

Abstract
Quantitative proteomics employing isobaric reagents has been established as a powerful tool for biological discovery. Current workflows often utilize a dedicated quantitative spectrum to improve quantitative accuracy and precision. A consequence of this approach is a dramatic reduction in the spectral acquisition rate, which necessitates the use of additional instrument time to achieve comprehensive proteomic depth. This work assesses the performance and benefits of online and real-time spectral identification in quantitative multiplexed workflows. A Real-Time Search (RTS) algorithm was implemented to identify fragment spectra within milliseconds as they are acquired using a probabilistic score and to trigger quantitative spectra only upon confident peptide identification. The RTS-MS3 was benchmarked against standard workflows using a complex two-proteome model of interference and a targeted 10-plex comparison of kinase abundance profiles. Applying the RTS-MS3 method provided the comprehensive characterization of a 10-plex proteome in 50% less acquisition time. These data indicate that the RTS-MS3 approach provides dramatic performance improvements for quantitative multiplexed experiments.
Keywords: proteomics, isobaric tag, multiplexing, real-time, online search, IAPI, SPS-MS3, TMT, iTRAQ
Graphical Abstract
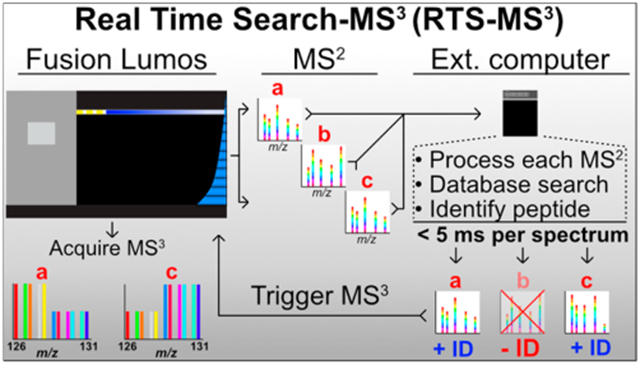
INTRODUCTION
Quantitative proteome characterization with isobaric reagents (e.g., TMT) is an established and compelling approach for comprehensive biological surveys of protein and post-translational modification dynamics.1–5 Significant advancements in these multiplexed workflows include the use of a dedicated quantitative spectrum (MS3), multifragment isolation (multinotch/SPS), and continuously evolving hardware (Orbitrap Fusion family), which have enabled broad applications of these workflows and routine use in discovery-driven experimentation. The standardized LC-MS workflow that incorporates these advancements leverages the widely used data-dependent methodologies to select, fragment, and sequence features of interest. To fully enable these multiplexed workflows, a quantitative MS3 scan, derived from the simultaneous isolation and fragmentation of multiple precursor fragment ions (SPS-MS3), is included for each interrogated feature. Although the incorporation of the SPS-MS3 is critical for accurate and precise quantitative profiling, it does necessitate longer ion injection times and higher resolution Orbitrap analysis. This, in-turn, results in reduced spectral acquisition rates which are countered by additional instrument acquisition time. Additionally, for multifraction data sets, nearly 75% of the acquired quantitative data are not retained in the finalized data set due to false-positive peptide identification and quantitative quality thresholds (Supp. Table S1). Finally, as the desire for statistical rigor and the interest in applying multiplexed workflows to clinically derived samples increase, the scope and numbers of samples will increase. The result being that experiments utilizing the existing workflows and incorporating hundreds of samples remain challenging.
Several publications have presented algorithms that were designed to improve the utility, robustness, and selectivity of data-dependent acquisition (DDA).6–8 These applications have operated on the external data system, through complicated operating system specific libraries, or on the internal instrument computer. These implementations focused primarily on improving the effectiveness of label-free MS2-based acquisition. Example applications included quantifying missing SILAC pairs and the resequencing of target peptides or post-translationally modified peptides.
Bailey et al. have presented an approach that modified the codebase of the instrument to enable fragment ion matching to inform downstream scan events.7 Although extremely flexible, modifications of the instrument codebase are limited by both hardware and software. The onboard instrument computer has not been designed for computationally intensive (both in terms of CPU and memory) tasks, such as peptide identification. This limitation required that the peptide assignment rely on the high-resolution and high mass accuracy matching of theoretical fragment ions to experimentally observed fragment ions. Furthermore, access to the instrument codebase is generally restricted by the manufacturer, limiting widespread adoption.
A more optimal solution would leverage the external data system to perform complex calculations and then dynamically interact with the instrument on an as-needed basis. Through their proof-of-concept manuscript, Graumann et al. utilized an OCX library to coordinate the interaction between a client application and the instrument.6 This provided the ability to test a specialized and restricted search algorithm for instant peptide assignment. However, the reliance on operating system specific implementations within the OCX library both limits and complicates any client implementation. Both publications effectively established that more advanced or intelligent data acquisition can be a powerful enhancement to data-dependent workflows. Nevertheless, in either case, the implementation of the algorithm was prohibitively complicated or did not provide sufficient justification when compared to traditional DDA-MS2 workflows for more widespread adoption.
Several publications have leveraged an application programming interface (API) to provide additional methodological control of targeted proteomics workflows.9,10 As described, the challenges associated with modifications to the onboard instrument codebase or OS-specific libraries are eliminated with API-based implementations. To our knowledge, no group has established the feasibility or application of an API based solution to data-dependent workflows, particularly in the context of sample multiplexing.
This work presents a framework for a Real-Time Search (RTS-MS3) and dynamic instrument control. A client application, executing on the external data system, receives MS2 spectra from the instrument, searches a complete protein database and assigns a probabilistic score to a peptide identification in real-time. Following confident peptide identification, the client application instructs the instrument to collect an SPS-MS3 spectrum with known and pure fragment ions. In the absence of a confident identification, no SPS-MS3 spectrum is collected, eliminating the increased time requirement. This results in greater spectral acquisition rates, improved quantitative accuracy, and more quantified peptides in less acquisition time. The RTS-MS3 application interfaces with the instrument through a new vendor-supplied instrument application programming interface (IAPI). The IAPI greatly simplifies the communication between a client application and the instrument and provides a robust and approachable method for more intelligent data acquisition. RTS-MS3 is not restricted by computational hardware or software as it operates on the external data system and implements a comprehensive peptide search in an average of 5 ms per MS2. We benchmarked the performance of the RTS-MS3 approach through a challenging two-proteome model of interference that permits the direct measurement of interference. We observe that the RTS-MS3 application can provide a 2-fold increase in the rate of data acquisition while reducing the amount of measurable interference. We utilized the improved performance of the RTS-MS3 approach to selectively characterize kinases in a 10-plex mixture of two cell lines. Finally, we assessed the performance of comprehensive proteome characterization by comparing results of 12 fractions acquired via 36 h of DDA-MS3 and 18 h RTS-MS3 acquisition. We observe that the RTS-MS3 acquisition quantified more proteins in 50% less time with excellent quantitative reproducibility.
EXPERIMENTAL PROCEDURES
RTS-MS3
The Real Time Search client application is composed of a graphical user and IAPI interface, written in C#, and a wrapper for a dynamic linked library, written in C++, containing the structures and methods for the computational and memory intensive operations. The IAPI libraries for the Orbitrap Fusion Lumos (ThermoFisher, San Jose, CA) were provided by the vendor. These libraries provide the base methods to dynamically interact with the instrument and at the time of publication the IAPI libraries were provided free of charge for any user.
The computational speed and efficient memory management of C++ was leveraged for the storage and access of the peptide library, spectral filtering and scoring, and the management of additional metadata. Protein sequences from a UniProt human (2014) and SGD S. cerevisae (2014) database were in silico digested into tryptic peptides with one missed cleavage allowed and a minimum and maximum length between 7 and 35 amino acids. The peptide database consists of a simple text file and is composed of one peptide sequence per line, along with a protein identifier and a boolean flag to indicate if the peptide should result in the submission of an MS3 upon confident identification. The human only peptide database contains greater than 1.57 M peptide sequences, the yeast database contains 380k peptide sequences, and a combined human/yeast peptide database contains greater than 1.95 M peptides. The C++ library also contains the structures and methods to rapidly manipulate, filter, and format spectral data for downstream scoring and peptide assignment.
Spectral scoring occurs via a cumulative binomial score.11 In brief, MS2 spectra are binned according to m/z and the top 9 most intense peaks are retained. Fragment ions derived from candidate peptides within the precursor mass tolerance (±10 ppm) are matched to the experimentally obtained spectra. A binomial probability is generated according to the number of matched fragments and the total number of fragments. Currently, no variable modifications are considered, although this could be added to a future implementation.
The graphical user interface, IAPI and C++ DLL wrapper were written in C#. The GUI presents several user-configurable options that control the parameters of the MS3 scan (Supp. Figure 1). The C# wrapper also mediates the communication and data transmission between the instrument and the C++ algorithms.
The general execution of the RTS client application occurs as follows: (1) the client application is initialized, user parameters are loaded, and the peptide database is parsed and loaded; (2) a data-dependent MS2 method is executed via the vendor supplied instrument control software; (3) MS2 are continually received, parsed, and scored by the RTS client; (4) an MS3 scan object, containing the relevant scan parameters is sent to the instrument; and (5) an quantitative MS3 is acquired.
We are currently working with the instrument manufacturer to make the RTS software available to the general community at no charge using the existing instrument API interface. Please check our Web site for information on running RTS on Orbitrap Fusion and Lumos instruments (https://gygi.med.harvard.edu/software).
Two-Proteome Interference Model
The two-proteome interference model consisted of tryptic yeast peptides (reduced and alkylated) labeled with TMT 11-plex regent using all but the 126 and 131c channel reporters. Yeast peptides were mixed at varying ratios across the remaining 9 channels for a total of 110 μg of yeast peptides: 3 channels with 6.66 μg, 3 channels with 10 μg, and 3 channels with 20 μg. The combined and labeled yeast peptides were mixed into a background of 1.1 mg of tryptic human peptides (reduced and alkylated) aliquoted evenly across the 11 TMT channels (100 μg per channel). Thus, the final amount of yeast peptides accounted for 10% of the total peptide mass in the two-proteome model.
Whole-Proteome 10-Plex Preparation
The sample preparation was performed as previously reported.12 HCT116 and HEK293 cells were homogenized in 8 M urea complemented with protease inhibitors (cOmplete, Roche) by 20 passes through a 21-gauge needle and vortexed for 2 min. The cell debris were separated by centrifugation at 21 130g for 15 min, and the supernatant was transferred to a new tube. The protein concentration was estimated using the BCA method, following the manufacturer’s instructions (ThermoFisher Scientific). Proteins were reduced using 5 mM TCEP for 30 min at room temperature and alkylated using 10 mM iodoacetamide for 30 min, in the dark. The excess iodoacetamide was quenched with 10 mM DTT for 15 min at room temperature, in the dark. The samples were cleaned using the methanol–chloroform precipitation method. The samples were resuspended in 200 mM EPPS, pH 8.5, and digested with Lys-C at a 100:1 protein-to-protease ratio and incubated overnight at room temperature. Trypsin was added at a 100:1 protein-to-protease ratio and the reaction was incubated for 6 h at 37 °C. After TMT labeling, the samples were combined and desalted. The pooled sample was vacuum centrifuged to near dryness and desalted before BpH-RP fractionation. For BpH-RP fractionation, an Agilent 300 extend C18 column was utilized to produce a total of 96 fractions, which were consolidated into 24 fractions, with 12 nonadjacent samples analyzed in the mass spectrometer.
Mass Spectrometry (MS) Analysis
All experiments were performed on an Orbitrap Fusion Lumos. For data-dependent experiments (MS2 and MS3) all instrument operational parameters were specified through the instrument method editor. Briefly, MS1 spectra were acquired at 120 K resolving power for a maximum of 100 ms in the Orbitrap, and features were filtered for monoisotopic peak assignment and a charge state greater than one. MS2 spectra were acquired either with a cycle time of two seconds (experiments with MS2 only acquisition) or by selecting the top 10 most abundant features (experiments with MS3 acquisition). MS2 spectra were acquired via collisional induced dissociation (CID), in the ion trap with an automatic gain control (AGC) of 20K, quadrupole isolation width of 0.5 m/z and a maximum ion time of 35 ms. For MS3 acquisition, a synchronous precursor selection (SPS) of 10 fragments ions was acquired for a maximum of 150 ms with an AGC of 50K and a normalized collision energy of 55.
For RTS-MS3 acquisition, the scan events were acquired as above, but no MS3 scan was specified in the method. MS2 spectra were acquired within a 2 s cycle time. MS2 spectra with a binomial score greater than or equal to 55 triggered the submission of an MS3 spectrum to the instrument. The parameters for the MS3 spectrum during a RTS experiment were the same as the DDA-MS3 except for the selection of the SPS ions. The fragments ions were filtered according to the following: (1) the intensities of the MS2 fragments must be greater than 5% of the base peak and (2) the fragments must match a b- or y-type ion from the predicted peptide.
Proteome Informatics
All acquired data were processed via a previously described inhouse informatics pipeline.13 Briefly, raw data were converted to mzXML, and spectra were identified with SEQUEST utilizing a fasta formatted database (UniProt human and SGD S. cerevisae, 2014) with common contaminants and reversed sequences appended. Spectral searches were done with the following parameters: 50 PPM precursor tolerance, fully tryptic peptides only, fragment ion tolerance of 0.9 Da with a static modification of TMT (+229.163 Da) and carbamidomethylation of cysteine (+57.021 Da). Oxidation of methionine (+15.995 Da) was specified as a variable modification. A false discovery rate of 1% was obtained via a linear discriminant analysis which utilized several features (XCorr, deltaCorr, PPM, missed cleavages, and charge state). Resulting peptides were further filtered to provide a 1% protein FDR. Proteins were collapsed into groups via the rules of parsimony. TMT reporter ion intensities were extracted and divided by reported noise values to derive TMT reporter ion signal/noise ratios.
The interference free index (IFI) was calculated for the two-proteome model of interference by dividing the sum of the signal/noise intensities of the interference only channels (126, 131c) by the sum of all channels. The resulting value represents the proportion of total reporter ion intensity attributable to interference. The proportion of interference is subtracted from 1 to provide the IFI.
RESULTS AND DISCUSSION
Comparing Default Acquisition Methodologies
Hybrid instrumentation, such as the Orbitrap Fusion Lumos, allows for impressive rates of spectral acquisition. This is accomplished by leveraging the parallel operation of various mass analyzers within the instrument. We first set out to benchmark the differences in acquisition rates with an identical method, with and without collection of the MS3 scans. The scan sequence of a typical DDA-MS2 is highlighted in Figure 1A and is composed of high-resolution precursor scans (FTMS1) that trigger n low-resolution fragment scans (ITMS2), with these scans occurring in parallel. The DDA-MS3 approach builds on the DDA-MS2 scan sequence by also acquiring a synchronous precursor selection (SPS) MS3 quantitative spectrum for each MS2. To account for ions lost to transmission and the selection of only a portion of the MS2 ion current, SPS-MS3 spectra generally have longer ion injection times. Also, the SPS-MS3 spectrum is acquired in the Orbitrap at a resolution of 50k in order to resolve the TMT reporter ions with a mass delta of 0.006 Da. These two parameters decrease the average MS2 acquisition rate per second from 15 Hz (DDA-MS2) to 4 Hz (DDA-MS3) (Figure 1B). Ultimately, the decrease in acquisition rate results in a significant reduction in acquired MS2 (Figure 1C ~ 100k DDA-MS2; 29k DDA-MS3; mean of triplicates) and a substantial reduction in the number of identified proteins (Figure 1D 4,800 DDA-MS2; 2,000 DDA-MS3). Finally, it was observed that the proportion of MS2 retained in the finalized data set was only 5% lower than the DDA-MS3 (Figure 1E) despite the 4-fold increase in the number of spectra acquired. This illustrates the breadth of peptides that are accessible with higher rates of acquisition but are not routinely characterized in a DDA-MS3 experiment. This suggests that improving the rate of acquisition for DDA-MS3 type experiments would result in the identification and quantification of additional peptides.
Figure 1.
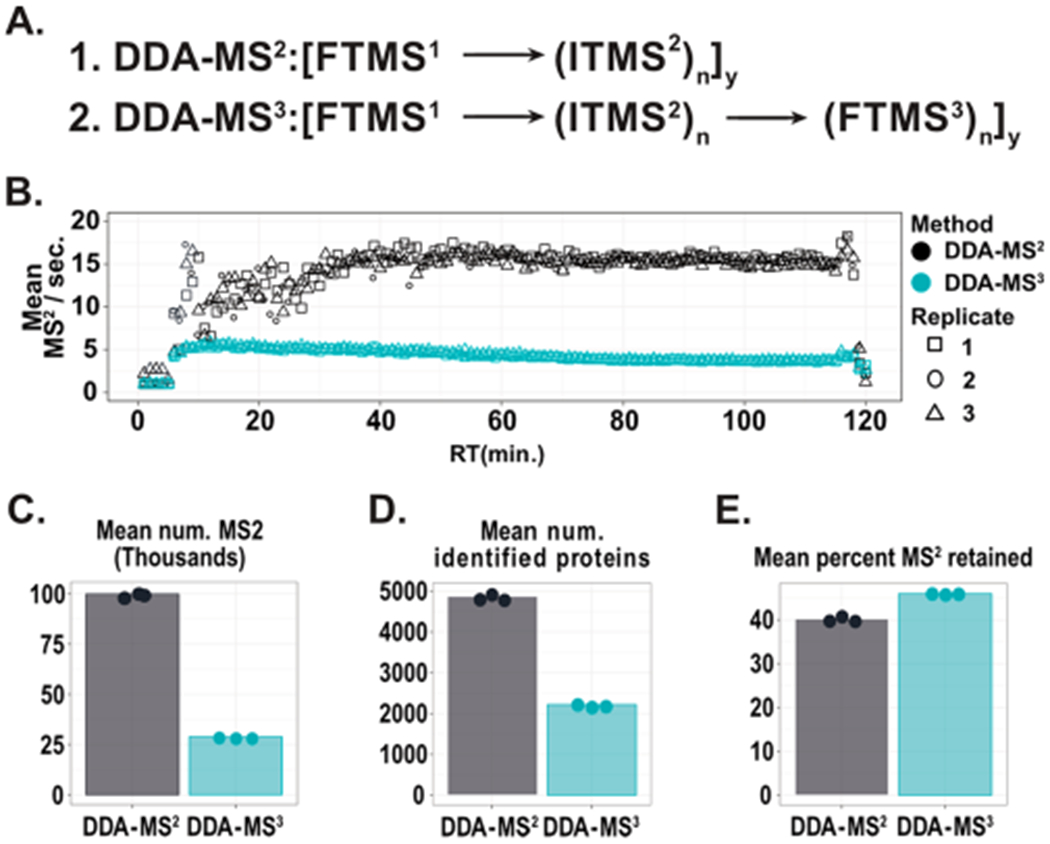
Comparing DDA-MS2 and DDA-MS3 acquisition. (A) The scan sequence of DDA-MS2 and DDA-MS3. (B) The acquisition rate for DDA-MS2 and DDA-MS3 methods were compared across 2-h, triplicate injections of TMT labeled human lysate. The only difference is including an MS3 scan for each MS2. The DDA-MS2 approach results in greater than a 3-fold increase in the number of MS2 spectra acquired per second. (C) Comparing scan metrics between DDA-MS2 (gray) and DDA-MS3 (blue). The substantial increase in MS2 acquisition rate for DDA-MS2 acquisition results in nearly a 4-fold increase in the total number of MS2 spectra acquired in 2 h. (D) As the number of MS2 spectra increases, we observe a greater than 2-fold increase in the number of identified proteins. (E) Although nearly 4 times more MS2 were acquired during the DDA-MS2 method, we observe only a slight decrease in the fraction of spectra retained following filtering. This highlights the impressive number of accessible peptides that are generally not interrogated through DDA-MS3 approaches.
Implementing a Real-Time-Search for Isobaric Workflows (RTS-MS3)
The RTS-MS3 client application, operating solely on the external data system, is composed of a C# wrapper for the graphical user interface and communication with the instrument and a C++ library for peptide database management and scoring. Broadly, the RTS-MS3 client listens for MS2 spectra from the instrument. After receipt of an MS2 spectrum, candidate peptides are scored according to a cumulative binomial probability approach. Peptides with a binomial score exceeding a configurable threshold will trigger the client application to submit quantitative MS3 spectra to the instrument for subsequent acquisition. This process repeats continually throughout the entirety of the analysis time (Figure 2A).
Figure 2.
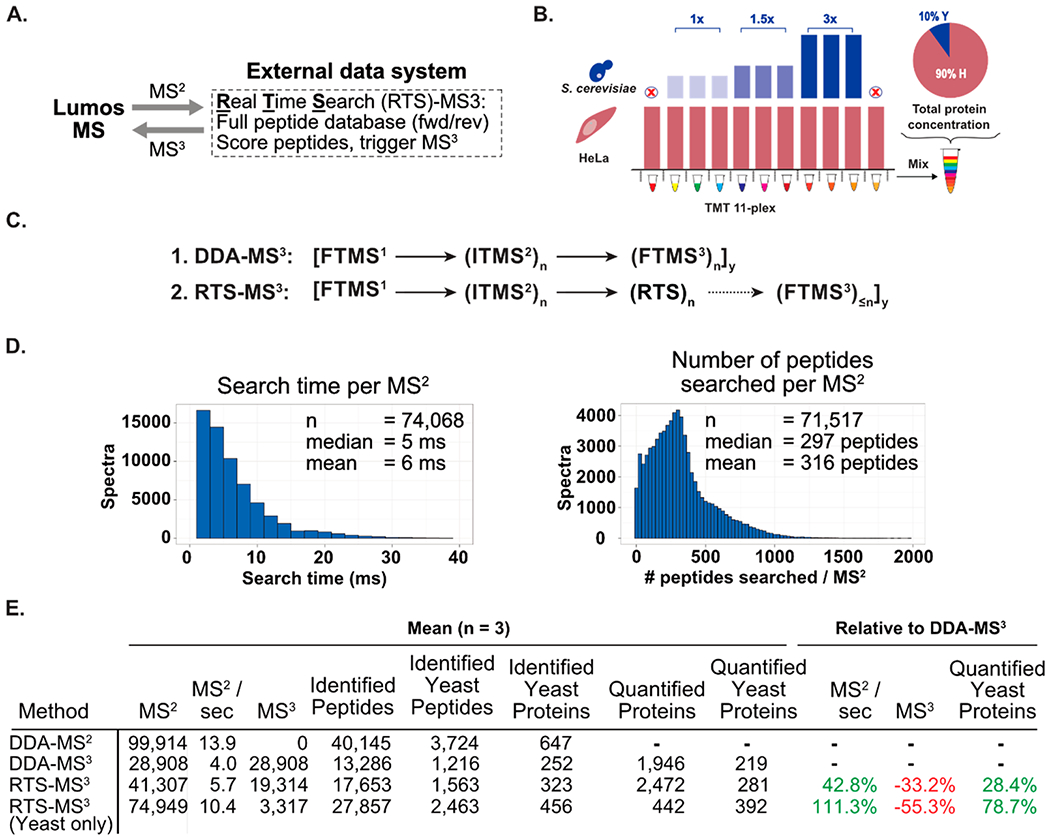
Active instrument engagement and prompt spectral identification dramatically improves the performance of MS3 quantitative proteomic workflows. (A) The RTS-MS3 approach incorporates a real-time spectral identification step utilizing a comprehensive peptide database to identify peptides prior to triggering quantitative MS3 spectra. (B) Yeast and HeLa whole cell lysate were prepared and labeled with TMT11-plex reagents. The yeast proteomes were mixed to produce ratios of 1:1.5:3 in biological triplicate with nine TMT reagents. An equal concentration of HeLa lysate was labeled with all 11 TMT reagents. The yeast mixture was combined with the human mixture to a final concentration of 10% yeast and 90% HeLa. The two TMT channels lacking yeast proteome provide direct measurement of interference. (C) The scan sequence for the RTS-MS3 approach incorporates the same scan events as a DDA-MS3 approach. However, prior to acquiring a quantitative MS3 scan, the RTS-MS3 algorithm is engaged to identify peptides from ITMS2 spectra in real-time. (D) For each interrogated peptide, the RTS-MS3 approach completed the real-time peptide identification in an average of 6 ms while searching an average of ~300 peptides. (E) Table comparing RTS and DDA methods. Data are triplicate runs of 120 min. RTS-MS3 method triggered MS3 for both human and yeast matched peptides. RTS-MS3 (Yeast only) triggered MS3 spectra only for peptides derived from yeast proteins.
Two-Proteome Model of Interference
To evaluate the performance of the RTS-MS3 approach, a challenging two-proteome model of interference was constructed (Figure 2B). Yeast whole cell lysate was prepared and labeled with nine Tandem Mass Tag reagents (TMT) and mixed at a ratio of 1:1.5:3 in triplicate. Separately, a mixture of 11 HeLa samples were labeled with TMT and mixed at an equal concentration. The final sample was constructed by adding yeast mixture to the HeLa mixture to a final concentration of 10%. Notably, this sample includes two channels that contain only human peptides and provide a direct measurement of interference.
RTS-MS3 Performance
The scan sequence of the RTS-MS3 approach is an extension of the standard DDA-MS3 method (Figure 1A) and leverages the low-resolution and high scan rate of the ion-trap (ITMS2) and the high-resolution and mass accuracy of the Orbitrap (FTMS1 and FTMS3). Following an MS2 acquisition, a real-time search is executed against the spectrum which provides a peptide sequence, protein reference, binomial score, and additional metadata (contaminant assignment, reverse entry, PPM mass error, etc.). A primary constraint for previous on-the-fly identification strategies was the time required for spectral processing/filtering, candidate selection, fragment matching, and spectral scoring. As current hybrid instruments can acquire tandem mass spectra at a rate greater than 40 MS2/second, the search time per MS2 was a primary design feature. The computational speed and memory management of C++ was leveraged for the more computationally critical aspects of the method. Accordingly, the average search time, for a database composed of more than 1.95 million yeast and human peptides, with a precursor mass tolerance of ±10 PPM was 6 ms (median 5 ms, Figure 2D, left). Remarkably, this search time was achieved with an average of nearly 300 candidate peptides searched per spectrum (Figure 2D, right). Finally, the acquisition of the RTS triggered MS3 routinely occurred less than one second after the acquisition of the parent MS2 (mean = 0.96, median = 0.69, Supporting Figure 2).
Metrics of the RTS-MS3 approach were determined through triplicate analysis of the previously described two-proteome sample. As described in Figure 1, the DDA-MS2 data sets illustrate the significant gain in data acquisition rate and the corresponding improvements to total peptide, total protein, and yeast peptide identifications. Implementing the RTS-MS3 approach resulted in a 33% decrease in the number of MS3 spectra acquired, as subthreshold, reverse, or contaminant peptides did not trigger MS3 spectra. This allowed for a 42% increase in the number of MS2 spectra acquired, resulting in nearly 30% more quantified yeast proteins (Figure 2E, third row). To further highlight the potential of the RTS-MS3 method, triplicate analyses of the yeast/human mixture were acquired with the same peptide database but with MS3 triggering permitted only upon confident yeast peptide identification. The result was that human, subthreshold, reverse, or contaminant peptides did not trigger MS3 spectra. When compared to the DDA-MS3 data set, the yeast only RTS-MS3 acquisition resulted in an even further reduction in the number of MS3 spectra acquired (−55%), which permitted the acquisition of over 2.5 times more MS2 spectra. The 111% increase in MS2 spectra resulted in a 79% in the number of quantified yeast proteins (Figure 2E, bottom row).
Reducing Isobaric Tag Interference through Direct Selection of Fragment Ions as SPS Precursors
As have been previously shown, the use of pure, identified fragments for an MS3 can reduce the quantity of interference, especially for lower abundance peptides.14 For the DDA-MS3 method, the n most abundant fragment ions are selected from the MS2 for subsequent inclusion and fragmentation during an MS3 acquisition (Supp. Figure 3). As the abundance of a precursor decreases, the likelihood that a higher-intensity, interfering fragment ion is present increases (Supp. Figure 4). The RTS-MS3 approach enables the dynamic and real-time selection of identified and pure fragment ions for quantitative MS3 spectra. This ultimately provides an additional reduction in the degree of observed interference. We utilized the yeast/human mixture to quantitatively assess the reduction of interference (Figure 3A). Overall, it is evident that the DDA-MS3 provides accurate and precise quantitative data with the RTS-MS3 providing a slight reduction in the average amount of observed interference. However, it has been previously shown that precursors of lower abundance will exhibit greater interference during DDA-MS3 acquistion.14 To assess the improvements of real-time fragment ion selection, the precursor abundance for all quantified peptides acquired through both methods was compared (Figure 3B). The range of abundances spans 5 orders of magnitude and within each binned abundance, the number of peptides quantified within the RTS-MS3 acquisition is ~2-fold higher. To compare the amount of interference within each method, we computed the mean interference free index (IFI) for all yeast peptides within each of the three precursor abundance bins (Figure 3C).15 Within each precursor bin, the mean IFI was higher for the RTS-MS3 peptides (higher IFI equals less interference). The reduction in interference was most obvious for peptides within the lowest abundance bin. Overall, it is apparent that the DDA-MS3 workflow provides excellent quantitative performance and dramatically reduces the effects of isobaric interference. However, the additional reductions in interference provided by the RTS-MS3 approach are simply a passive benefit of the approach and can ensure that as acquisition rates and the depth of proteome characterization improve, quantitative measurements remain highly accurate and precise.
Figure 3.
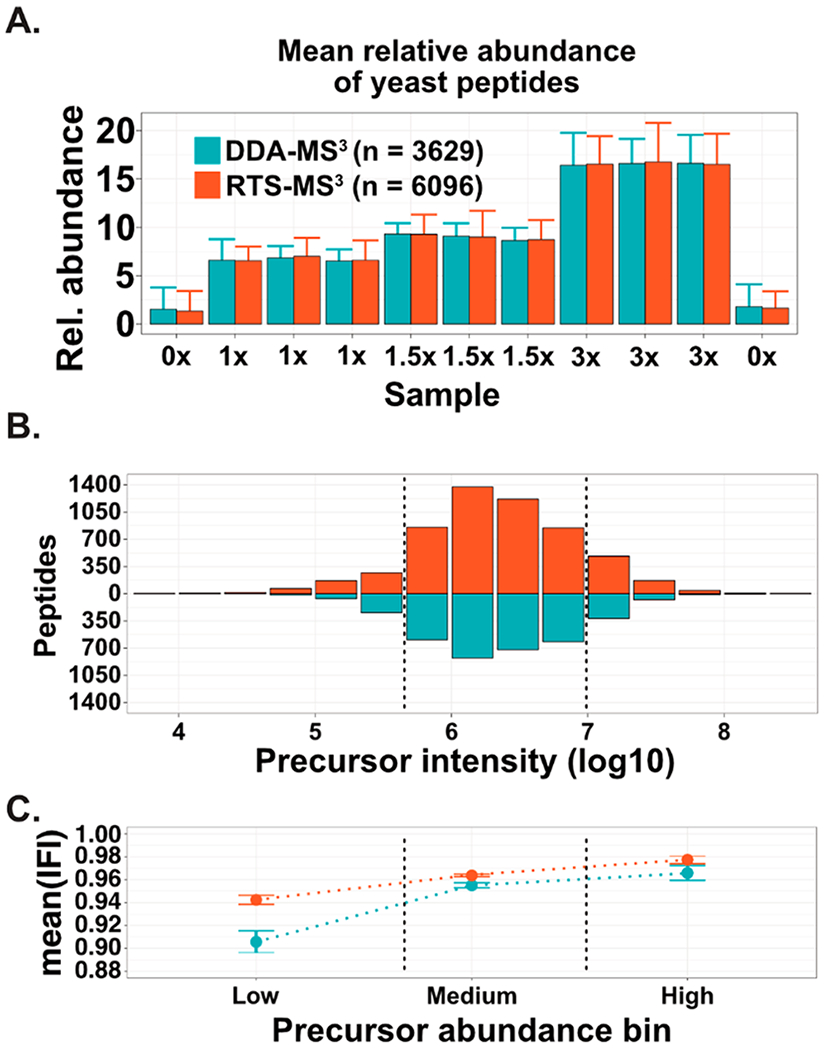
RTS-MS3 utilizes precursor specific fragment ions for improved quantitative performance. (A) A DDA-MS3 and RTS-MS3 approach was utilized to characterize a two-proteome model of interference in triplicate. Both approaches resulted in accurate and precise quantitation, mirroring the expected distribution of signal. A slight reduction in the average intensity of the interference only channels (“0x”) was observed. (B) The distribution of precursor abundances (log10) for the yeast peptides from the DDA-MS3 and RTS-MS3 acquisition were compared. In each of the abundance bins, approximately 2-fold more peptides were quantified during the RTS-MS3 experiments. (C) The mean interference-free index (points, bars represent standard error of the mean, IFI equal to 1.0 is no interference) for all peptides was computed for each precursor abundance bin and method. For each bin, the RTS-MS3 peptides were characterized with less interference (higher IFI). This is especially evident for the lower abundance precursors.
Improved Characterization of Subproteomes
As described above, implementing the RTS-MS3 methodology for a yeast-specific analysis resulted in nearly a 2-fold increase in the number of quantified proteins. To further evaluate the performance of the RTS-MS3 approach for characterizing subproteome populations, a target list of 454 kinase proteins was specified and utilized to characterize a sample composed of biological quintuplicates of HCT116 and HEK293T cultures in a TMT 10-plex (Figure 4A). This sample was analyzed in technical triplicate for 2 h per replicate with both a DDA-MS3 and RTS-MS3 method. For the RTS-MS3 method, only peptides that exceeded the score threshold and were assigned to one of the target kinases triggered a quantitative MS3 spectrum. This approach resulted in the characterization of 2-fold more total kinases (DDA-MS3: 43 kinases; RTS-MS3: 85 kinases). The number of quantified kinases and the number of kinases quantified across the n replicates is also approximately 2-fold higher within the RTS-MS3 data set (Figure 4B). Notably, the RTS-MS3 acquisition resulted in the characterization of 53 kinases not quantified in the DDA-MS3 analysis. The mean quantitative profiles for the 32 kinases identified through both methods correlate well (r = 0.95), with the RTS-MS3 data exhibiting a slightly higher dynamic range for several proteins (Figure 4D). The 53 RTS-MS3 uniquely quantified proteins were investigated; with four proteins exhibiting significantly different expression between the HCT116 and HEK quintuplicates (Two-sided Student’s t test, Benjamini–Hochberg corrected p-value <0.05 and fold-change greater/less than 2, Figure 4E,F).
Figure 4.
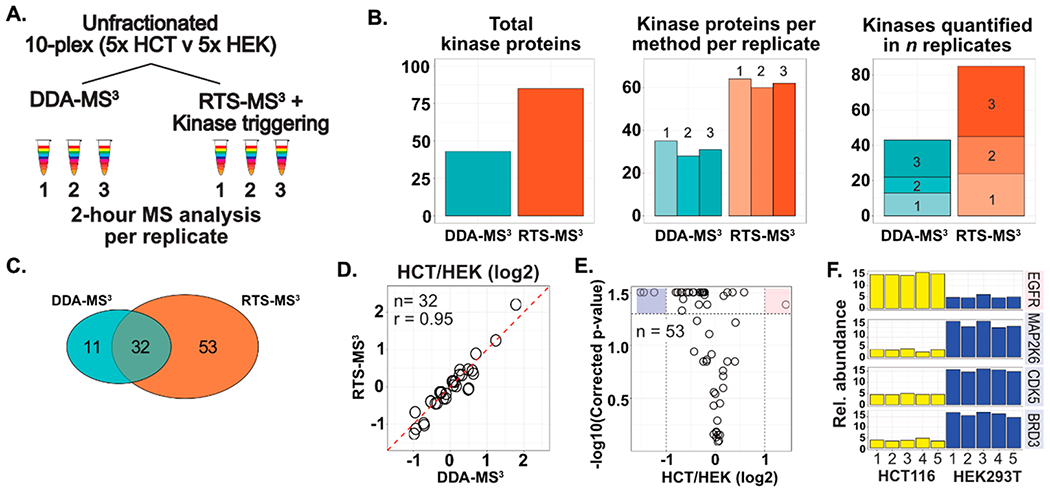
Improving the characterization of subproteomes in complex unfractionated samples. (A) Quintuplicates of HCT116 and HEK293T cells were prepared for multiplexed characterization as a single TMT10plex sample. The 5v5 mixture was analyzed with both a DDA-MS3 and an RTS-MS3 based approach for 2 h, in triplicate. For the RTS-MS3 acquisition, proteins annotated as a kinase were selectively targeted for quantitation. Only identified peptides corresponding to these proteins triggered a quantitative MS3 spectra. (B) The RTS-MS3 approach quantified nearly 2-fold more kinase proteins in 2 h of unfractionated acquisition, when compared to the DDA-MS3 approach. This was consistently observed across each of the triplicate analyses with over 2-fold more kinase proteins quantified across all replicates. (C) The RTS-MS3 analysis provided excellent coverage of kinases identified in the DDA-MS3 acquisition with comparable quantification (D). (E) The quantitative profiles of the 53 kinases uniquely quantified in the RTS-MS3 data set were assessed for significant changes in expression. F) Four kinases (EGFR, MAP2K6, CDK5, and BRD3) were observed to have statistically significant differences in expression between the HCT116 and HEK cell lines.
RTS-MS3 for Comprehensive Proteome Characterization
Comprehensive proteome characterization of multiple samples via isobaric labeling is increasingly common for routine biological discovery. Additionally, the size and scale of these projects are also expanding to include additional treatments, replicates, and controls. The general workflow for multiplexed samples includes offline and orthogonal sample fractionation followed by the analysis of 12 fractions with 3 h of data acquisition per fraction per multiplexed experiment. The 36 h of total analysis time has been empirically optimized to balance instrument time and depth of proteome recovery. However, as the demand for greater numbers of samples increases, the bottleneck in data production becomes data acquisition. Therefore, a reduction in the amount of analysis time per multiplexed experiment would greatly offset the increasing sample demand. The previously described 10-plex sample composed of quintuplicate HCT116 and HEK293T cultures was further processed by fractionation into 12 fractions (Figure 5A). Each fraction was analyzed with two methods: (1) DDA-MS3 (3 h acquisition per fraction) and (2) RTS-MS3 (1.5 h acquisition per fraction). The RTS-MS3 acquisition utilized a complete human proteome for spectral identification and included common contaminants and reverse sequences. Additionally, the number of quantified peptides per protein was logged during the analysis in order to limit the degree of repeat protein quantification. Specifically, after four peptides assigned to a particular protein had triggered a quantitative MS3 spectrum, no peptides assigned to the same protein would trigger additional MS3 spectra.
Figure 5.
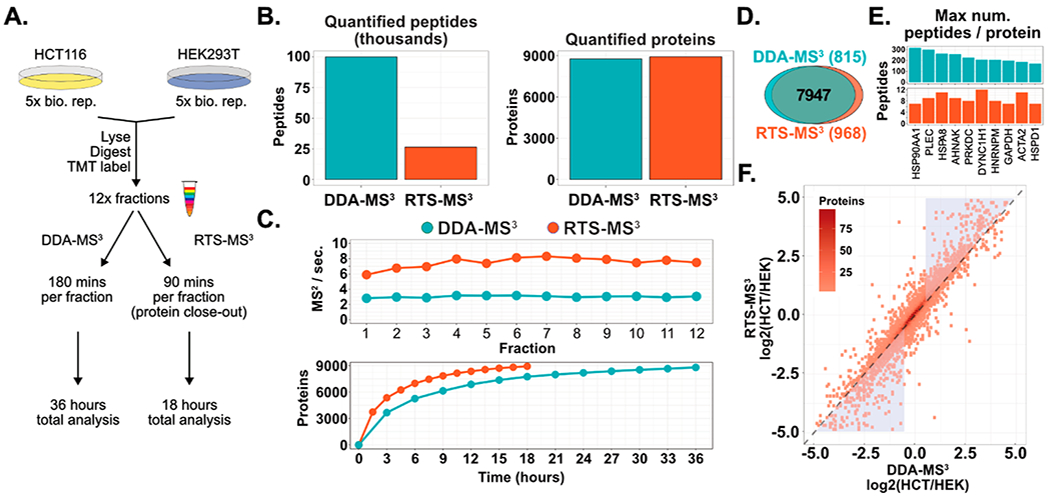
Reducing the acquisition time of whole proteome characterization with RTS-MS3 and protein close-out. (A) Quintuplicates of HCT116 and HEK293T (from Figure 4) were further prepared for whole proteome analysis through offline fractionation (12 fractions). Each fraction was characterized twice: (1) with a DDA-MS3 method for 180 min and (2) with the RTS-MS3 method for 90 min and proteins were excluded from further analysis after 4 peptides had been quantified. The DDA-MS3 method resulted in a total of 36 h of acquisition and the RTS-MS3 was acquired for a total of 18 h. (B) Utilizing the RTS-MS3 approach permitted the cataloguing of peptides and the ability to selectively trigger quantitative MS3 spectra or to forego additional protein quantitation through additional peptide quantification. This resulted in a 4-fold decrease in the number of peptides quantified in the RTS-MS3 data set. Yet, the RTS-MS3 approach quantified more proteins (8915) in 18 h than the DDA-MS3 approach (8,762) in 36 h. (C) The acquisition rate with the RTS-MS3 approach is approximately 2.5 times higher than the DDA-MS3 acquisition. This increase in duty cycle results in the quantification of more proteins across each fraction in half the acquisition time. (D) The RTS-MS3 provided more uniquely quantified proteins. (E) The ability to selectively trigger MS3 spectra dramatically reduces the acquisition of redundant quantified peptides. (F) The mean HCT116/HEK ratio for all proteins quantified in both data sets was compared. Although the data were collected in 50% less time, the RTS-MS3 provided comparable protein quantification to the DDA-MS3data set (r = 0.91). Notably, the RTS-MS3 approach shows greater dynamic range for highly dynamic proteins (blue triangles).
Not surprisingly, we observed that the final RTS-MS3 data set was composed of only 26 367 quantified peptides, nearly a 4-fold reduction in peptides compared to the DDA-MS3 method (99 899 peptides). The DDA-MS3 method produced a deep proteome, quantifying 8762 proteins in 36 h of analysis time. Remarkably, the RTS-MS3 quantified 8915 proteins in only 18 h of analysis time (Figure 5B). This improvement in proteome depth, with only 50% of the acquisition time, can be attributed to the increase in acquisition rate across all 12 fractions (Figure 5C, top). The DDA-MS3 provided a near constant acquisition rate of 3 Hz, whereas the RTS-MS3 method achieves a peak of 8 Hz (mean 7.5 Hz). Correspondingly, the RTS-MS3 approach quantified more proteins in each fraction (Figure 5C, bottom) and more uniquely quantified proteins (Figure 5D). As described above, the RTS-MS3 logged the number of quantified peptides per protein in an effort to further improve the acquisition rate by limiting the number of replicate peptides subjected to quantitative MS3 analysis. Figure 5E illustrates the 10 proteins from the DDA-MS3 data set with the highest number of quantified peptides. For 56 proteins, the DDA-MS3 method acquired more than one hundred peptides for the same protein, with 7 high abundance proteins having several hundred or more peptides per protein. Conversely, the RTS-MS3 data set resulted in only 6 proteins with more than 10 peptides per protein (average 3 peptides per protein, all proteins). The quality of the RTS-MS3 derived quantitative profiles was assessed to determine the effects, if any, from the RTS method or the reduced analysis time. The ratios (HCT116/HEK, log 2) between 7947 proteins quantified by both methods were compared. A strong correlation was observed (r = 0.91) between the quantitative profiles, indicating excellent reproducibility (Figure 5F). Interestingly, a noticeable shift in ratios was observed for proteins that exhibited highly dynamic expression between the cells (triangles, Figure 5F). For the majority of these proteins, the measured ratio between the HCT116 and HEK cell lines was extended following the RTS-MS3 acquisition. This extension of the dynamic range is likely due to the reduction in interference provide by the real-time selection of precursor specific fragment ions and further highlights the benefits of this method.
CONCLUSIONS
The RTS-MS3 approach makes use of the IAPI to dynamically include dedicated MS3 spectra only under specific conditions. Matching a peptide to an MS2 spectrum in real time opens several new possibilities. For example, SPS ions can be selected based on actual b- and y-type ions, specific gene sets (GO categories) can be targeted, and closing out proteins that already have sufficient quantitative information can be performed. In the future, we plan to introduce real-time machine learning to classify true and false positives during the analysis and incorporate post-translational modifications into the workflow. Finally, dedicated MS3 scans can be employed as highly specialized quantification scans with targeted or highly optimized parameters. For example, we are working to convert the TOMAHAQ assay for targeted quantitative proteomics to a similar platform.14
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Gygi laboratory, including E. Huttlin and M. Jedrychowski for valuable discussions. We thank C. Rose (Genentech Inc.) for his contributions to an earlier implementation. We thank D. Bailey, G. McAlister and V. Zabrouskov (ThermoFisher) for continued instrument and API support. This work was supported in part by DK098285 to JAP and NIH grant (HG096745) to SPG.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.8b00899.
Comprehensive proteome characterization retains only a portion of the acquired data after filtering, graphical user interface of the RTS client application, distribution of time (seconds) between acquisition of RTS triggered MS3 and preceding parent MS2, selection of identified fragment ions improves MS3 accuracy and precision, RTS-MS3 improves the quantitative performance of lower abundance peptides (PDF)
Two-proteome model of interference quantitative protein data (XLSX)
Quantitative protein data for kinase characterization (XLSX)
Quantitative protein data for deep-proteome comparison (XLSX)
The mass spectrometry data has been deposited to MassIVE and can be accessed at: ftp://massive.ucsd.edu
The authors declare no competing financial interest.
REFERENCES
- (1).Ross PL; Huang YN; Marchese JN; Williamson B; Parker K; Hattan S; Khainovski N; Pillai S; Dey S; Daniels S; et al. Multiplexed Protein Quantitation in Saccharomyces Cerevisiae Using Amine-Reactive Isobaric Tagging Reagents. Mol. Cell. Proteomics 2004, 3 (12), 1154–1169. [DOI] [PubMed] [Google Scholar]
- (2).Thompson A; Schäfer J; Kuhn K; Kienle S; Schwarz J; Schmidt G; Neumann T; Hamon C Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Anal. Chem 2003, 75 (8), 1895–1904. [DOI] [PubMed] [Google Scholar]
- (3).Kazak L; Chouchani ET; Jedrychowski MP; Erickson BK ; Shinoda K; Cohen P; Vetrivelan R; Lu GZ; Laznik-Bogoslavski D; Hasenfuss SC; et al. A Creatine-Driven Substrate Cycle Enhances Energy Expenditure and Thermogenesis in Beige Fat. Cell 2015, 163 (3), 643–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Chick JM; Munger SC; Simecek P; Huttlin EL; Choi K; Gatti DM; Raghupathy N; Svenson KL; Churchill GA; Gygi SP Defining the Consequences of Genetic Variation on a Proteome-Wide Scale. Nature 2016, 534 (7608), 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chouchani ET; Kazak L; Jedrychowski MP; Lu GZ; Erickson BK; Szpyt J; Pierce KA; Laznik-Bogoslavski D; Vetrivelan R; Clish CB; et al. Mitochondrial ROS Regulate Thermogenic Energy Expenditure and Sulfenylation of UCP1. Nature 2016, 532 (7597), 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Graumann J; Scheltema RA; Zhang Y; Cox J; Mann M A Framework for Intelligent Data Acquisition and Real-Time Database Searching for Shotgun Proteomics. Mol. Cell Proteomics 2012, 11 (3). DOI: 10.1074/mcp.M111.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bailey DJ; Rose CM; McAlister GC; Brumbaugh J; Yu P; Wenger CD; Westphall MS; Thomson JA; Coon JJ Instant Spectral Assignment for Advanced Decision Tree-Driven Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (22), 8411–8416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bailey DJ; McDevitt MT; Westphall MS; Pagliarini DJ; Coon JJ Intelligent Data Acquisition Blends Targeted and Discovery Methods. J. Proteome Res 2014, 13 (4), 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gallien S; Kim SY; Domon B Large-Scale Targeted Proteomics Using Internal Standard Triggered-Parallel Reaction Monitoring (IS-PRM). Mol. Cell. Proteomics 2015, 14 (6), 1630–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kaufmann A; Walker S Improved Performance of Multiplexed Targeted Tandem Mass Spectrometry Scans Using Customized Q Orbitrap Data Acquisition. Rapid Commun. Mass Spectrom 2016, 30 (9), 1131–1138. [DOI] [PubMed] [Google Scholar]
- (11).Beausoleil SA; Villén J; Gerber SA; Rush J; Gygi SP A Probability-Based Approach for High-Throughput Protein Phosphorylation Analysis and Site Localization. Nat. Biotechnol 2006, 24 (10), 1285–1292. [DOI] [PubMed] [Google Scholar]
- (12).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)Proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res 2018, 17 (6), 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Nightingale K; Lin K-M; Ravenhill BJ; Davies C; Nobre L; Fielding CA; Ruckova E; Fletcher-Etherington A; Soday L; Nichols H; et al. High-Definition Analysis of Host Protein Stability during Human Cytomegalovirus Infection Reveals Antiviral Factors and Viral Evasion Mechanisms. Cell Host Microbe 2018, 24 (3), 447–460.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Erickson BK; Rose CM; Braun CR; Erickson AR; Knott J; McAlister GC; Wühr M; Paulo JA; Everley RA; Gygi SP A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 2017, 65, 361–370, DOI: 10.1016/j.molcel.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Paulo JA; O’Connell JD; Gygi SP A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom 2016, 27 (10), 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.