. Engl. J. Med. 370 (20), 1899–908. 10.1056/NEJMoa1313122. [DOI] [PubMed] [Google Scholar]
- Pena I.; Pilar Manzano M.; Cantizani J.; Kessler A.; Alonso-Padilla J.; Bardera A. I.; Alvarez E.; Colmenarejo G.; Cotillo I.; Roquero I.; de Dios-Anton F.; Barroso V.; Rodriguez A.; Gray D. W.; Navarro M.; Kumar V.; Sherstnev A.; Drewry D. H.; Brown J. R.; Fiandor J. M.; Julio Martin J. (2015) New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: an open resource. Sci. Rep. 5, 8771. 10.1038/srep08771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S.; Roach S. L.; Barnes S. W.; Hoepfner D.; Walker J. R.; Chatterjee A. K.; Neitz R. J.; Arkin M. R.; McNamara C. W.; Ballard J.; Lai Y.; Fu Y.; Molteni V.; Yeh V.; McKerrow J. H.; Glynne R. J.; Supek F. (2015) Utilizing Chemical Genomics to Identify Cytochrome b as a Novel Drug Target for Chagas Disease. PLoS Pathog. 11 (7), e1005058 10.1371/journal.ppat.1005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukes J.; Guilbride D. L.; Votypka J.; Zikova A.; Benne R.; Englund P. T. (2002) Kinetoplast DNA network: evolution of an improbable structure. Eukaryotic Cell 1 (4), 495–502. 10.1128/EC.1.4.495-502.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckel W.; Thauer R. K. (2018) Flavin-Based Electron Bifurcation, A New Mechanism of Biological Energy Coupling. Chem. Rev. 118 (7), 3862–3886. 10.1021/acs.chemrev.7b00707. [DOI] [PubMed] [Google Scholar]
- di Rago J. P.; Colson A. M. (1988) Molecular basis for resistance to antimycin and diuron, Q-cycle inhibitors acting at the Qi site in the mitochondrial ubiquinol-cytochrome c reductase in Saccharomyces cerevisiae. J. Biol. Chem. 263 (25), 12564–70. [PubMed] [Google Scholar]
- Clarkson A. B. Jr.; Bienen E. J.; Pollakis G.; Grady R. W. (1989) Respiration of bloodstream forms of the parasite Trypanosoma brucei brucei is dependent on a plant-like alternative oxidase. J. Biol. Chem. 264 (30), 17770–17776. [PubMed] [Google Scholar]
- Zhang Z.; Huang L.; Shulmeister V. M.; Chi Y. I.; Kim K. K.; Hung L. W.; Crofts A. R.; Berry E. A.; Kim S. H. (1998) Electron transfer by domain movement in cytochrome bc1. Nature 392 (6677), 677–84. 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- Schrödinger . (2018) Schrödinger Small Molecule Drug Discovery suite, Schrödinger, New York.
- MacLean L. M.; Thomas J.; Lewis M. D.; Cotillo I.; Gray D. W. (2018) Development of Trypanosoma cruzi in vitro assays to identify compounds suitable for progression in Chagas’ disease drug discovery. PLoS Neglected Trop. Dis. 12 (7), e0006612. 10.1371/journal.pntd.0006612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley J.; Brand S.; Voice M.; Caballero I.; Calvo D.; Read K. D. (2015) Development of a Fluorescence-based Trypanosoma cruzi CYP51 Inhibition Assay for Effective Compound Triaging in Drug Discovery Programmes for Chagas Disease. PLoS Neglected Trop. Dis. 9 (9), e0004014 10.1371/journal.pntd.0004014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Thomas M.; Patterson S.; Crouch S.; De Rycker M.; Lowe R.; Gresham S.; Urbaniak M. D.; Otto T. D.; Stojanovski L.; Simeons F. R. C.; Manthri S.; MacLean L. M.; Zuccotto F.; Homeyer N.; Pflaumer H.; Boesche M.; Sastry L.; Connolly P.; Albrecht S.; Berriman M.; Drewes G.; Gray D. W.; Ghidelli-Disse S.; Dixon S.; Fiandor J. M.; Wyatt P. G.; Ferguson M. A. J.; Fairlamb A. H.; Miles T. J.; Read K. D.; Gilbert I. H. (2018) Cyclin-dependent kinase 12 is a drug target for visceral leishmaniasis. Nature 560 (7717), 192–197. 10.1038/s41586-018-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie S.; Brand S.; Thomas M.; De Rycker M.; Chung C. W.; Pena I.; Bingham R. P.; Bueren-Calabuig J. A.; Cantizani J.; Cebrian D.; Craggs P. D.; Ferguson L.; Goswami P.; Hobrath J.; Howe J.; Jeacock L.; Ko E. J.; Korczynska J.; MacLean L.; Manthri S.; Martinez M. S.; Mata-Cantero L.; Moniz S.; Nuhs A.; Osuna-Cabello M.; Pinto E.; Riley J.; Robinson S.; Rowland P.; Simeons F. R. C.; Shishikura Y.; Spinks D.; Stojanovski L.; Thomas J.; Thompson S.; Viayna Gaza E.; Wall R. J.; Zuccotto F.; Horn D.; Ferguson M. A. J.; Fairlamb A. H.; Fiandor J. M.; Martin J.; Gray D. W.; Miles T. J.; Gilbert I. H.; Read K. D.; Marco M.; Wyatt P. G. (2019) Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci. U. S. A. 116 (19), 9318–9323. 10.1073/pnas.1820175116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall R. J.; Rico E.; Lukac I.; Zuccotto F.; Elg S.; Gilbert I. H.; Freund Y.; Alley M. R. K.; Field M. C.; Wyllie S.; Horn D. (2018) Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc. Natl. Acad. Sci. U. S. A. 115 (38), 9616–9621. 10.1073/pnas.1807915115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare S.; Nagle A. S.; Biggart A.; Lai Y. H.; Liang F.; Davis L. C.; Barnes S. W.; Mathison C. J.; Myburgh E.; Gao M. Y.; Gillespie J. R.; Liu X.; Tan J. L.; Stinson M.; Rivera I. C.; Ballard J.; Yeh V.; Groessl T.; Federe G.; Koh H. X.; Venable J. D.; Bursulaya B.; Shapiro M.; Mishra P. K.; Spraggon G.; Brock A.; Mottram J. C.; Buckner F. S.; Rao S. P.; Wen B. G.; Walker J. R.; Tuntland T.; Molteni V.; Glynne R. J.; Supek F. (2016) Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537 (7619), 229–233. 10.1038/nature19339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes M. L.; Avery V. M. (2015) Development and application of a sensitive, phenotypic, high-throughput image-based assay to identify compound activity against Trypanosoma cruzi amastigotes. Int. J. Parasitol.: Drugs Drug Resist. 5 (3), 215–28. 10.1016/j.ijpddr.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher N.; Meunier B. (2008) Molecular basis of resistance to cytochrome bc1 inhibitors. FEMS Yeast Res. 8 (2), 183–92. 10.1111/j.1567-1364.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- Abrahams K. A.; Cox J. A.; Spivey V. L.; Loman N. J.; Pallen M. J.; Constantinidou C.; Fernandez R.; Alemparte C.; Remuinan M. J.; Barros D.; Ballell L.; Besra G. S. (2012) Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS One 7 (12), e52951 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foo C. S.; Lupien A.; Kienle M.; Vocat A.; Benjak A. (2018) Arylvinylpiperazine Amides, a New Class of Potent Inhibitors Targeting QcrB of Mycobacterium tuberculosis. mBio 9 (5), e01276. 10.1128/mBio.01276-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. K.; Urgaonkar S.; Cortese J. F.; Gamo F. J.; Garcia-Bustos J. F.; Lafuente M. J.; Patel V.; Ross L.; Coleman B. I.; Derbyshire E. R.; Clish C. B.; Serrano A. E.; Cromwell M.; Barker R. H. Jr.; Dvorin J. D.; Duraisingh M. T.; Wirth D. F.; Clardy J.; Mazitschek R. (2011) Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum cytochrome bc1 inhibitors. Chem. Biol. 18 (12), 1602–10. 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam T. G.; McNamara C. W.; Bopp S.; Dharia N. V.; Meister S.; Bonamy G. M.; Plouffe D. M.; Kato N.; McCormack S.; Bursulaya B.; Ke H.; Vaidya A. B.; Schultz P. G.; Winzeler E. A. (2011) A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem. Biol. 6 (11), 1214–22. 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani S.; Araki S.; Yamaguchi T.; Takii Y.; Ohshima T.; Matsuo N. (2002) Biological properties of the novel fungicide cyazofamid against Phytophthora infestans on tomato and Pseudoperonospora cubensis on cucumber. Pest Manage. Sci. 58 (2), 139–45. 10.1002/ps.430. [DOI] [PubMed] [Google Scholar]
- Lynch M. (2007) The origins of genome architecture, Sinauer, Sunderland, Massachusetts. [Google Scholar]
- Goodman C. D.; Siregar J. E.; Mollard V.; Vega-Rodriguez J.; Syafruddin D.; Matsuoka H.; Matsuzaki M.; Toyama T.; Sturm A.; Cozijnsen A.; Jacobs-Lorena M.; Kita K.; Marzuki S.; McFadden G. I. (2016) Parasites resistant to the antimalarial atovaquone fail to transmit by mosquitoes. Science (Washington, DC, U. S.) 352 (6283), 349–53. 10.1126/science.aad9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trypobase Consortium. http://www.ipb.csic.es/trypobase/index.html.
- Goyard S.; Segawa H.; Gordon J.; Showalter M.; Duncan R.; Turco S. J.; Beverley S. M. (2003) An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol. Biochem. Parasitol. 130 (1), 31–42. 10.1016/S0166-6851(03)00142-7. [DOI] [PubMed] [Google Scholar]
- Roberts A. J.; Torrie L. S.; Wyllie S.; Fairlamb A. H. (2014) Biochemical and genetic characterization of Trypanosoma cruzi N-myristoyltransferase. Biochem. J. 459 (2), 323–32. 10.1042/BJ20131033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter K. J.; Le Quesne S. A.; Fairlamb A. H. (1994) Identification and biosynthesis of N1,N9-bis(glutathionyl)aminopropylcadaverine (homotrypanothione) in Trypanosoma cruzi. Eur. J. Biochem. 226 (3), 1019–1027. 10.1111/j.1432-1033.1994.t01-1-01019.x. [DOI] [PubMed] [Google Scholar]
- Jones D. C.; Hallyburton I.; Stojanovski L.; Read K. D.; Frearson J. A.; Fairlamb A. H. (2010) Identification of a kappa-opioid agonist as a potent and selective lead for drug development against human African trypanosomiasis. Biochem. Pharmacol. 80 (10), 1478–86. 10.1016/j.bcp.2010.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun R.; Schonenberger (1979) Cultivation and in vitro cloning or procyclic culture forms of Trypanosoma brucei in a semi-defined medium. Short communication. Acta tropica 36 (3), 289–292. [PubMed] [Google Scholar]
- De Rycker M.; Thomas J.; Riley J.; Brough S. J.; Miles T. J.; Gray D. W. (2016) Identification of Trypanocidal Activity for Known Clinical Compounds Using a New Trypanosoma cruzi Hit-Discovery Screening Cascade. PLoS Neglected Trop. Dis. 10 (4), e0004584 10.1371/journal.pntd.0004584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M.; Hallyburton I.; Thomas J.; Campbell L.; Wyllie S.; Joshi D.; Cameron S.; Gilbert I. H.; Wyatt P. G.; Frearson J. A.; Fairlamb A. H.; Gray D. W. (2013) Comparison of a high-throughput high-content intracellular Leishmania donovani assay with an axenic amastigote assay. Antimicrob. Agents Chemother. 57 (7), 2913–22. 10.1128/AAC.02398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B.; Salzberg S. L. (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9 (4), 357–9. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25 (16), 2078–9. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2011) A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27 (21), 2987–93. 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T.; Harris S. R.; Berriman M.; Parkhill J.; McQuillan J. A. (2012) Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28 (4), 464–9. 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturm N. R.; Simpson L. (1990) Kinetoplast DNA minicircles encode guide RNAs for editing of cytochrome oxidase subunit III mRNA. Cell 61 (5), 879–84. 10.1016/0092-8674(90)90198-N. [DOI] [PubMed] [Google Scholar]
- Sali A.; Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234 (3), 779–815. 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Webb B.; Sali A. (2016) Comparative Protein Structure Modeling Using MODELLER. Current protocols in bioinformatics 54, 1–37. 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C.; Higgins D. G.; Heringa J. (2000) T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302 (1), 205–17. 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Moss D. S.; Thornton J. M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26 (2), 283–291. 10.1107/S0021889892009944. [DOI] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47 (7), 1739–49. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Huang L. S.; Cobessi D.; Tung E. Y.; Berry E. A. (2005) Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: a new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol. 351 (3), 573–97. 10.1016/j.jmb.2005.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger . (2018) Schrödinger Desmond Molecular Dynamics System, Schrödinger.
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. (2016) OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 12 (1), 281–96. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Berweger C. D.; van Gunsteren W. F.; Müller-Plathe F. (1995) Force field parametrization by weak coupling. Re-engineering SPC water. Chem. Phys. Lett. 232 (5), 429–436. 10.1016/0009-2614(94)01391-8. [DOI] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Brand S.; Ko E. J.; Viayna E.; Thompson S.; Spinks D.; Thomas M.; Sandberg L.; Francisco A. F.; Jayawardhana S.; Smith V. C.; Jansen C.; De Rycker M.; Thomas J.; MacLean L.; Osuna-Cabello M.; Riley J.; Scullion P.; Stojanovski L.; Simeons F. R. C.; Epemolu O.; Shishikura Y.; Crouch S. D.; Bakshi T. S.; Nixon C. J.; Reid I. H.; Hill A. P.; Underwood T. Z.; Hindley S. J.; Robinson S. A.; Kelly J. M.; Fiandor J. M.; Wyatt P. G.; Marco M.; Miles T. J.; Read K. D.; Gilbert I. H. (2017) Discovery and Optimization of 5-Amino-1,2,3-triazole-4-carboxamide Series against Trypanosoma cruzi. J. Med. Chem. 60 (17), 7284–7299. 10.1021/acs.jmedchem.7b00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding M. G.; di Rago J. P.; Trumpower B. L. (2006) Investigating the Qn site of the cytochrome bc1 complex in Saccharomyces cerevisiae with mutants resistant to ilicicolin H, a novel Qn site inhibitor. J. Biol. Chem. 281 (47), 36036–43. 10.1074/jbc.M608026200. [DOI] [PubMed] [Google Scholar]
- Waterhouse A. M.; Procter J. B.; Martin D. M.; Clamp M.; Barton G. J. (2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25 (9), 1189–1191. 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.