
Figure 5.
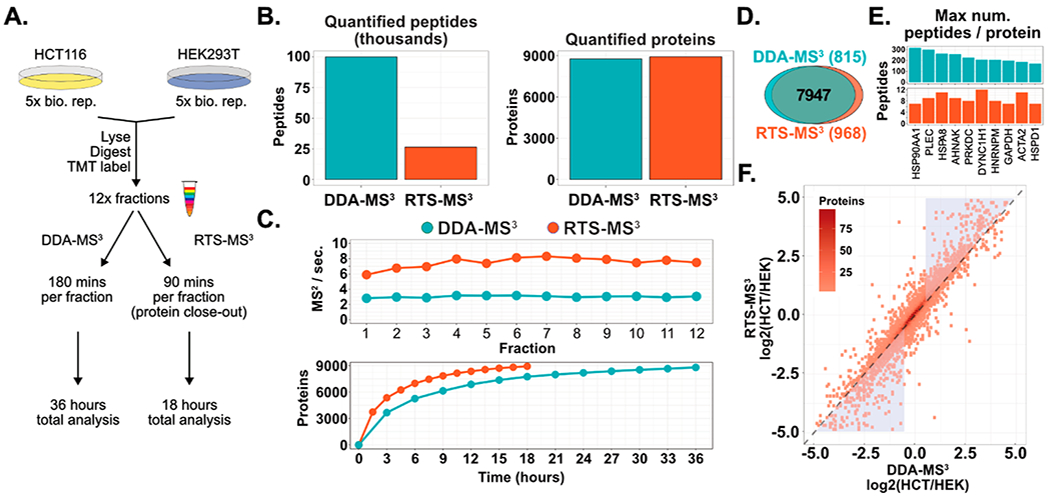
Reducing the acquisition time of whole proteome characterization with RTS-MS3 and protein close-out. (A) Quintuplicates of HCT116 and HEK293T (from Figure 4) were further prepared for whole proteome analysis through offline fractionation (12 fractions). Each fraction was characterized twice: (1) with a DDA-MS3 method for 180 min and (2) with the RTS-MS3 method for 90 min and proteins were excluded from further analysis after 4 peptides had been quantified. The DDA-MS3 method resulted in a total of 36 h of acquisition and the RTS-MS3 was acquired for a total of 18 h. (B) Utilizing the RTS-MS3 approach permitted the cataloguing of peptides and the ability to selectively trigger quantitative MS3 spectra or to forego additional protein quantitation through additional peptide quantification. This resulted in a 4-fold decrease in the number of peptides quantified in the RTS-MS3 data set. Yet, the RTS-MS3 approach quantified more proteins (8915) in 18 h than the DDA-MS3 approach (8,762) in 36 h. (C) The acquisition rate with the RTS-MS3 approach is approximately 2.5 times higher than the DDA-MS3 acquisition. This increase in duty cycle results in the quantification of more proteins across each fraction in half the acquisition time. (D) The RTS-MS3 provided more uniquely quantified proteins. (E) The ability to selectively trigger MS3 spectra dramatically reduces the acquisition of redundant quantified peptides. (F) The mean HCT116/HEK ratio for all proteins quantified in both data sets was compared. Although the data were collected in 50% less time, the RTS-MS3 provided comparable protein quantification to the DDA-MS3data set (r = 0.91). Notably, the RTS-MS3 approach shows greater dynamic range for highly dynamic proteins (blue triangles).