

Abstract
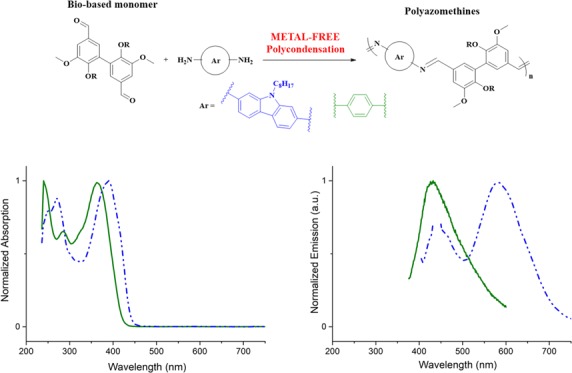
Divanillin was synthesized in high yield and purity using Laccase from Trametes versicolor. It was then polymerized with benzene-1,4-diamine and 2,7-diaminocarbazole to form polyazomethines. Polymerizations were performed under microwave irradiation and without transition-metal-based catalysts. These biobased conjugated polyazomethines present a broad fluorescence spectrum ranging from 400 to 600 nm. Depending on the co-monomer used, polyazomethines with molar masses of around 10 kg·mol–1 and with electronic gaps ranging from 2.66 to 2.85 eV were obtained. Furthermore, time-dependent density functional theory (TD-DFT) calculations were performed to corroborate the experimental results.
1. Introduction
Thanks to recent works on the selective oxidative coupling of phenolic molecules via enzymatic catalysis, a biobased platform of lignin-derived aromatics is now available.1,2 Among them, vanillin has a large potential, as it is one of the only phenolic compounds industrially available from biorefinery.3 Vanillin can be easily dimerized from C–C coupling using an environmental-friendly process as was demonstrated by Llevot et al.4 Such divanillin was used as a biobased aromatic building block for designing semi-aromatic polymers exhibiting different thermomechanical properties.5 However, divanillin monomer has also the potential to be valorized in the field of organic electronics.6 Indeed, in addition to the two phenolic functions, such derivative possesses two aldehyde moieties that can be involved in condensation reactions, for instance with amino groups, leading to polyazomethines. Polyazomethines exhibit many advantages, as they can be obtained through metal-free polycondensation with water as the only byproduct.7−12 Moreover, polyazomethines can be synthesized under microwave irradiation, which brings the benefit of being faster and more efficient than conventional heating.13,14 π-Conjugated polyazomethines present a good thermal resistance and are stable in air.15,16 They possess electronic properties similar to those of poly(p-phenylene vinylene) (PPV), which can be tuned to exhibit a high absolute fluorescence quantum yield when doped and exhibit a good chemical and electrochemical resistance.9,15−22 Interestingly, such polymers could be prepared by selecting diamino-functionalized monomers vs bis-aldehyde divanillin.
In this work, we describe the synthesis of a divanillin monomer substituted by alkyl chains and its subsequent polycondensation with 1,4-benzene diamine or 2,7-carbazole diamine. The optical and electrochemical properties of the so-formed polyazomethines were also investigated.
2. Results and Discussion
6,6-Dihydroxy-5,5-dimethoxy-[1,1-biphenyl]-3,3-dicarboxaldehyde (divanillin or DV) was synthesized from vanillin at room temperature using the procedure already described by us (Scheme 1).1 In 1H NMR (Figure S1), the signal at 9.7 ppm corresponds to the two aldehyde protons and the signals at 7.5 and 7.2 ppm to the aromatic ones. The signal at 3.8 ppm corresponds to the methoxy groups. The structure of DV was also attested by attenuated total reflectance Fourier transform infrared (ATR-FTIR) (Figure S2).
Scheme 1. Alkylation of Divanillin with 2-Ethylhexyl Side Chains (DVEH) and Polymerization with p-Phenylenediamine (P1) and Diamino Carbazole (P2).

To improve the solubility of DV and polyazomethines thereof in classical organic solvents, the latter was alkylated.9,23,24 Etherification reaction was performed in dimethyl sulfoxide (DMSO) in the presence of KOH and 2-ethylhexyl bromide to give divanillin ethyl hexylated or DVEH (Scheme 1). The alkylation reaction was assessed by 1H NMR spectroscopy and ATR-FTIR (Figures S3 and S4); nuclear magnetic resonance (NMR) signals around 3.81 ppm and the signals between 0.69 and 1.39 ppm confirmed the completion of the alkylation reaction.
The divanillin-based monomer (DVEH) was polymerized at the stoichiometry ratio 1:1 both with commercially available benzene-1,4-diamine and 2,7-diaminocarbazole to give, respectively, P1 and P2 (synthesized according to the literature)12 (Scheme 1). The polymerization reaction performed under microwave irradiation is usually faster than conventional heating.25,26 Silica was added to the reaction medium to remove water and shift the equilibrium toward the polymer formation. P1A and P2 were obtained after 4 h of reaction and purified by precipitation in methanol. Interestingly, the use of silica allowed a simple recovery process of the polyazomethine through filtration before precipitation. However, silica is not necessary to carry out the polymerization reaction. Indeed, the synthesis of the polyazomethine was also performed without silica, in just 5 min instead of 4 h (P1B). The crude polyazomethine was then dissolved in methylene chloride; methanol was added and then the solvent was evaporated using a rotary evaporator. The powder obtained was rinsed with methanol, giving the final product P1C.
The polyazomethines were characterized by NMR and size exclusion chromatography (SEC). 1H NMR signal at 8.5 ppm clearly attested the formation of azomethine bond (−CH=N−) (Figures S5 and S6). All the molecular characteristics and thermal and optical properties of DVEH-based polyazomethines are reported in Table 1.
Table 1. Molar Masses and Thermal and Optical Properties of Divanillin-Based Polyazomethines.
| polyazomethines | co-monomer of DVEH | experimental conditionsa | M̅nb [g·mol–1] | M̅wb [g·mol–1] | Đb |
 b b
|
Tdc [°C] | absorptiond,e λsolmax(λfilm) [nm] | emissiond,e λsolmax(λsol) [nm] |
|---|---|---|---|---|---|---|---|---|---|
| P1A | NH2–Ph–NH2 | 4 h, silica, purified | 10 600 | 21 900 | 2.1 | 18 | 395 | 285–365 (290–370) | 430 (460) |
| P1B | NH2–Ph–NH2 | 5 min, no silica, crude | 5700 | 11 500 | 2 | 9 | 285–365 (290–370) | ||
| P1C | NH2–Ph–NH2 | 5 min, no silica, purified | 10 000 | 20 000 | 2 | 17 | |||
| P2 | NH2–Cbz–NH2 | 4 h, silica, purified | 6200 | 15 400 | 2.5 | 7 | 401 | 270–390 (ND) | 580 (ND) |
Under microwave irradiation at 130 °C, in toluene with APTS as a catalyst.
Determined by size exclusion chromatography (SEC) relative to polystyrene standards in tetrahydrofuran (THF) at 40 °C.
Decomposition temperature at 20% weight loss, evaluated under N2 at a heating rate of 10 °C ·min–1.
Determined in dichloromethane solution.
Determined on films obtained by drop-casting on quartz.
SEC traces show discrete peaks at low molar masses, according to a polycondensation pathway with heavy monomers (Figures S7 and S8). Both polymers have a dispersity close to 2, which is consistent with a step-growth mechanism (at high conversion). The two families of polyazomethines present apparent number-average molar masses M̅n of 10 600 and 6200 g·mol–1, respectively, for P1A and P2 for a reaction time of 4 h. For the polyazomethines P1, a similar molar mass was obtained in just 5 min of reaction after purification. Interestingly, postcondensation probably occurred during the purification, resulting in a higher molar mass (10 000 g/mol with 70% yield) compared to the crude. It confirms that postcondensation occurs during solvent evaporation.
The thermal properties of polyazomethines were determined by thermogravimetric analysis (TGA) under N2 atmosphere at a heating rate of 10 °C·min–1. All polyazomethines present good thermal stability with a degradation temperature of around 405 °C (Figures S9 and S10). The diamine used does not have a significant impact on the thermal stability. In addition, differential scanning calorimetry (DSC) analyses carried out between −80 and 200 °C did not show any specific transition neither glass transition nor crystallinity behavior, indicating the very rigid structure of these polyazomethines and their probable amorphous state (see Figures S11 and S12).
All the polyazomethines were characterized by UV–vis absorption and fluorescence spectroscopy analyses. As shown in Figure 1, the carbazole-based polyazomethine’s main absorption band is red-shifted as compared to that of the phenyl-based derivative from 364 to 390 nm. This behavior could be due to a stronger electron-rich/electron-poor coupling in the case of the carbazolylene derivative or to a more planar conformation increasing the conjugation length. It is even amplified in fluorescence spectra where the shift can reach 150 nm. This is coherent with the higher molar attenuation coefficient of carbazole compared to the that of bisvanillin.27 Both P1 and P2 have a weak emission, with a fluorescence quantum yield below 2% in the solution.
Figure 1.
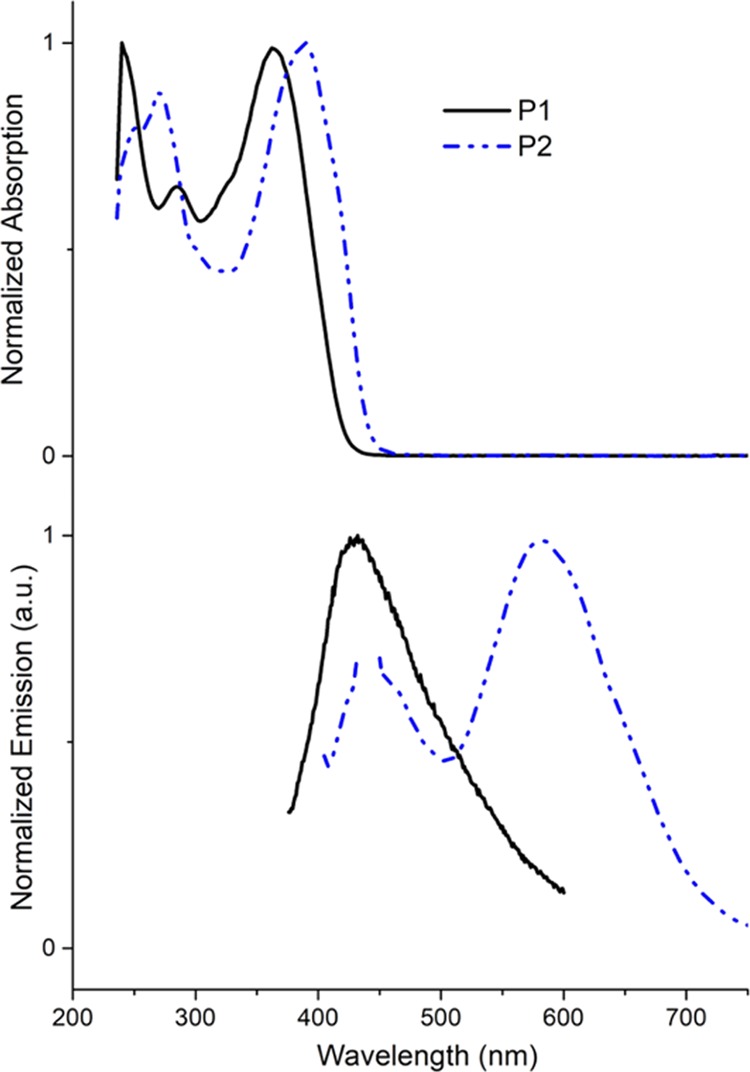
Absorption spectra (top) and emission spectra (excited at 360 nm, bottom) of the two DVEH-based polyazomethines P1A and P2 in methylene chloride.
Electrochemical features of these polyazomethines were determined by cyclic voltammetry using dichloromethane solutions of tetrabutylammonium hexafluorophosphate (TBAPF6) as an electrolyte, silver as a reference electrode, and platinum as a working and counter electrodes. Finally, the material was solubilized in the electrolyte solution at a concentration of 0.1 g·L-1 (Figures S13 and S14). The values of the energy levels are indicated in Table 2.
Table 2. Physical Properties of Divanillin-Based Polyazomethines.
| polyazomethine | HOMO [eV]a | LUMO [eV]a | Egelec [eV]b | Egopt [eV]c | HOMOd | LUMOd | EGd |
|---|---|---|---|---|---|---|---|
| P1A | –5.21 | –2.36 | 2.85 | 3.02 | –5.500 | –1.911 | 3.58 |
| P2 | –5.18 | –2.50 | 2.68 | 2.85 | –5.203 | –1.808 | 3.39 |
Estimated from the oxidation and reduction potentials measured by cyclic voltammetry in CH2Cl2 solution.
Calculated by the difference between oxidation and reduction potentials.
Calculated by the Tauc method.
Calculated by B3LYP/6-31(d,p) level of theory.
The slight differences between the electrochemical and optical band gaps can be explained by the fact that the redox peaks result from localized sites rather than from the conjugated backbone.28 For the 1,4-diaminobenzene-based polyazomethines, both optical and electrochemical gaps are comparable within experimental errors, with a value of around 2.67 eV for the electrochemical gap and 2.96 eV for the optical one.
In the case of carbazole-based polyazomethine, these values reach, respectively, 2.66 and 2.85 eV. These gap reductions are coherent with the red-shift observed in both absorption and emission spectra.
Density-functional theory (DFT) calculations were carried out on DV-Ph and DV-Cbz derivatives at B3LYP/6-31 (d,p) level of theory with chloroform as an implicit solvent conductor-like polarizable continuum model (CPCM). Different angles in both molecules were first estimated (Figures S15 and S16 and Table S1). The choice of DFT functional employed is based on the closeness of the calculated electronic band gap (EG = EHOMO – ELUMO) of monomers (as a function of different DFT functionals employed, see Table S2 for more details) with respect to the experimental values determined by cyclic voltammetry (Table S3). DV-Cbz derivative shows lower EG compared to that of DV-Ph derivative as reported in Table 2.
Furthermore, Khun’s model was employed to compute the electronic band gap at polymer limit (eq 1).29,30
| 1 |
When n → ∞, n is the number of monomer units in the oligomer, E0 is to electronic transition energy when N = 1, N is the number of double bonds along the shortest conjugated pathway between terminal carbon atoms for oligomers of size n (Figure S17). Dk is the force constant that represents the strength of coupling between single and double bonds, implicitly linked to the efficiency of connection between the units in an oligomer. By plotting EG as a function of the number of double bonds (N), it exhibits higher π-conjugation of DV-Cbz derivative, suggesting a better delocalization of π electrons31 with respect to DV-Ph derivatives, with the values of Dk as 0.818 and 0.748, respectively (Table S4). These results are in agreement with the experimental data.
3. Conclusions
The synthesis of different polyazomethines embedding divanillin biobased platform was performed. Divanillin was synthesized and alkylated and then polymerized with two different diamines: 1,4-benzene diamine and an alkylated 2,7-diaminocarbazole. The polymerization was performed under microwave irradiation, without any metallic catalyst and with water as the only byproduct. Remarkably, all polyazomethines present a broad emission at the beginning of the visible range. The carbazole-based polyazomethine is red-shifted compared to the phenyl-based one in the case of both absorption and emission. This bathochromic shift is expected as the carbazole moiety is more electron-donating than the phenyl one. Interestingly, the methodology followed meets some challenges for a “green chemistry” such as lowering the number of steps, avoiding transition-metal catalyst, and use of biobased monomers to quote a few. In addition, these new polyazomethines have promising properties (confirmed by time-dependent DFT (TD-DFT) calculations) for optoelectronic applications like an organic light-emitting diode (OLED) for example.
These encouraging results pave the way for a whole family of biobased π-conjugated polyazomethines. Optical properties can be enhanced by optimizing the structure and/or by playing on doping. The extension to other π-conjugated moieties is currently investigated. Furthermore, for a better understanding of optical and electrical properties, small model trimers and dimers will be isolated and fully characterized. All these new developments will be discussed in forthcoming papers.
4. Experimental Section
4.1. Materials
Vanillin (>97%), laccase from Trametes versicolor, and 2-ethyl hexyl bromide (95%) were obtained from Sigma-Aldrich. para-Toluene sulfonic acid (PTSA, 99%) was purchased from TCI. Silica gel (pore size 60 Å, 230–400 mesh particle size, particle size 40–63 μm) was obtained from Honeywell Fluka. All products and solvents (reagent grade) were used as received except otherwise mentioned. The solvents were of reagent grade quality and purified wherever necessary according to the methods reported in the literature. Flash chromatography was performed on a Grace Reveleris apparatus, employing silica cartridges from Grace. Cyclohexane: ethyl acetate gradients were used as eluents. The detection was performed through ELSD and UV detectors at 254 and 280 nm. The reactions under microwave irradiation were performed on a Discover-SP from CEM, with the temperature measured by infrared; the power of the apparatus is constantly adjusted to reach and then stay at the set temperature.
4.2. Characterization Techniques
1H, 13C, and 1H–13C HSQC NMR measurements were performed with a Bruker Avance 400 spectrometer (400.20 and 100.63 MHz for 1H and 13C, respectively) at room temperature using a deuterated solvent.
IR spectra were recorded with a Bruker Tensor 27 spectrometer using a 0.6 mm diameter beam. The samples were analyzed with the attenuated total reflexion (ATR) method.
High-resolution mass spectroscopy analyses were performed on an AutoSpec-Waters spectrometer (EI).
Optical absorption spectra were obtained with a UV–vis spectrophotometer (UV-3600, Shimadzu). Photoluminescence spectra were obtained from a spectrofluorometer (Fluoromax-4, Horiba Scientific).
Molar masses of polymers P1A, P1B, and P1C measured in THF were determined by size exclusion chromatography (SEC) using a three-column set of Resipore Agilent: one guard column Resipore Agilent PL1113-1300, then two-column Resipore Agilent PL1113-6300, connected in series, and calibrated with narrow polystyrene standards from Polymer Laboratories using both refractometric (GPS 2155) and UV detectors (Viscotek). THF was used as an eluent (0.8 mL·min–1) and trichlorobenzene as a flow marker (0.15%) at 30 °C.
Molar masses of polymer P2 measured in THF were determined by size exclusion chromatography (SEC) using a three-column set of TSK gel TOSOH (G2000, G3000, G4000 with pore sizes of 20, 75, and 200 Å, respectively, connected in series) calibrated with narrow polystyrene standards from Polymer Laboratories using both refractometric and UV detectors (Varian). THF was used as eluent (1 mL/min) and trichlorobenzene as a flow marker at 40 °C.
Molar masses of polymer samples in CHCl3 were measured by SEC at 40 °C with THF as an eluent, using a Viscotek VE2001-GPC. Polymer Laboratories-Varian (one guard column and three columns based on cross-linked polystyrene, pore sizes = 200, 75, and 20 Å), and PS standards were used for calibration.
TGA was performed on a TA-Q50, from 25 to 600–700 °C with the heating of 10 °C·min–1 under nitrogen flow.
DSC analysis was performed on a TA instrument under a helium flow, with an LN2 cooling and modulated with ±0.64 °C every 60 s. The sample was heated at 10 °C·min–1 and cooled down at 5 °C·min–1.
Electrochemical measurements and highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) calculations were performed in a solution. A solution of 0.1 g·L–1 of the investigated polymer in CH2Cl2 with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as an electrolyte was prepared. Cyclic voltammetry (CV) measurements were then performed on the solution using silver wire as the reference electrode and platinum as the working and counter electrodes. A solution of ferrocene (1 mM in the same solvent) was prepared in the same conditions and the redox potential of Fc/Fc+ vs Ag (EFc/Fc+ Ag) was measured, to be used as a reference for calibration.
4.3. Synthesis of 6,6′-Dihydroxy-5,5′-dimethoxy[1,1′-biphenyl]-3,3′-dicarboxaldehyde (Divanillin or DV)
A solution of vanillin in acetone was added to acetate buffer saturated in oxygen with laccase from T. versicolor. In these conditions, the dimer formed precipitates and can be recovered by simple filtration. The filtrate was then simply reloaded in vanillin and oxygen to start again the synthesis. Yield: 85%.
1H NMR (400.20 MHz, (CD3)2SO, ppm): d 9.69 (s, 2 H); 7.57 (d, J = 1.9 Hz, 2H); 7.16 (d, J = 1.9 Hz, 2H); 3.76 (s, 6H). 13C NMR (100.63 MHz, (CD3)2SO, ppm): 191.2, 150.4, 148.16, 128.2, 127.8, 124.6, 109.2, 56.0.
FTIR (ATR, cm–1): ν = 3220, 1676, 1587, 1413, 1245, 1127, 750.
4.4. Synthesis of 6,6′-Bis-2-ethylhexyl-5,5′-dimethoxy-[1,1′-biphenyl]-3,3′-dicarboxaldehyde (DVEH)
In a dried and nitrogen flushed 100 mL glassware, divanillin (2 g, 6.62 mmol) was solubilized in 20 mL of previously dried DMSO. Then, KOH (0.89 mg, 15.9 mmol) was added to the reaction mixture, which was heated at 80 °C for 2 h. Then, 2.2 equiv of 2-ethylhexyl bromide was added to the reaction mixture and heated for 12 more hours. The reaction mixture was then poured into 300 mL of water and extracted with 100 mL of diethyl ether three times. The crude product recovered after the diethyl ether evaporation was purified with flash chromatography in a mixture of cyclohexane/ethyl acetate (95/5).
1H NMR (400.20 MHz, CDCl3, ppm): d 9.89 (s, 2H); 7.46 (dd, J = 8.7, 1.9 Hz, 4H); 3.95 (s, 6H); 3.81 (m, 4H); 1.39 1.06 (m, 18H); 0.80 (t, J = 7 Hz, 6H); 0.69 (t, J = 7.4 Hz, 6H). 13C NMR (100.63 MHz, CDCl3, ppm): 91.1, 153.6, 152.1, 132.0, 131.6, 128.6, 109.7, 75.2, 55.9, 40.4, 30.3, 29.1, 23.5, 22.9, 14.1, 11.0.
FTIR (ATR, cm–1): ν = 2918, 2851, 1697, 1574, 1458, 1386, 1275, 1220, 1127, 1042, 995, 860, 763, 622.
HRMS (EI+, m/z) [M]+ calcd (%) for C32H46O6Na: 549.3186, found 549.3163.
4.5. Synthesis of Polyazomethines P1A and P2
In a 10 mL microwave-dedicated glassware, a stoichiometric amount of p-phenylenediamine or 2,7-diaminocarbazole and the previously synthesized DVEH is put in a suspension of 5 mL of toluene. Silica (300 mg) was then added with the catalytic amount of PTSA and the reaction heated at 130 °C for 4 h using microwave irradiation. The crude polymer was then precipitated in methanol to afford the final polymers unless specified otherwise.
4.6. Synthesis of Polyazomethine P1B and P1C
In a 10 mL microwave-dedicated glassware, a stoichiometric amount of p-phenylenediamine and the previously synthesized DVEH is put in the suspension of 5 mL of toluene. A catalytic amount of PTSA was then added and the reaction heated at 130 °C for 5 min using microwave irradiation. The solvent is then removed from the crude mixture to give the crude polymer (P1B). This crude is then dissolved in a minimum amount of methylene chloride, and 100 mL of methanol was added. The solution turns cloudy. The solvents are then evaporated using a rotary evaporator to obtain a yellow powder, which is rinsed with methanol to give the final polymer P1C.
4.7. Characterization of P1
1H NMR (400.20 MHz, CDCl3, ppm): d 9.89 (s, 0.07H); 8.42 (s, 2H); 7.66 (s, 1.9H); 7.47 (m, 0.2H); 7.37 (m, 1.7H); 6.98 (m, 0.1H); 6.72 (m, 0.2H); 3.99 (s, 6.8H); 3.79 (m, 4.2H); 1.42, 1.02 (m, 21.2H); 0.85–0.77 (m, 6.6H); 0.76–0.68 (m, 6.4H).
13C NMR (100.63 MHz, CDCl3, ppm): d 191.3, 153.6, 152.3, 136.0, 132.2, 131.8, 128.4, 122.0, 112.4, 109.9, 109.6, 90.3, 73.6, 56.1, 32.1, 30.3, 29.8, 29.5, 25.9, 25.9, 22.8, 14.3.
FTIR (ATR, cm–1): ν = 2924, 2851, 1697, 1581, 1508, 1464, 1264, 1128, 1025, 800, 725, 615.
SEC, 1H NMR, and TGA traces are available in the Supporting Information.
4.8. Characterization of P2
1H NMR (400.20 MHz, CDCl3, ppm): 9.93 (s, 0.03H); 8.55 (m, 2H); 8.05 (m, 1.83H); 7.74 (s, 1.78H); 7.43 (m, 2.77H); 7.13 (m, 2H); 4.55 (s, 1H); 4.03 (s, 6.36H); 3.81 (m, 3.88H); 2.33 (m, 2.23H); 1.95 (m, 2.54H); 1.43 0.96 (m, 50.13H), 0.79 (m, 19.13H).
13C NMR (100.63 MHz, CDCl3, ppm): d 159.3, 153.7, 152.2, 150.2, 149.6, 143.5, 140.0, 132.4, 131.7, 127.2, 122.3, 120.9, 120.7, 120.4, 112.1, 109.8, 109.4, 104.9, 102.2, 75.3, 72.1, 56.1, 40.6, 39.2, 33.8, 31.9, 30.5, 29.6, 29.5, 29.3, 29.3, 27.1, 23.7, 23.2, 22.7, 14.3, 14.2, 11.2.
FT IR (ATR, cm–1): ν = 2924, 2851, 1734, 1581, 1446, 1373, 1264, 1227, 1336, 1049, 970, 872, 788, 653.
SEC, 1H NMR, and TGA traces are available in the Supporting Information.
4.9. Computational Methods
All computational details are presented in the Supporting Information.
Acknowledgments
The authors would like to acknowledge CESAMO (Characterization platform of Université de Bordeaux) for mass spectrometry. Computational time is provided by Université de Bordeaux. The authors would like to acknowledge Dr. Luca Muccioli, Dr. Frédéric Castet, and Dr. Katarzyna Brymora for technical discussion on computational approach.
Glossary
Abbreviations Used
- DMSO
dimethyl sulfoxide
- DSC
dynamic scanning calorimetry
- HOMO
highest occupied molecular orbital
- LUMO
lowest unoccupied molecular orbital
- NMR
nuclear magnetic resonance
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04181.
Computational details; 1H NMR (top) and 13C NMR (bottom) spectra of DV; ATR-FTIR spectrum of DVEH; SEC traces of P1A, P1B, and P1C; cyclic voltammograms (left-reduction, right-oxidation) of P1A; stable conformers obtained from the PES scans for θ1, θ2, and θ3 and relative energies (ΔE in kcal/mol); electronic (EG) and optical (EVert) gaps (in eV); and maximum absorption wavelengths (PDF)
Author Present Address
† Arkema, Centre de production de Feuchy, BP 70029, 62051 Saint-Laurent-Blangy Cedex, France (G.G.).
Author Contributions
§ G.G. and L.G. have contributed equally to this work and must be considered as “first authors”.
Author Contributions
The manuscript was written through contributions of all authors, and they have given approval to the final version of the manuscript.
This work was funded by Solvay Company, Région Nouvelle Aquitaine and also the French State grant ANR-10-LABX-0042-AMADEus managed by the French National Research Agency under the initiative of excellence IdEx Bordeaux program (reference ANR-10-IDEX-0003-02).
The authors declare no competing financial interest.
Supplementary Material
References
- Llevot A.; Grau E.; Carlotti S.; Grelier S.; Cramail H. Renewable (semi)aromatic polyesters from symmetrical vanillin-based dimers. Polym. Chem. 2015, 6, 6058–6066. 10.1039/C5PY00824G. [DOI] [Google Scholar]
- Kobayashi S.; Makino A. Enzymatic Polymer Synthesis: An Opportunity for Green Polymer Chemistry. Chem. Rev. 2009, 109, 5288–5353. 10.1021/cr900165z. [DOI] [PubMed] [Google Scholar]
- Fache M.; Boutevin B.; Caillol S. Epoxy thermosets from model mixtures of the lignin-to-vanillin process. Green Chem. 2016, 18, 712–725. 10.1039/C5GC01070E. [DOI] [Google Scholar]
- Llevot A.; Grau E.; Carlotti S.; Grelier S.; Cramail H. Selective laccase-catalyzed dimerization of phenolic compounds derived from lignin: Towards original symmetrical bio-based (bis) aromatic monomers. J. Mol. Catal. B: Enzym. 2016, 125, 34–41. 10.1016/j.molcatb.2015.12.006. [DOI] [Google Scholar]
- Llevot A.; Grau E.; Carlotti S.; Grelier S.; Cramail H. From Lignin-derived Aromatic Compounds to Novel Biobased Polymers. Macromol. Rapid Commun. 2016, 37, 9–28. 10.1002/marc.201500474. [DOI] [PubMed] [Google Scholar]
- Gaur M.; Lohani J.; Balakrishnan V. R.; Raghunathan P.; Eswaran S. V. Dehydrodivanillin: Multi-dimensional NMR Spectral Studies, Surface Morphology and Electrical Characteristics of Thin Films. Bull. Korean Chem. Soc. 2009, 30, 2895–2898. 10.5012/bkcs.2009.30.12.2895. [DOI] [Google Scholar]
- Adams R.; Bullock J. E.; Wilson W. C. Contribution to the structure of benzidine. J. Am. Chem. Soc. 1923, 45, 521–527. 10.1021/ja01655a032. [DOI] [Google Scholar]
- Marvel C. S.; Hill H. W. Polyazines. J. Am. Chem. Soc. 1950, 72, 4819–4820. 10.1021/ja01166a516. [DOI] [Google Scholar]
- Yang C. J.; Jenekhe S. A. Conjugated aromatic poly(azomethines). 1. Characterization of structure, electronic spectra, and processing of thin films from soluble complexes. Chem. Mater. 1991, 3, 878–887. 10.1021/cm00017a025. [DOI] [Google Scholar]
- Yang C. J.; Jenekhe S. A. Effect of Structure on refracrive Index of Conjugated Polyimines. Chem. Mater. 1994, 6, 196–203. 10.1021/cm00038a016. [DOI] [Google Scholar]
- Yang C.-J.; Jenekhe S. A. Conjugated Aromatic Polyimines. 2. Synthesis, Structure, and Properties of New Aromatic Polyazomethines. Macromolecules 1995, 28, 1180–1196. 10.1021/ma00108a054. [DOI] [Google Scholar]
- Garbay G.; Muccioli L.; Pavlopoulou E.; Hanifa A.; Hadziioannou G.; Brochon C.; Cloutet E. Carbazole-based π-conjugated polyazomethines: Effects of catenation and comonomer insertion on optoelectronic features. Polymer 2017, 119, 274–284. 10.1016/j.polymer.2017.05.039. [DOI] [Google Scholar]
- Lidström P.; Tierney J.; Wathey B.; Westman J. Microwave assisted organic synthesis—a review. Tetrahedron 2001, 57, 9225–9283. 10.1016/S0040-4020(01)00906-1. [DOI] [Google Scholar]
- Kappe C. O. Controlled microwave heating in modern organic synthesis. Angew. Chem., Int. Ed. 2004, 43, 6250–6284. 10.1002/anie.200400655. [DOI] [PubMed] [Google Scholar]
- Barik S.; Skene W. G. Turning-on the Quenched Fluorescence of Azomethines through Structural Modifications. Eur. J. Org. Chem. 2013, 2013, 2563–2572. 10.1002/ejoc.201201502. [DOI] [Google Scholar]
- Thomas O.; Inganäs O.; Andersson M. R. Synthesis and Properties of a Soluble Conjugated Poly(azomethine) with High Molecular Weight. Macromolecules 1998, 31, 2676–2678. 10.1021/ma9701090. [DOI] [Google Scholar]
- Wang C.; Shieh S.; LeGoff E.; Kanatzidis M. G. Synthesis and characterization of a new conjugated aromatic poly(azomethine) derivative based on the 3′,4′-dibutyl-α-terthiophene building block. Macromolecules 1996, 29, 3147–3156. 10.1021/ma9514131. [DOI] [Google Scholar]
- Barik S.; Bletzacker T.; Skene W. G. π-Conjugated Fluorescent Azomethine Copolymers: Opto-Electronic, Halochromic, and Doping Properties. Macromolecules 2012, 45, 1165–1173. 10.1021/ma2024304. [DOI] [Google Scholar]
- Barik S.; Friedland S.; Skene W. G. Understanding the reversible anodic behaviour and fluorescence properties of fluorenylazomethines—A structure-property study. Can. J. Chem. 2010, 88, 945–953. 10.1139/V10-080. [DOI] [Google Scholar]
- Barik S.; Skene W. G. A fluorescent all-fluorene polyazomethine—towards soluble conjugated polymers exhibiting high fluorescence and electrochromic properties. Polym. Chem. 2011, 2, 1091. 10.1039/c0py00394h. [DOI] [Google Scholar]
- Iwan A.; Palewicz M.; Chuchmała A.; Gorecki L.; Sikora A.; Mazurek B.; Pasciak G. Opto(electrical) properties of new aromatic polyazomethines with fluorene moieties in the main chain for polymeric photovoltaic devices. Synth. Met. 2012, 162, 143–153. 10.1016/j.synthmet.2011.11.024. [DOI] [Google Scholar]
- Tsai F.-C.; Chang C.-C.; Liu C.-L.; Chen W.-C.; Jenekhe S. A. New thiophene - linked conjugated poly (azomethine)s: Theoretical electronic structure, synthesis, and properties. Macromolecules 2005, 38, 1958–1966. 10.1021/ma048112o. [DOI] [Google Scholar]
- Lee K.-S.; Won J. C.; Jung J. C. Synthesis and characterization of processable conducting polyazomethines. Die Makromol. Chem. 1989, 190, 1547–1552. 10.1002/macp.1989.021900706. [DOI] [Google Scholar]
- Reinhardt B. A.; Unroe M. R. Preparation of aromatic Schiff base polymers with oxydecyl pendants for increased solubility. Polym. Prepr. 1990, 620–621. [Google Scholar]
- Kuwabara J.; Yasuda T.; Choi S. J.; Lu W.; Yamazaki K.; Kagaya S.; Han L.; Kanbara T. Direct arylation polycondensation: A promising method for the synthesis of highly pure, high-molecular-weight conjugated polymers needed for improving the performance of organic photovoltaics. Adv. Funct. Mater. 2014, 24, 3226–3233. 10.1002/adfm.201302851. [DOI] [Google Scholar]
- Nayak S. N.; Bhasin C. P.; Nayak M. G. A review on microwave-assisted transesterification processes using various catalytic and non-catalytic systems. Renewable Energy 2019, 143, 1366–1387. 10.1016/j.renene.2019.05.056. [DOI] [Google Scholar]
- Walba H. K.; Branch G. E. K. The Absorption Spectra of Some N-Substituted p-Aminotriphenylmethyl Ions. J. Am. Chem. Soc. 1951, 73, 3341–3348. 10.1021/ja01151a102. [DOI] [Google Scholar]
- Boudreault P.-L. T.; Najari A.; Leclerc M. Processable Low-Bandgap Polymers for Photovoltaic Applications. Chem. Mater. 2011, 23, 456–469. 10.1021/cm1021855. [DOI] [Google Scholar]
- Torras J.; Casanovas J.; Alemán C. Reviewing Extrapolation Procedures of the Electronic Properties on the π-Conjugated Polymer Limit. J. Phys. Chem. A 2012, 116, 7571–7583. 10.1021/jp303584b. [DOI] [PubMed] [Google Scholar]
- Gierschner J.; Cornil J.; Egelhaaf H.-J. Optical Bandgaps of π-Conjugated Organic Materials at the Polymer Limit: Experiment and Theory. Adv. Mater. 2007, 19, 173–191. 10.1002/adma.200600277. [DOI] [Google Scholar]
- Wykes M.; Milián-Medina B.; Gierschner J. Computational engineering of low bandgap copolymers. Front. Chem. 2013, 1, 35. 10.3389/fchem.2013.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.