

SUMMARY
Tumors that overexpress the MYC oncogene are frequently aneuploid, a state associated with highly aggressive cancers and tumor evolution. However, how MYC causes aneuploidy is not well understood. Here, we show that MYC overexpression induces mitotic spindle assembly defects and chromosomal instability (CIN) through effects on microtubule nucleation and organization. Attenuating MYC expression reverses mitotic defects, even in established tumor cell lines, indicating an ongoing role for MYC in CIN. MYC reprograms mitotic gene expression, and we identify TPX2 to be permissive for spindle assembly in MYC-high cells. TPX2 depletion blocks mitotic progression, induces cell death, and prevents tumor growth. Further elevating TPX2 expression reduces mitotic defects in MYC-high cells. MYC and TPX2 expression may be useful bio-markers to stratify patients for anti-mitotic therapies. Our studies implicate MYC as a regulator of mitosis and suggest that blocking MYC activity can attenuate the emergence of CIN and tumor evolution.
Graphical Abstract
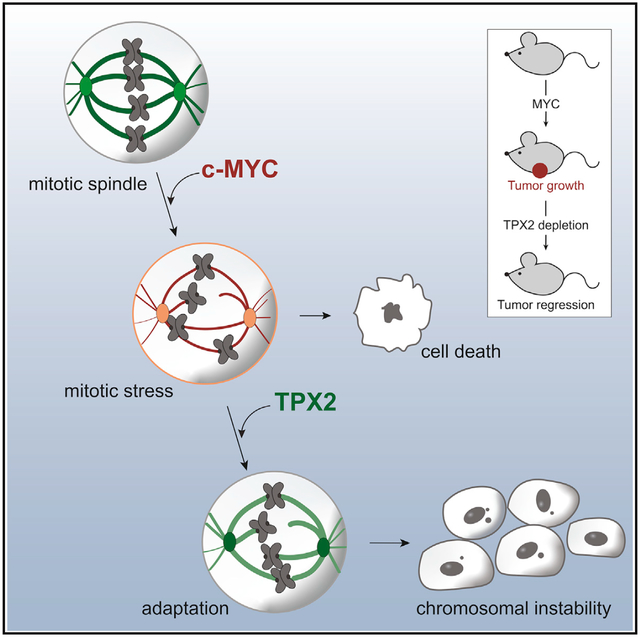
In Brief
Rohrberg et al. identify a reversible role of the MYC oncogene for inducing chromosomal instability by inducing error-prone mitosis. MYC-high tumor cells rely on the mitotic regulator TPX2 to survive the altered mitotic program, revealing a synthetic-lethal interaction between MYC overexpression and TPX2 loss as a potential therapeutic strategy.
INTRODUCTION
Aneuploidy, a state of abnormal chromosome number, is a hallmark of cancer, with >70% of common solid tumors found to be aneuploid (Boveri, 2008; Cimini, 2008). Aneuploidy is frequently caused by chromosomal instability (CIN), chromosome missegregation that leads to chromosome loss or gain (Lengauer et al., 1997; Thompson and Compton, 2008). CIN is a major driver of tumor evolution and promotes drug resistance and metastasis (Bakhoum et al., 2018; Greaves, 2015; Turajlic and Swanton, 2017); however, the major mechanisms that induce CIN remain poorly understood.
The MYC oncogene is frequently overexpressed in a wide variety of aggressive and metastatic tumors and has been associated with aneuploidy (Felsher and Bishop, 1999a; Karlsson et al., 2003; McCormack et al., 1998; Soucek and Evan, 2010). One of the key biological functions of MYC is its ability to facilitate entry and progression through G1 and S phases of the cell cycle by regulating gene transcription (Bretones et al., 2015). However, whether MYC also affects mitotic progression and induces CIN is unclear. We and others have found that cells with elevated MYC activity are sensitive to mitotic interruption such as treatment with microtubule-targeting agents, mitotic kinase inhibitors, or small interfering RNA (siRNA)-mediated depletion of spindle-related genes (Dauch et al., 2016; Goga et al., 2007; Horiuchi et al., 2012; Kessler et al., 2012; Littler et al., 2019; Martins et al., 2015; Menssen et al., 2007; Pereira et al., 2017; Topham et al., 2015). However, a molecular mechanism for the synthetic-lethal interactions of MYC with mitotic regulators is missing. Clarifying such a mechanism could reveal novel treatment strategies for aggressive MYC-overexpressing cancers.
Chromosome segregation is mediated by the mitotic spindle, while spindle error detection occurs through the spindle assembly checkpoint (SAC). The SAC delays chromosome segregation until appropriate attachments of chromosomes to spindle microtubules are established (Joglekar, 2016). In cancer cells, where CIN is common, chromosomes frequently missegregate as a result of microtubule-chromosome attachment errors that are not detected by the SAC (Bakhoum et al., 2009). Various defects in spindle formation can cause attachment errors and CIN (Cimini, 2008). One key mediator of spindle formation is the microtubule-binding protein TPX2, which is overexpressed in many aggressive human tumors, and its overexpression is highly correlated with CIN (Carter et al., 2006; Hu et al., 2012). However, the mechanisms of TPX2 deregulation and its specific role in CIN formation remain unclear (Carter et al., 2006; Neumayer et al., 2014). Here, we identify the MYC oncogene to reversibly induce CIN in various cellular models through effects on mitotic spindle formation. Using gene expression data and genomic functional screening approaches, we identify TPX2 as an important factor for the survival of cells with MYC overexpression.
RESULTS
MYC Overexpression Delays Mitotic Progression and Causes CIN
Although MYC overexpression has been frequently associated with more rapid cellular proliferation, its role in regulating mitosis remains poorly understood. We sought to determine whether MYC alters mitotic progression in human non-transformed epithelial cell lines. We engineered human retinal pigment epithelium (RPE-1) cells to constitutively overexpress MYC (RPE-MYC) or a control plasmid (RPE-NEO) (Goga et al., 2007; Yang et al., 2010). We performed immunofluorescence on fixed cells to examine kinetochore and chromosome localization. MYC overexpression was associated with an increased percentage of cells with misaligned chromosomes in metaphase and lagging chromosomes in anaphase (Figures 1A and 1B). Lagging chromosomes often form micronuclei in the subsequent S phase (Cimini et al., 2002). Consistent with this, we observed more micronucleated RPE-MYC cells compared to RPE-NEO cells (Figures 1A and 1B). We next asked whether MYC alters mitotic progression. We performed time-lapse microscopy of H2B-mCherry-expressing cells to compare the time from chromosome condensation to the onset of chromosome segregation, an established assay for mitotic timing (Knouse et al., 2018; Meraldi et al., 2004; Stolz et al., 2010) (Figure 1C). RPE-MYC cells took more time to reach anaphase onset compared to RPE-NEO cells (Figure 1D). Anaphase onset can be delayed by the SAC. To check whether SAC function is affected by MYC, we probed for Mad1 localization (Kuhn and Dumont, 2017; Maldonado and Kapoor, 2011). Mad1 localized to kinetochores in RPE-MYC cells, indicating a functional SAC (Figure S1A). Thus, an increase in mitotic errors, a prometaphase delay, and a robust Mad1 staining suggest an MYC-induced activation of the SAC.
Figure 1. MYC Reversibly Induces CIN.
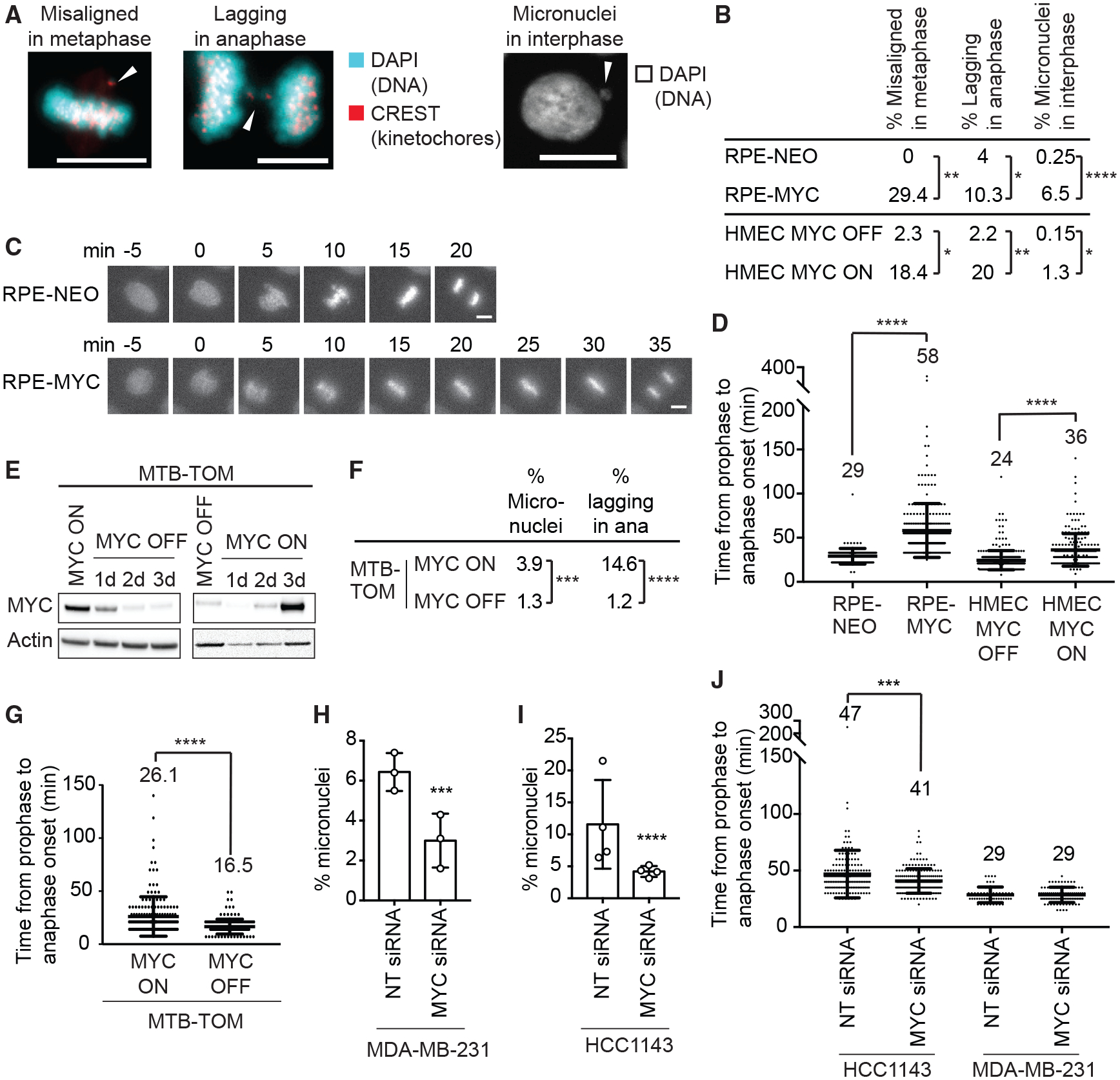
(A) Images of mitotic defects in RPE-MYC cells. Arrows indicate misaligned and missegregated chromosomes. Scale bars, 10 μm.
(B) Percentage of mitotic defects. HMEC MYC ON, MYC activated for 3 days. Fisher’s exact test, n = 100–300 mitotic cells and n = 800–1,000 cells for micronuclei from 3 independent experiments.
(C) Fluorescent time-lapse images of RPE-NEO and RPE-MYC cells expressing H2B-mCherry. Scale bars, 10 μm.
(D) Time from chromosome condensation to anaphase onset (average time shown above each plot). Mean ± SD. Unpaired t test, n = 140–380 from 3 independent experiments.
(E) Western blot analysis of MYC in MTB-TOM cells grown in the presence (MYC ON) and absence of doxycycline (MYC OFF).
(F) Percentage of mitotic defects in MTB-TOM MYC ON and MYC OFF (3 days) cells. Fisher’s exact test, n = 1,628 and 520 for micronuclei and 178 and 164 for mitotic errors, 3 independent experiments.
(G) Time from chromosome condensation to anaphase onset of MTB-TOM cells expressing H2B-mCherry (average time shown above each plot). Mean ± SD. t test, n = 178 and 164, 3 independent experiments.
(H and I) Percentage of micronucleated MDA-MB-231 (H) and HCC1143 (I) cells 3 days after transfection with non-targeting (NT) or MYC siRNA. Mean ± SD. Fisher’s exact test, n = 628–1,056, 3 independent experiments. See also Figures S1H–S1L.
(J) Time from chromosome condensation to anaphase onset 3 days after treatment with NT or MYC siRNA (average time shown above each plot). Mean ± SD. Unpaired t test, n = 171–179, 3 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Long-term MYC overexpression could lead to genomic changes and the accumulation of mutations, hindering the ability to discern direct versus indirect effects. To examine the temporal dependence on MYC to elicit mitotic errors, we tested early passage non-immortalized human mammary epithelial cells (HMECs) that express an MYC-estrogen receptor (ER) transgene, allowing for transient activation of MYC following treatment with 4-hydroxytamoxifen (TAM) (Horiuchi et al., 2016). Activation of MYC in HMECs for 3 days (MYC ON) increased the number of cells with misaligned and lagging chromosomes and enhanced micronuclei formation (Figure 1B). MYC activation also delayed anaphase onset in H2B-mCherry-expressing HMECs, confirming the results observed in RPE1 cells (Figure 1D). Thus, unlike the ability of MYC to accelerate transition through the G1/S checkpoint (Amati et al., 1998; Ryl et al., 2017; Sheen and Dickson, 2002), we find that increased MYC activity delays progression through mitosis and induces CIN.
MYC-Induced CIN Is Reversible
To test whether MYC-induced CIN is reversible, we derived a cell line from an MYC-driven transgenic mouse model of triple-negative breast cancer, in which MYC overexpression is inducible with doxycycline (MMTV-rtTA TetO-Myc [MTBTOM]) (Camarda et al., 2016; D’Cruz et al., 2001; Pfefferle et al., 2013). We immortalized MTB-TOM cells in the presence of doxycycline to ensure MYC overexpression. Expression of the human MYC transgene was drastically reduced 2 days after doxycycline withdrawal (Figure 1E; see STAR Methods for characterization). We determined whether MTB-TOM cells grown in the presence of doxycycline (MYC ON) display CIN. A total of 15% of MYC ON MTB-TOM cells had lagging chromosomes, and 4% were micronucleated, similar to the frequency found in RPE-MYC cells (Figure 1F). We next tested whether turning off MYC expression decreases CIN. The number of lagging chromosomes and micronucleated cells decreased to 1.2% and 1.3%, respectively (MYC OFF; Figure 1F). To test whether the MYC-induced anaphase delay observed in RPE-1 cells and HMECs is reversible, we performed time-lapse microscopy of H2B-mCherry-expressing MTB-TOM cells. Turning off MYC expression reduced the time for transition from prophase to anaphase on average by ~9 min, suggesting an earlier satisfaction of the SAC and a reduction in chromosome attachment errors (Figure 1G).
We next wondered whether the attenuation of MYC overexpression also reverses CIN in established human tumor cell lines. We quantified the number of micronucleated cells and measured mitotic timing in two MTB-TOM cell lines, HCC1143 and MDA-MB-231, that exhibit elevated MYC expression (Horiuchi et al., 2012) (Figure S3F). Both cell lines demonstrated chromosomal instability, with 6.7% of MDAMB-231 cells and 11.6% of HCC1143 cells being micronucleated (Figure 1H). Depleting MYC with a pool of siRNAs reduced the number of micronucleated cells to 3.1% and 4.3%, respectively, suggesting that MYC-induced CIN is partially reversible (Figures 1H, 1I, S1H, and S1J). MYC depletion also reduced time to anaphase onset in HCC1143 cells, indicating an earlier satisfaction of the SAC (Figure 1J). Untreated MDA-MB-231 cells completed mitosis very quickly (29 min), and MYC depletion did not further accelerate the process (Figure 1J). However, MDA-MB-231 cells have been reported to have a weakened SAC, providing an explanation for the early anaphase onset even in the presence of considerable CIN (6.7% micronucleated cells) (Riffell et al., 2009). To exclude off-target effects, we also tested two individual siRNAs targeting MYC in HCC1143 cells. Single siRNAs were less efficient to deplete MYC; however, even partial MYC depletion resulted in fewer micronuclei (Figures S1F–S1H). Thus, we observed a correlation between the level of MYC expression and percentage of micronucleated cells across several cell lines and depletion experiments (Figure S1I). Those data, together with the data from other MYC-high and -low cell lines (i.e., RPE-MYCs versus RPE-NEOs, HMECs, MTB-TOMs), argue that MYC reversibly induces mitotic defects. Attenuation of MYC expression can reverse CIN, even in established, genetically complex cancer cell lines, suggesting an ongoing role for MYC in the persistence of CIN.
MYC Overexpression Impairs Mitotic Spindle Formation
Multiple kinds of mitotic defects can lead to chromosome attachment errors and CIN (Thompson et al., 2010). We performed nocodazole washout assays to determine which aspects of mitotic spindle assembly are affected by MYC. Nocodazole incubation depolymerizes microtubules and arrests cells in prometaphase (Cavazza et al., 2016). Upon washout, microtubules nucleate from both centrosomal and non-centrosomal sites to reform the spindle (Figure 2A) (Petry and Vale, 2015). 2 min after washout, we observed more total microtubule nucleation sites and, in particular, more microtubules nucleated from non-centrosomal sites in RPE-MYC cells compared to control cells (Figures 2B and S2A). MYC overexpression also inhibited the coalescence of microtubule asters even 30 min after washout (Figure 2B). Consequently, chromosome alignment was impaired: only 30% of RPE-MYC cells had aligned chromosomes 30 min after washout compared to 83% of RPE-NEO cells (with an additional 4.6% ±2.6% of RPE-NEO cells having completed anaphase, n = 3) (Figure 2C). We next tested whether short-term MYC activation also attenuates spindle reassembly in HMECs. We observed similar defects of microtubule assembly and chromosome alignment in HMEC MYC ON, indicating a specific effect of MYC on mitotic spindle formation (Figures S2B–S2D).
Figure 2. MYC Impairs Mitotic Spindle Formation.
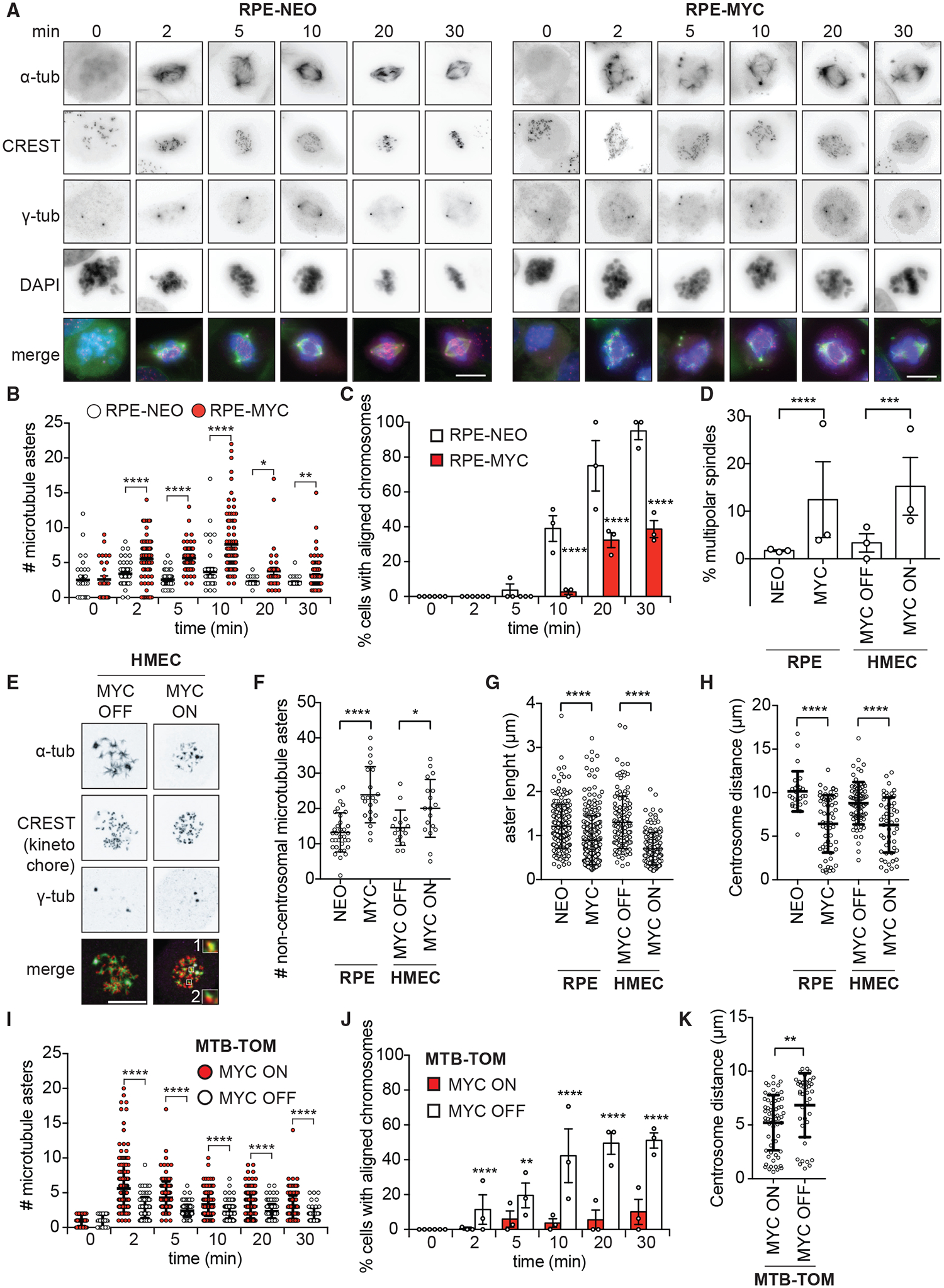
(A–C) Nocodazole washout assay in RPE-NEO and RPE-MYC cells.
(A) Representative images. Scale bars, 10 μm.
(B) Number of microtubule foci/asters. Mean ± SEM t test, n = 26–99, 3 independent experiments. See also Figure S2A.
(C) Percentage of cells with aligned chromosomes. Mean ± SEM. Fisher’s exact test, n = 13–139, 3 independent experiments.
(D) Percentage of multipolar spindles. HMEC MYC ON, MYC activated for 3 days. Mean ± SEM. Fisher’s exact test, n = 97–315, 3 independent experiments. See also Figures S2E and S2F.
(E–G) Confocal microscopy after nocodazole washout.
(E) Representative images of HMEC cells 2 min after washout. Scale bar, 10 μm. Insets show a 3-fold magnification of microtubule asters nucleating at non-centrosomal sites colocalizing with (1) or nucleating next to kinetochores (2).
(F) Number of microtubule asters at non-centrosomal sites 2 min after washout. Mean ± SD. Unpaired t test, n = 16–35, 2 independent experiments.
(G) Length of microtubule asters 5 min after washout. Mean ± SD. Fisher’s exact test, n = 139–286, 2 independent experiments.
(H) Centrosome distance 30 min after nocodazole washout. Mean ± SD. Unpaired t test, n = 26–73, at least 3 independent experiments.
(I–K) Nocodazole washout assay in MTB-TOM MYC ON and MYC OFF (3 days).
(I) Number of microtubule asters. Mean ± SEM. t test, n = 47–208, 3 independent experiments. See also Figure S2G.
(J) Percentage of cells with aligned chromosomes. Mean ± SEM. Fisher’s exact test, n = 81–209 cells, 3 independent experiments.
(K) Centrosome distance 30 min after washout. Mean ± S.D. Unpaired t test, n = 41 and 63 cells, 3 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
The increased number of microtubule asters observed in MYC-high cells could be the result of increased centrosome numbers (Sugihara et al., 2004) or of increased microtubule nucleation from non-centrosomal sites. To test the former possibility, we examined untreated cells by immunofluorescence microscopy. We found an 11% and a 12% increase in the frequency of multipolar spindles in RPE-MYC and HMEC MYC ON, respectively, compared to control cells (Figure 2D). Confocal microscopy revealed that 31% and 39% of those multipolar spindles contained >2 centrioles per pole in RPE-MYC and HMEC MYC ON, respectively (Figures S2E and S2F). A small number of bipolar RPE-MYC cells (0.7%) also had extra free centrioles, suggesting that MYC causes centriole amplification. Although we observed an 11% (RPE-MYC) and a 12% (HMEC MYC ON) increase in multipolar spindles due to MYC overexpression or activation, this difference cannot account for the spindle reformation defects we observed in >70% of cells in the nocodazole washout assay. We next used confocal microscopy to reveal the localization of microtubule nucleation sites after nocodazole washout. We observed more microtubule nucleation sites at non-centrosomal sites in RPE-MYC and in HMECs MYC ON compared to control cells (Figures 2E and 2F).
Confocal microscopy after nocodazole washout also revealed shorter microtubule asters in MYC-high cells compared to control cells, suggesting that MYC impairs microtubule growth (Figure 2G). Growing microtubules are required to push apart centrosomes during spindle formation. Thus, MYC-induced defects in microtubule growth may impair centrosome segregation. To test whether centrosome segregation was affected by MYC, we measured centrosome distance 30 min after washout. We found that centrosomes were positioned closer together in RPE-MYC cells and HMECs MYC ON compared to control cells, which is consistent with the observed defects in microtubule aster length (Figure 2H).
We next asked whether MYC depletion can rescue spindle-formation defects. Spindle re-assembly after nocodazole washout in doxycycline-treated MTB-TOM cells (MYC ON) resembled the phenotype of RPE-MYC cells. A high number of microtubule asters emanated from non-centrosomal sites, and chromosome alignment was slow (Figures 2I, 2J, and S2G). Switching off MYC expression rescued spindle-formation defects. The number of microtubule nucleation sites decreased, nucleation was found predominantly at the centrosomes, chromosome alignment was accelerated, and centrosome segregation increased (Figures 2I–2K). We next examined the reversibility of spindle defects in MYC-high human breast cancer cell lines MDA-MB-231 and HCC1143. MYC depletion using siRNA partially reversed microtubule nucleation defects, aster growth, and chromosome alignment, demonstrating the reversibility of MYC-induced mitotic errors (Figures S2H–S2K). MYC influences several steps of spindle assembly and mitotic progression, resulting in a delay of bipolar spindle formation and chromosome alignment. Attenuating MYC expression, even in established MYC-driven tumor cells, reverses spindle-formation defects.
MYC Regulates Genes Involved in Spindle Formation
We next investigated how MYC may induce mitotic aberrations. MYC is a transcription factor that binds to active promoters of direct target genes (Chen et al., 2008; Lin et al., 2012; Nie et al., 2012; Walz et al., 2014). To investigate whether the mitotic defects are dependent on the transcriptional activity of MYC, we tested whether a transcriptionally inactive MYC mutant can increase the time from prophase to anaphase onset in previously described Rat1a cells (Schwinkendorf and Gallant, 2009). Wild-type Rat1a cells took an average of 13 min to proceed from prophase to anaphase onset (Figure 3B). Overexpressing mouse MYC in Rat1a cells that have had the endogenous rat MYC gene knocked out (Rat1a myc−/−) (Bush et al., 1998) caused a significant delay in anaphase onset (21.4 min) (Figures 3A and 3B) and micronuclei formation, indicating CIN (Figure 3C). In contrast, expression of the transcriptionally inactive mutant MYCDMBII in Rat1a myc−/− cells did not alter mitotic timing or micronuclei formation, indicating that the transcriptional function of MYC is required to induce mitotic defects (Figures 3A–3C).
Figure 3. MYC Regulates the Expression of Mitotic Spindle Genes.
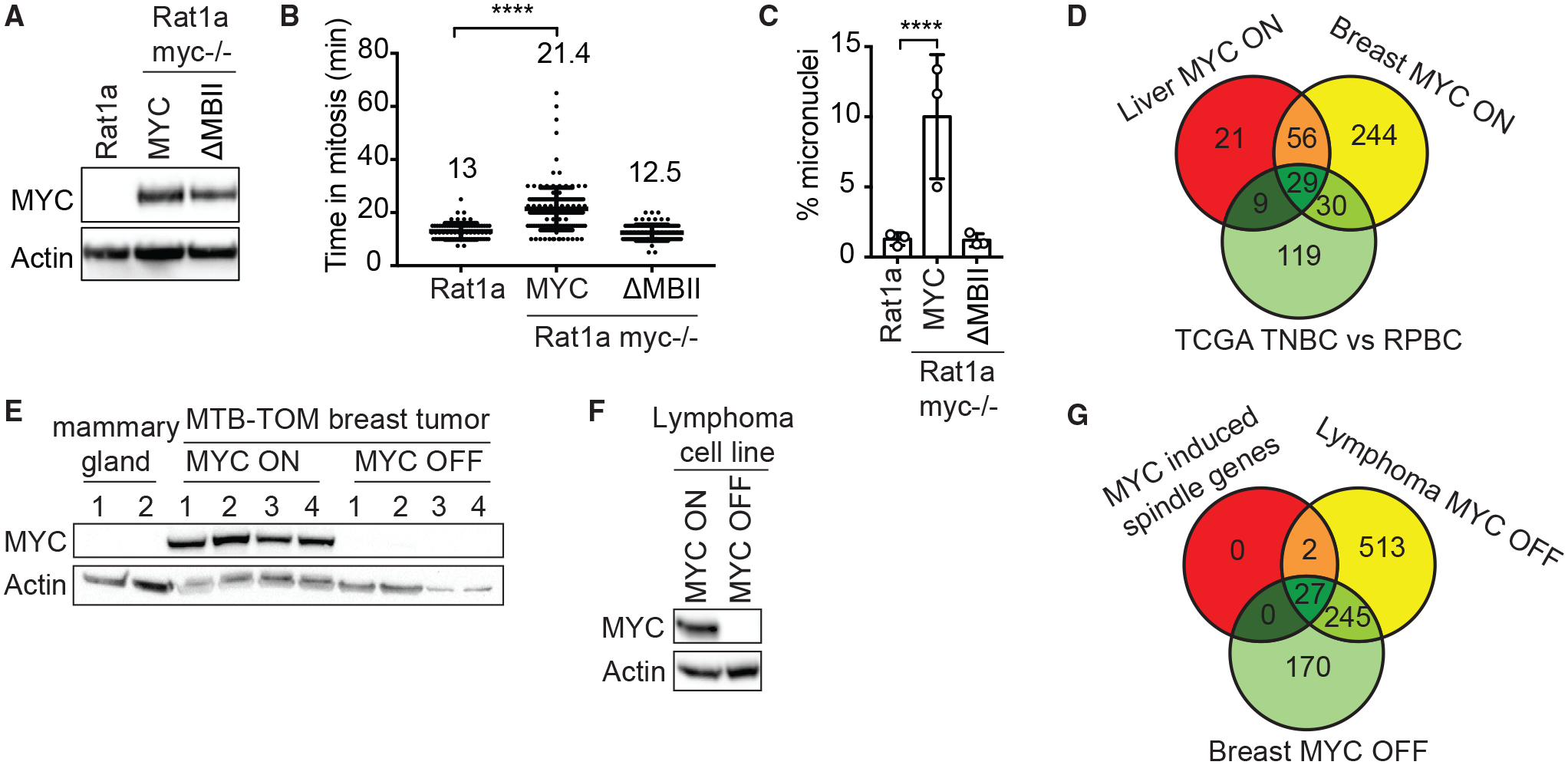
(A–C) Rat1a and Rat1a myc−/− cells expressing mouse wild-type MYC or the transcriptionally inactive mutant MYCDMBII.
(A) Western blot analysis of murine MYC; endogenous rat MYC expression was not detected.
(B) Time from chromosome condensation to anaphase onset. Mean ± SD. Unpaired t test, n = 78–167, 3 independent experiments.
(C) Percentage of micronucleated cells. Mean ± SD. Fisher’s exact test, n = 730–1,478, 3 independent experiments.
(D) Deregulated mitotic spindle genes in murine MTB-TOM breast and liver tumors compared to the corresponding normal tissue and in human TNBC compared to RPBC (TCGA data). Log2 fold change >1, FDR < 0.05.
(E) Western blot analysis of MYC in MTB-TOM tumors (MYC ON, n = 4), tumors off doxycycline for 3 days (MYC OFF, n = 4) and normal mammary gland (n = 2).
(F) Western blot of MYC in Eμ-tTA/TRE-MYC lymphoma cells in the absence (MYC ON) and presence (MYC OFF) of doxycycline for 3 days.
(G) Deregulated mitotic spindle genes when MYC expression is turned off in MTB-TOM breast tumors and Eμ-tTA/TRE-MYC lymphoma cells compared to the 29 MYC-induced spindle genes identified in (D). Log2 fold change > –1, FDR < 0.05. See also Table S2.
****p < 0.0001.
Next, we wondered which mitotic genes could be transcriptionally affected by MYC. We analyzed the expression of 1,452 genes associated with the Gene Ontology terms kinetochore, microtubule, mitosis, and mitotic spindle (called spindle genes) in two transgenic tumor mouse model systems. Tumors are driven by conditionally expressed MYC in the LAP-tTA × TetO-MYC (LT2-MYC) bi-transgenic liver tumor mouse model (Anderton et al., 2017; Shachaf et al., 2004) and the transgenic mouse model of MTB-TOM (D’Cruz et al., 2001). A total of 115 spindle genes were differentially expressed in mouse liver tumors and 359 were differentially expressed in breast tumors compared to normal tissue (Table S2). We next examined human breast cancer patient data from The Cancer Genome Atlas (TCGA). Previously, as a subset, receptor-triple negative breast cancer (TNBC) tumors were found to express elevated MYC compared to receptor-positive breast cancer (RPBC) tumors (Cancer Genome Atlas Network, 2012; Horiuchi et al., 2012). By comparing TNBC and RPBC tumors, we found that 191 spindle genes were differentially expressed (Table S2). The expression of 29 spindle genes was altered across all 3 datasets, suggesting that those may mediate the MYC-induced spindle defects (Figure 3D). We next wondered whether spindle gene expression is reversible. To test this, we briefly turned off MYC expression in four MTB-TOM tumors (Figure 3E) and in a cell line from the double-transgenic Eμ-tTA/TRE-MYC lymphoma mouse model (Felsher and Bishop, 1999b; Goga et al., 2007) (Figure 3F). Depletion of MYC for 3 days reversed the expression of 27 of the 29 spindle genes (Figure 3G; Table S1). To determine whether those genes are direct transcriptional targets of MYC, we analyzed chromatin immunoprecipitation sequencing (ChIP-seq) data from 2 prior studies (Sabò et al., 2014; Walz et al., 2014). Among the 24 genes that were present in both datasets, 20 were found to have MYC bound to promoters in at least 3 of 4 cell types (Table S1). These data indicate that MYC alters the transcription of multiple mitotic spindle genes, many of which have been shown to regulate microtubule behavior and spindle assembly.
Elevated TPX2 Expression Facilitates Mitotic Completion in MYC-High Cells
All or a subset of the MYC-regulated spindle genes may contribute to the observed mitotic aberrations. TPX2, BIRC5 (survivin), and KIF11 (Eg5) proteins have been previously found to interact with the spindle. TPX2 is the main factor that facilitates microtubule nucleation at chromosomes (Gruss et al., 2002), regulates spindle pole formation via Eg5 (Eibes et al., 2018; Ma et al., 2011), and its localization depends on survivin (Xia et al., 2008). Our observations that MYC-high activity leads to spindle abnormalities and that MYC actively regulates the expression of spindle genes led us to hypothesize that MYC-high cells may depend on the expression of these genes. If this were true, then targeting these genes may lead to therapeutically beneficial synthetic-lethal interactions in the context of MYC-high cells. To elucidate whether MYC-high cells depend on TPX2, survivin, or Eg5, we performed siRNA knockdown in RPE-NEO and RPE-MYC cells and measured cell death (Figure 4A). We included several control genes—MAP7, PDS5B, and LMNA—whose expression was not altered in an MYC-dependent manner, but they do have a function in mitosis (Carretero et al., 2013; Dechat et al., 2007; Faire et al., 1999; Gallaud et al., 2014; Heald and McKeon, 1990). We also examined STMN1, a protein that destabilizes microtubules, and NPM1, whose expression was MYC dependent in four of five RNA sequencing (RNA-seq) datasets (Table S2) and plays a role in centrosome duplication (Okuda, 2002; Wittmann et al., 2004). As a positive control, we included treatment with the cyclin-dependent kinase 1 (CDK1) inhibitor purvalanol A, which has previously been shown to be synthetic-lethal with MYC overexpression (Goga et al., 2007). Depletion of TPX2, survivin, and Eg5 markedly reduced the viability of RPE-MYC cells while minimally affecting RPE-NEO cells (Figure 4A).
Figure 4. TPX2 Expression Correlates with MYC and Its Depletion Kills MYC-High Cells.
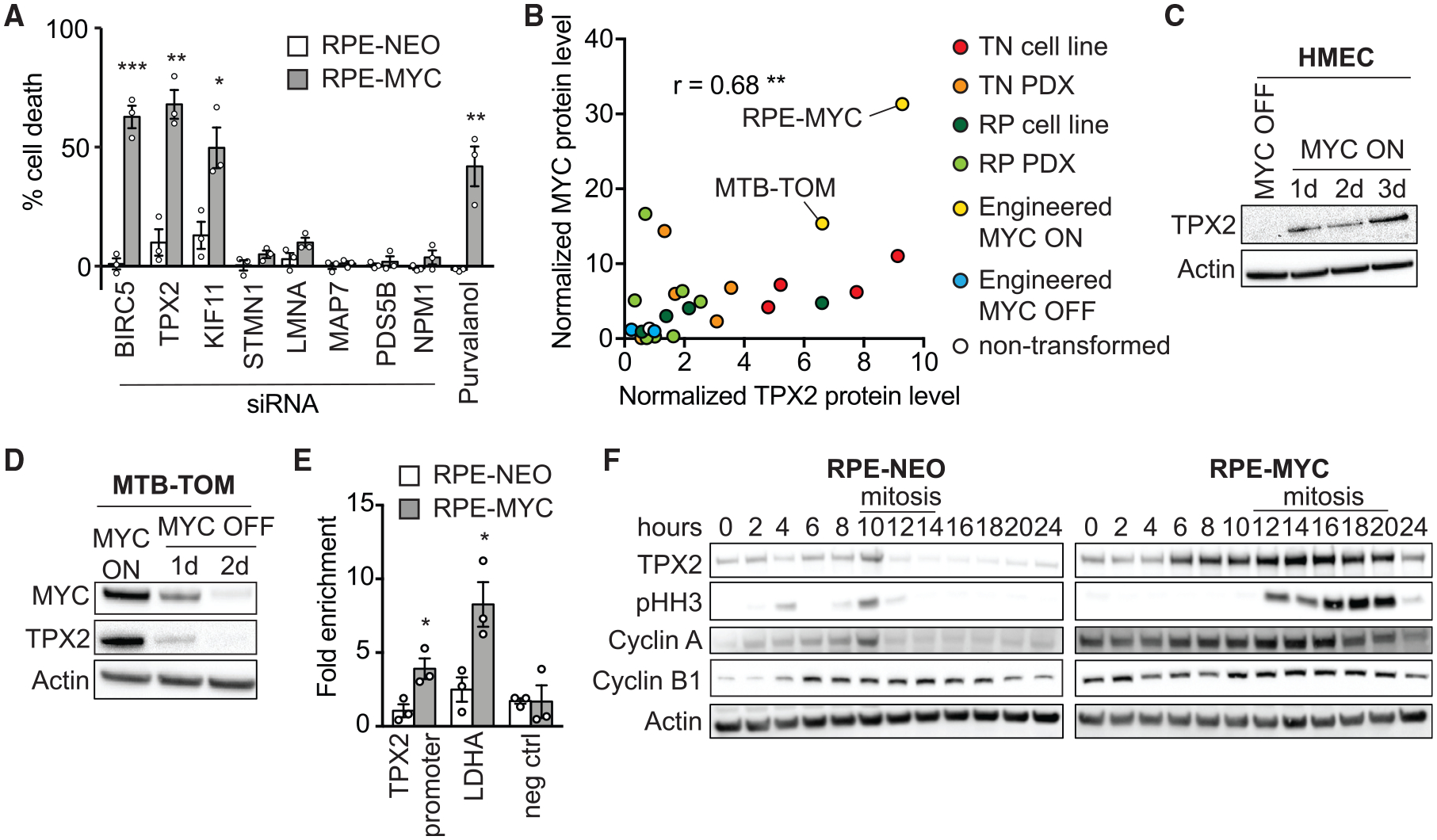
(A) Percentage of cell death in RPE-NEO (white bars) and RPE-MYC cells (gray bars) 3 days after siRNA treatment normalized to control siRNA, and after treatment with 10 μM purvalanol A normalized to DMSO. Bars, mean ± SEM. t test, n = 3.
(B) Correlation of MYC and TPX2 protein levels in RPE-MYC and MTB-TOM (engineered MYC ON, yellow), RPE-NEO, and MTB-TOM off doxycycline for 2 days (engineered MYC OFF, blue), 4 triple negative breast cancer (TNBC) cell lines (red), 4 receptor-positive breast cancer (RPBC) cell lines (dark green), 5 patient-derived xenograft (PDX) TNBC tumors (orange), 7 RPBC PDX tumors (light green), and non-transformed mouse mammary gland (white). See also Figures S3C–S3F. The correlation coefficient was computed using Pearson correlation.
(C) Western blot of TPX2 in HMEC MYC ON and HMEC MYC OFF.
(D) Western blot of TPX2 and MYC in MTB-TOM MYC ON and MYC OFF.
(E) ChIP of MYC in RPE-NEO and RPE-MYC followed by qPCR against the promoter region of TPX2 and LDHA (positive control) and an upstream region of the LDHA promoter (negative control). Mean ± SEM. Unpaired t test, n = 3. See also Figure S3B.
(F) Western blot of TPX2, pHH3, Cyclin A, and Cyclin B1 in RPE-NEO and RPE-MYC cells; time after release from a double thymidine block is indicated. Mitotic cell rounding observed by light microscopy is indicated with line. See also Figure S3G.
*p < 0.05, **p < 0.01, ***p < 0.001.
We focused on the microtubule-binding protein TPX2 because it is required for the initiation of microtubule growth from non-centrosomal sites, controls centrosome movement, and is important for efficient mitotic completion (Eibes et al., 2018; Gruss et al., 2001; Petry, 2016). We speculated that it may be a central player in regulating the MYC-induced mitotic aberrations. We sought to explore in more detail the connection between MYC and TPX2 expression. In addition to finding TPX2 mRNA upregulation in various MYC-driven tumor contexts (Figures 3D and 3G; Table S1), we found a significant co-occurrence of MYC and TPX2 mRNA in primary human breast tumors (cBioportal; Figure S3A). We next wondered whether MYC and TPX2 protein levels also correlate. We performed western blot analysis of MTB-TOM cells in the absence and presence of doxycycline (to regulate MYC expression), non-tumor mouse mammary gland, tumor tissue from 12 breast cancer patient-derived xenograft (PDX) models and various cell lines, including RPE-NEO, RPE-MYC, and 8 TNBC and RPBC cell lines (Figures S3C–S3F). We found a significant co-occurrence of TPX2 and MYC protein levels. Non-transformed tissues expressed low levels of both proteins, while aggressive cancer cells expressed high levels (Figure 4B). Furthermore, TPX2 expression was rapidly and reversibly induced when MYC activity was regulated in HMECs and MTB-TOM tumors, which is consistent with a direct role for MYC in regulating TPX2 expression (Figures 4C and 4D). To validate that TPX2 is a transcriptional target of MYC, we analyzed publicly available ChIP-seq data from the Encode project. MYC and its binding partner MAX are bound to the promoter region of TPX2 in multiple cell lines (Figure S3B). To confirm that TPX2 is a target of MYC in RPE-1 cells, we performed ChIP followed by qPCR against the promoter region of TPX2 and lactate dehydrogenase A (LDHA) (positive control) and an upstream sequence of LDHA (negative control) (Figure 4E). We found MYC bound to the promoter region of TPX2 in RPE-MYC, but much less so in RPE-NEO cells, indicating that TPX2 is transcriptionally regulated by MYC. These data, together with data from Walz et al. (2014) and Sabò et al. (2014) (Table S1), strongly suggest that TPX2 is a direct transcriptional target of MYC.
TPX2 expression increases through the cell cycle and peaks in late G2 and mitosis and is degraded upon mitotic exit (Gruss et al., 2002). To examine TPX2 protein expression in MYC-high cells, we synchronized cells at the G1/S boundary by a double thymidine block and released them to harvest lysates at 2-h intervals post-release. RPE-NEO and RPE-MYC cells entered mitosis at ~10 and 12 h, respectively, as evidenced by histone H3 phosphorylation, increased cyclin A and B levels, and cell rounding observed by phase microscopy (Figures 4F and S3G). In RPE-MYC cells, more TPX2 protein is found at every cell-cycle stage and is not fully degraded at the end of mitosis, demonstrating that TPX2 protein expression is elevated in MYC-high cells.
TPX2 Depletion Is Synthetic-Lethal with MYC
TPX2 depletion selectively caused cell death in RPE-MYC, but not in RPE-NEO cells (Figure 4A). We confirmed TPX2 knockdown efficiency by western blot (Figure 5A). To further evaluate whether TPX2 induces apoptotic cell death preferentially in MYC-high cells, we monitored poly (ADP-ribose) polymerase (PARP) cleavage as a marker of apoptosis. Notably, PARP cleavage 3 days after TPX2 knockdown was induced in RPE-MYC, but not in RPE-NEO cells (Figure 5A), and together with morphologic changes (Figure 5B) demonstrated cell death. To confirm the specificity of the siRNA effect, we generated an allelic series of three doxycycline-inducible short hairpin RNAs (shRNAs) directed against TPX2. The greatest TPX2 knockdown induced the highest amount of cell death, whereas partial depletion induced less, indicating specificity and dose-dependent effects (Figures S4A and S4B).
Figure 5. TPX2 Is Required for the Survival of MYC-High Cells.
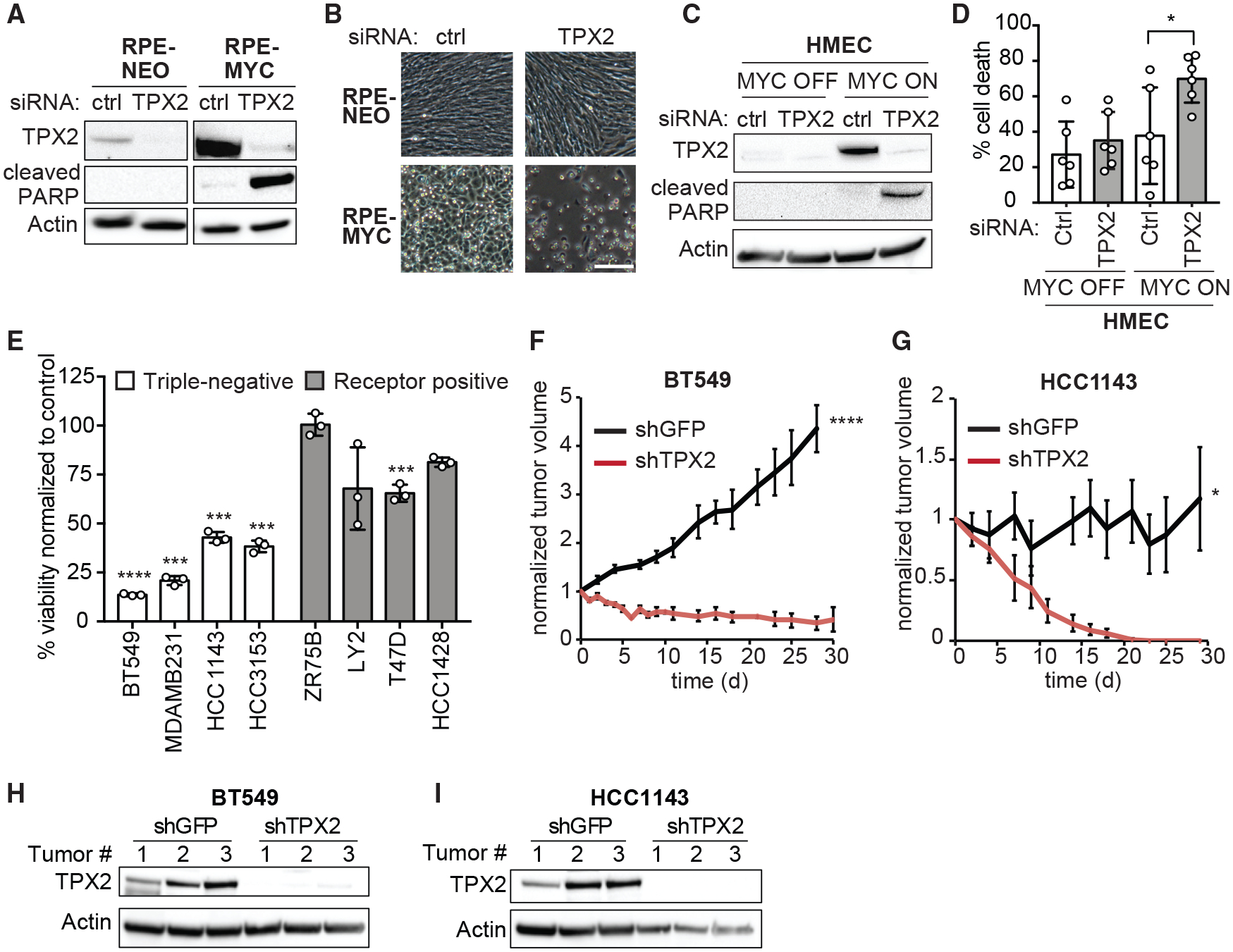
(A and B) Western blot of TPX2 and cleaved PARP (A) and micrographs (B) of RPE-NEO and RPE-MYC cells 3 days after transfection with control (ctrl) or TPX2 siRNA. Scale bar, 100 μm.
(C and D) Western blot of TPX2 and cleaved PARP (C) and percentage of cell death (D) of HMEC MYC OFF and HMEC MYC ON 3 days after transfection with control (ctrl) or TPX2 siRNA. Mean ± S.D., t test, n = 6. See also Figure S4C.
(E) Percent viability of breast cancer cell lines three days after transfection with TPX2 siRNA normalized to control siRNA. Mean ± SEM, t test, n = 3. See also Figure S4D.
(F and G) Relative volume of BT549 (F) and HCC1143 (G) xenograft tumors expressing doxycycline inducible shRNA against TPX2 (shTPX2) or GFP (shGFP). Mean ± SEM. BT549 shGFP (n = 7), shTPX2 (n = 7), HCC1143 shGFP (n = 6), and HCC1143 shTPX2 (n = 5). Unpaired t test. See also Figures S4E–S4F.
(H and I) Western blot of TPX2 in BT549 (H) and HCC1143 (I) xenograft tumors at endpoint (n = 3).
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We next tested whether the MYC-TPX2 synthetic-lethality also occurs in other cell lines. Knockdown of TPX2 in HMECs induced PARP cleavage only after MYC activation (Figure 5C). Bright-field microscopy and quantification of cell death confirmed cell death in HMECs with activated MYC (Figures 5D and S4C). Next, we tested whether breast cancer cell lines with varying MYC levels are sensitive to TPX2 loss. We treated eight cell lines with control or TPX2 siRNA and measured cell viability and PARP cleavage. The loss of TPX2 reduced viability by ~50% to 80% in MYC-high TNBC cell lines (Figure 5E). In contrast, the viability of only one RPBC cell line, T47D, decreased ~30% after TPX2 knockdown, while the other RPBC cell lines were less sensitive. Similarly, PARP cleavage was observed in all TNBC cell lines, mildly in the RPBC cell line ZR75B, and strongly in the RPBC cell line T47D (Figure S4D). Despite being receptor positive, T47D cells have relatively high levels of MYC and TPX2, providing a possible explanation for the sensitivity of those cells to TPX2 depletion (Figure S3F).
The most stringent test of a synthetic-lethal interaction is to determine whether it can block in vivo tumor formation. Since a selective small-molecule inhibitor of TPX2 does not yet exist, we sought to test whether TPX2 depletion can regress MYC-high xenograft tumors. We generated two TNBC cell lines (BT549 and HCC1143) that express a doxycycline-inducible shRNA against TPX2 (shTPX2) or GFP as control (shGFP). Doxycycline treatment induced TPX2 knockdown and cell death in cultured cells (Figures S4E and S4F). Similarly, after allowing tumors to form, BT549 tumors shrank rapidly upon the expression of shTPX2, and the survival of all mice was prolonged (Figures 5F and S4G) compared to shGFP expression. Likewise, HCC1143 tumors completely disappeared 20 days after the induction of shTPX2 expression, indicating that TPX2 is required for tumor survival in vivo (Figure 5G). Western blot analysis showed that doxycycline administration depleted TPX2 in tumors expressing shTPX2 and not in tumors expressing shGFP (Figures 5H and 5I). Thus, TPX2 is a synthetic-lethal interaction partner of MYC, and its depletion can regress established tumors.
High TPX2 Expression Is Required for Spindle Assembly in Cells that Overexpress MYC
We sought to determine how TPX2 depletion differentially affects MYC-high versus low cells. We found that TPX2 localization was not altered upon MYC overexpression. TPX2 localized to the nucleus in interphase and to spindle pole microtubules in mitosis in RPE-NEO and RPE-MYC cells, as reported previously (Gruss et al., 2002) (Figures S5A and S5B). It has been shown that TPX2 loss causes spindle-formation failure in HeLa cells, which reportedly express high levels of MYC (Cappellen et al., 2007; Garrett et al., 2002; Gruss et al., 2002). However, the consequences of TPX2 loss in non-transformed MYC-low cells are not established. To understand whether TPX2 depletion differentially affects MYC-high versus -low cells, we performed live-cell imaging of cells expressing the FUCCI (fluorescent ubiquitination-based cell-cycle indicator) cell-cycle reporter (Sakaue-Sawano et al., 2008) (Figure 6A). After TPX2 knockdown, the majority (55%) of RPE-MYC cells died in mitosis after a prolonged arrest (Figure 6B), and 23% of the arrested cells slipped into G1 and subsequently died. Only ~20% of RPE-MYC cells survived 48 h of siRNA treatment compared to 92% of RPE-NEO cells (Figure 6B). DNA content analysis confirmed a partial G2 arrest in RPE-NEO cells and dramatic cell death in RPE-MYC cells (observed as cells with <2N DNA content) after TPX2 knockdown (Figure 6C). Furthermore, ~25% of RPE-MYC cells accumulated >4N DNA content, consistent with the observed mitotic slippage followed by possible endoreduplication (Figure 6B).
Figure 6. TPX2 Protects Mitotic Spindle Function in MYC-High Cells.
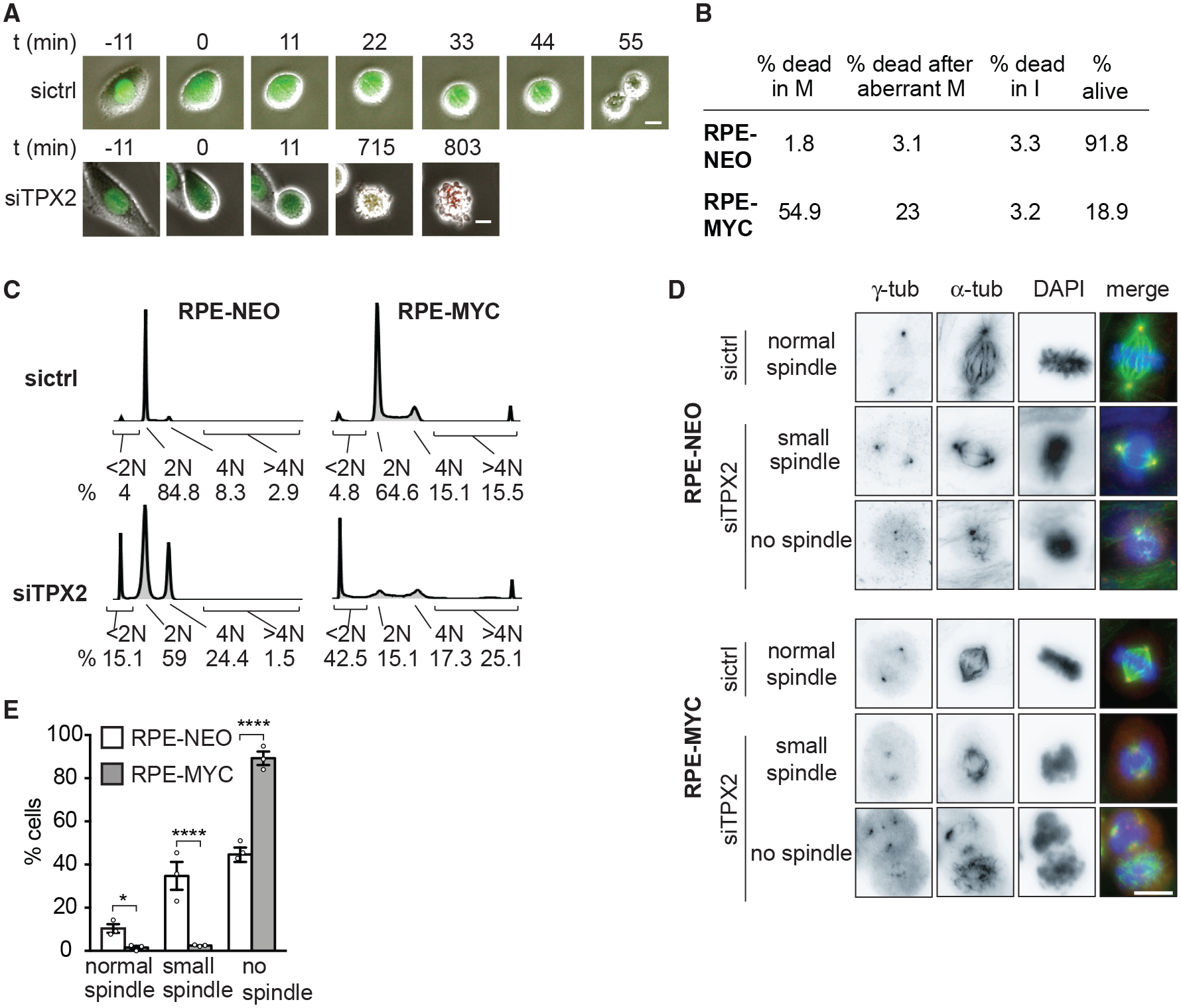
(A) Time-lapse images of RPE-MYC cells expressing the FUCCI cell-cycle marker 12 h after transfection with control (sictrl) or TPX2 (siTPX2) siRNA. Fluorescence and phase-contrast images were overlaid. Scale bars, 20 μm.
(B) Percentage of cells undergoing cell death in mitosis (M), after aberrant mitosis and in interphase (I) 12–24 h after TPX2 knockdown in RPE-NEO and RPE-MYC cells. n = 3.
(C) Cell-cycle profiles 48 h after siRNA treatment with percentage of dead cells (<2N), cells in G1 (2N), and G2-M (4N) phases of the cell cycle and cells with >4N DNA content.
(D) Images of RPE-NEO (top) and RPE-MYC (bottom) cells 24 h after transfection. Scale bar, 10 μm.
(E) Percentage of mitotic cells with normal, small, or no spindles 24 h after transfection with siTPX2. Mean ± SEM. Fisher’s exact test, n = 36–126 mitotic cells, 3 independent experiments. See also Figure S5.
*p < 0.05, **p < 0.01, ****p < 0.0001.
We next examined the effects of TPX2 depletion on mitotic spindle regrowth after nocodazole washout (Figure 6D). Only 4% of mitotic RPE-MYC cells formed a spindle (1.5% formed normal spindles and 2.5% formed small spindles) 24 h after TPX2 knockdown compared to 45% of mitotic RPE-NEO cells (10.4% normal spindles and 34.8% small spindles) (Figures 6D and 6E). We next examined spindle structures with confocal microscopy in an asynchronous growing cell population following TPX2 depletion (Figures S5C–S5E). None of the mitotic RPE-MYC cells formed a normal-appearing spindle: 17.4% formed a small spindle, while the remainder of the cells did not form any spindle 18 h after TPX2 depletion (Figure S5D). RPE-NEO cells were affected to a lesser extent: 81.25% formed a small spindle and only 6.25% did not form a spindle (Figure S5D). Thus, our data suggest that MYC-high cells rely on high levels of TPX2 to efficiently build the mitotic spindle. In contrast, non-transformed, low-MYC cells are less sensitive to TPX2 depletion, as even very low TPX2 levels appear to be sufficient for spindle assembly and progression through mitosis.
High TPX2 Expression Is Protective for Cells that Overexpress MYC
We next sought to determine whether the observed high levels of TPX2 cause spindle-formation defects, or rather, whether they are necessary for the survival of MYC-high cells. We tested several doxycycline-inducible shRNAs against TPX2 in RPE-MYC cells and identified one that partially lowers TPX2 expression to a level similar to that of RPE-NEO cells and that did not induce appreciable cell death (Figures 7A and 7B). To assess the consequences of partial TPX2 depletion, we tested time in mitosis, frequency of micronuclei formation, and spindle assembly. Partially lowering TPX2 levels to endogenous levels in RPE-MYC cells further increased the time in mitosis from 39 min in control cells to 67 min in TPX2-depleted cells (Figure 7C). It also increased the percentage of micronucleated cells from 7.2% to 13.7%, indicating increased CIN (Figure 7D). Whereas partially lowering TPX2 did not affect the number of microtubule nucleation sites after nocodazole washout, it did delay chromosome alignment and centrosome positioning. At 90 min after washout, <5% of cells had aligned chromosomes compared to 32% of control cells, and centrosomes were positioned closer together (Figures 7E–7H). These data indicate that lowering TPX2 expression to endogenous levels does not rescue MYC-induced mitotic defects, but rather worsens the defects.
Figure 7. TPX2 Overexpression Is Necessary for MYC-High Cells to Progress through Mitosis.
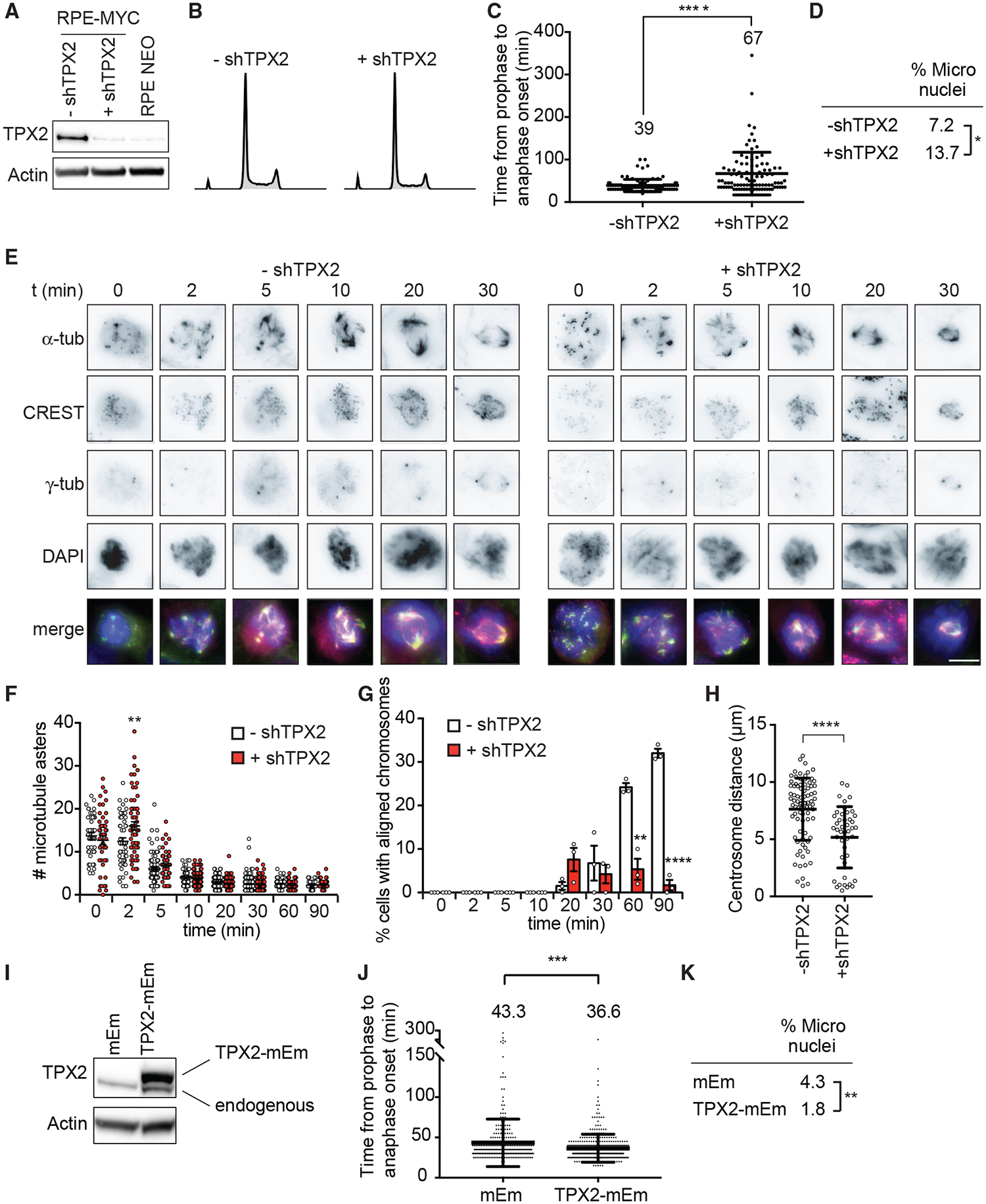
(A–H) RPE-MYC cells expressing doxycycline-inducible shRNA against TPX2 in the absence (−shTPX2) and presence (+shTPX2) of doxycycline for 4 days.
(A) Western blot analysis of TPX2.
(B) Cell-cycle profiles.
(C) Time from nuclear envelope breakdown to anaphase onset quantified from time-lapse microscopy experiments. Mean ± SD. t test, n = 164 and 164, 3 independent experiments.
(D) Percentage of micronucleated cells. Fisher’s exact test, n = 223 and 182, 3 independent experiments.
(E–H) Nocodazole wash-out assay.
(E) Representative images. Scale bar, 10 μm.
(F) Number of microtubule asters. Mean ± SEM. Unpaired t test, n = 45–117, 3 independent experiments.
(G) Percentage of cells with aligned chromosomes. Mean ± SEM. Fisher’s exact test, n = 45–117, 3 independent experiments.
(H) Centrosome distance 90 min after washout. Mean ± SEM. Unpaired t test, n = 20–84, 3 independent experiments.
(I–K) RPE-MYC cells expressing TPX2-mEmerald (TPX2mEm) or empty vector (mEm).
(I) Western blot of TPX2. Exogenous TPX2-mEm is shifted upward.
(J) Time from nuclear envelope breakdown to anaphase onset from time-lapse microscopy experiments. Mean ± SD. t test, n = 307 and 383, 3 independent experiments.
(K) Percentage of micronucleated cells. Fisher’s exact test, n = 1,579 and 2,008, 3 independent experiments.
*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We hypothesized that high TPX2 expression is required for the completion of mitosis and survival of MYC-high cells. To test this hypothesis, we asked whether further increasing TPX2 levels in MYC-high cells rescues aspects of mitotic progression and CIN. We transduced RPE-MYC cells with a lentivirus to stably express a TPX2-mEmerald fusion protein (TPX2-mEm) or a lentivirus carrying the empty mEmerald vector (mEm) (Figure 7I). Overexpression of TPX2-mEm partially rescued the mitotic delay caused by MYC overexpression (Figure 7J) and reduced the frequency of micronucleated cells, indicating that additional TPX2 reduces CIN (Figure 7K). In summary, TPX2 overexpression in MYC-high cells prevents rather than causes mitotic spindle defects, thus protecting cells from deleterious amounts of CIN and cell death.
DISCUSSION
We show that MYC overexpression reversibly induces and maintains CIN, providing insights into how MYC contributes to aneuploidy, tumorigenesis, and tumor evolution. MYC affects various aspects of spindle assembly by deregulating multiple mitotic spindle genes, many or all of which may contribute to the observed defects. For example, motor proteins, such as Eg5 or KIF15, facilitate microtubule aster coalescence and spindle pole separation; STMN1 and KIF2C regulate microtubule dynamics; and the centromeric protein CENP-F is important for kinetochore-microtubule attachment formation (Musinipally et al., 2013; Talapatra et al., 2015; Tanenbaum et al., 2009; Wittmann et al., 2004). Further studies are needed to evaluate which of these genes contribute to the mitotic spindle defects of MYC-high cells.
Here, we show that high TPX2 expression is required for spindle assembly of MYC-high cells. Several mitotic functions of TPX2 are mediated by its role as an Aurora kinase A (AURKA) activator (Bayliss et al., 2003; Eyers et al., 2003; Kufer et al., 2002). In fact, AURKA inhibition was shown to be synthetic-lethal with MYC; however, MYC degradation in interphase rather than a mitotic role has been proposed to cause cell death (Dauch et al., 2016; Gustafson et al., 2014). TPX2 also recruits Eg5 to microtubules, where it regulates microtubule density and spindle length. TPX2 also acts as a scaffold protein for the chromosomal passenger complex, where it interacts with survivin (BIRC5) and activates AURKB (Aguirre-Portolés et al., 2012; Bird and Hyman, 2008; Iyer and Tsai, 2012). To understand which functions of TPX2 are important for the observed MYC-TPX2 dependency, expression of TPX2 mutants with distinct effects on spindle assembly and mitotic function would be an interesting area for future research. Besides spindle-associated genes, MYC also deregulates the expression of apoptotic proteins that may influence the cellular response to the loss of TPX2 (Goga et al., 2007; Horiuchi et al., 2012; Topham et al., 2015).
The reversibility of MYC-induced spindle abnormalities indicates that MYC overexpression not only initiates but also maintains CIN. Increased MYC expression has been observed in early tumor metastasis (Lawson et al., 2015) and drug-resistant cancers (Singleton et al., 2017), situations that are associated with CIN and tumor evolution. Several small-molecule inhibitors that modulate MYC transcriptional activity are undergoing preclinical development and evaluation (Delmore et al., 2011; Horiuchi et al., 2014; Struntz et al., 2019; Wang et al., 2013; Zeng et al., 2018). We postulate that the inhibition of MYC and thereby CIN may be a useful strategy to prevent tumor evolution, drug resistance, and tumor relapse. Since TPX2 protein structure is largely intrinsically disordered, the development of anti-cancer drugs to directly inhibit TPX2 activity may pose a challenge (Zhang et al., 2017). Identifying other synthetic-lethal dependencies between MYC and spindle-associated genes should lead to further understanding of MYC-induced mitotic spindle stress and may permit the development of novel treatment strategies for MYC-overexpressing cancers. For example, inhibitors against Eg5 and other mitotic proteins are available and are being tested in clinical trials (Dominguez-Brauer et al., 2015; Owens, 2013). Finally, we propose that the expression of MYC and TPX2 in tumors may be a useful biomarker to stratify patients for new anti-mitotic therapies.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Lead Contact Andrei Goga (andrei.goga@ucsf.edu). Further information and requests for resources and reagents should be directed to and will be fulfilled by Andrei Goga. All unique/stable reagents (e.g., plasmids and cell lines) generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement, as needed.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In Vivo animal studies
All protocols described in this section regarding mouse studies were approved by the UCSF Institutional Animal Care and Use Committee. The study complied with all relevant ethical regulations. The ethical end point for tumor experiments was reached when a tumor reached ≥ 2 cm in any single dimension.
MTB-TOM (MMTV-rtTA/TetO-MYC) mice (D’Cruz et al., 2001) were bred and maintained off of doxycycline. At 12–15 weeks of age, female mice were fed doxycycline chow (200 mg/kg doxycycline, Bio-Serv) to induce MYC expression and tumorigenesis. Mice were monitored daily for tumor growth by inspection and caliper measurement in two dimensions. For RNA sequencing, when tumors reached 1 cm in any single dimension, tumors were either flash-frozen in liquid nitrogen or mice were taken off doxycycline for three days to switch off MYC expression. Littermates were randomly assigned to experimental groups. All samples used to generate patient-derived xenograft (PDX) tumors were obtained from DeRose et al. (2011). For cell line xenografts, HCC1143 and BT549 cells expressing Tet-pLKO-puro-shTPX2 no. 1 or Tet-pLKO-puro-shGFP were used. Xenografts were generated as follows: 3 mm3 PDX tumor chunks or 1 × 106 cells were transplanted into the cleared mammary fat pads of 4-week-old female NOD/SCID mice (Taconic Biosciences). For cell line xenograft studies, once the tumors reached 1 cm3, mice were fed doxycycline chow (200 mg/kg, Bio-Serv) to induce shTPX2 or shGFP expression. Tumor growth was monitored three times a week by caliper measurement in two dimensions. Mice were euthanized after 30 d of treatment or after tumors reached 2 cm in any dimension. Tumor volume was normalized to the volume before shRNA induction (Figures 5F and 5G).
Cell lines
The female breast cancer cell lines MDA-MB-231 and LY2 were grown in DMEM supplemented with 10% FBS and 10 U/ml penicillin, 10 mg/ml streptomycin at 37°C. HCC1143, BT549, HCC3153, HCC1428, T47D, ZR75B breast cancer cell lines were grown in RPMI supplemented with 10% FBS and 10 U/ml penicillin, 10 mg/ml streptomycin at 37°C. RPE-NEO and RPE-MYC (female) were grown in DMEM supplemented with 10% FBS and 10 U/ml penicillin, 10 mg/ml streptomycin at 37°C (Goga et al., 2007). RPE-NEO were not used beyond passage 16. Primary human mammary epithelial cells expressing MYC-ER and an shRNA targeting the p16 isoform-encoding sequence of CDKN2A (HMEC) were cultured as described (Horiuchi et al., 2016). Although expression of p16 shRNA delays senescence, the cells are not immortalized and undergo spontaneous senescence when continuously cultured. Therefore, cells were not used beyond 12 passages after derivation. HMEC cells were treated with 500 nM 4-hydroxytamoxifen (TAM) to induce MYC activation. T cell lymphoma cells from the conditional bi-transgenic model (Felsher and Bishop, 1999b) were infected with recombinant retrovirus expressing pMIG-BCL2-GFP, sorted for GFP expression and cultured in DMEM supplemented with 10% FBS and 10 U/ml penicillin, 10 mg/ml streptomycin at 37°C (Goga et al., 2007). Rat1 a cells were grown in DMEM supplemented with 10% FBS and 10 u/ml penicillin, 10 mg/ml streptomycin at 37°C as previously described (Schwinkendorf and Gallant, 2009).
All cell lines were continuously tested negative for mycoplasma contamination using PCR.
Generation and characterization of the MTB-TOM cell line
A tumor from an MTB-TOM (MMTV-rtTA/TetO-MYC) mouse was removed, weight determined, chopped and incubated in 5 ml/g resuspension medium (RPMI 1640 supplemented with 10 mM HEPES pH 7.4, 2.5% FBS, 1 μg/ml doxycycline and 1 mg/ml collagenase IV (Sigma)) for one hour at 37°C and 200 rpm shaking. Then, the cells were washed four times with wash medium (RPMI 1640 supplemented with 10 mM HEPES and 2.5% FBS) with centrifugation at 1200 rpm for 2 min and 3 quick spins (1200 rpm for 5 s). Cells were plated into PyMT medium (DMEM/F12 supplemented with 5 μg/ml Insulin, 1 μg/ml Hydrocortisone, 10 ng/ml EGF, 10% FBS, 10 U/ml penicillin, 10 mg/ml streptomycin, 50 μg/ml Gentamycin, 2 mM Glutamine, 10 mM HEPES pH 7.4, 1 μg/ml doxycycline) at 37°C. 1×106 cells were passaged every 3 days. After passage 10, cells were grown in DMEM (DMEM, 10% FBS, 10 U/ml penicillin, 10 mg/ml streptomycin) supplemented with 1 μg/ml doxycycline for another 10 passages. To switch off MYC expression, cells were grown in the absence of doxycycline for three days. In the absence of doxycycline, MTB-TOM cells continued to proliferate with a slightly decreased proliferation rate and a small amount of cell death suggesting that the effects of MYC overexpression are reversible (Figures S1B and S1C). Reportedly, withdrawal of doxycycline leads to an increase in endogenous MYC in MTB-TOM tumors (D’Cruz et al., 2001). Thus, we determined whether turning off expression of the human MYC transgene results in a corresponding increase in expression of the endogenous mouse MYC gene. Upon doxycycline withdrawal, endogenous murine MYC expression increased over time, detectable by qPCR and as a slightly upward shifted band by western blot that was shown to correspond to mouse MYC (Figures S1C and S1D) (Lehmann et al., 2012). We did not exceed three days incubation without doxycycline for the analysis of these cells to ensure low endogenous MYC expression.
METHOD DETAILS
Lentiviral constructs and cloning
The FUCCI plasmids pCDII-EF-MCS containing mKO2-hCdt1(30/120) and mAG-hGeminin(1/110) have been described previously (Sakaue-Sawano et al., 2008) (GenBank accession number AB370332 and AB370333). H2B-mCherry pLenti6/V5-DEST has been described previously (Pemble et al., 2017). For inducible shRNA expression, the following short hairpin sequences were cloned into the Tet-PLKO-puro vector as published (Wiederschain et al., 2009): shTPX2 no. 1: 5′-CCGGGAACAATCCATTCCGTCAAATCT CGAGATTTGACGGAATGGATTGTTCTTTTT –3′; shTPX2 no. 2: 5′- CCGGCTAATCTTCAGCAAGCTATTGCTCGAGCAATAGCTTG CTGAAGATTAGTTTTT –3′; shTPX2 no.3: 5′- CCGGTCCAGACCTTGCCCTACTAAGCTCGAGCTTAGTAGGGCAAGGTCTGGATTTTT –3′. For control shRNA against GFP with the following sequence was used: 5′-CCGGTACAACAGCCACAACGTCTATCTCGACATAGACGTTGTGGCTGTTGTATTTTTG –3′. For overexpressing TPX2 a shRNA-resistant (shRes) human TPX2 mutant was generated by overlapping PCR using the following primers to insert the indicated silent mutations into the shRNA target sequence: forward primer 5′ -CAGAAAAAGAAAATCTTGTGGAGCAATCTATCCCATCAAACGCTTGTTCTTCCCTGGAAG- 3′, reverse primer 5′ -CTTCCAGGGAAGAACAAGCGTTTGATGGGATAGATTGCTCCACAAGATTTTCTTTTTCTG- 3′. The shRes TPX2 mutant was cloned into pHR-mEmerald using MluI and BamHI restriction sites.
Lentiviral infection
293T cells were grown in DMEM medium with high glucose supplemented with 10% FBS and penicillin/streptomycin. Virus was produced in 10 cm plates and 1 mL supernatant with 8 μg/ml polybrene was used to infect 100,000 cells for 12 hours. Cells were selected for five days with 0.5 μg/ml puromycin. Cells expressing a fluorescent protein (FUCCI, H2B-mCherry and TPX2-mEmerald) were sorted using fluorescence associated cell sorting on a BD FACSAria II flow cytometer (Becton Dickinson).
Transfection and siRNA treatment
Cells were transfected using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher), according to the manufacturer’s instructions. All siRNAs were purchased from GE Dharmacon (ON-TARGETplus, four siRNAs per gene). For screening spindle genes in RPE-NEO and RPE-MYC (Figure 4A), 50 nM siRNA was used. For any other TPX2 knock down experiment 1.7 nM siRNA was used. For MYC knock down 30 nM siRNA was used. To verify specificity of the MYC knock down, the single siRNA sequences J-003282-23-0002 and J-003282-24-0002 were used (Dharmacon).
MTB-TOM growth curve and quantitative PCR
To determine the effects of c-Myc loss on MTB-TOM cell proliferation, cells were seeded in 6-well plates at 7.5 × 104 (Dox-On) or 2 × 105 (Dox-Off) cells/well. Media and doxycycline (1 μg/ml) or vehicle were replaced daily, and cells were harvested at 24, 48, 72 h. Cell counts were determined using the Countess Automated Cell Counter (Life Technologies) according to the manufacturer’s instructions.
For quantitative PCR, 2 × 105 cells were seeded into a 6-well plate in presence of 1 mg/ml doxycycline or vehicle. RNA was purified using the RNeasy kit (QIAGEN). 1 μg total RNA was used for reverse transcription using the iScript cDNA synthesis kit (Bio-Rad). Relative expression of human c-Myc, mouse c-Myc, and mouse GAPDH was analyzed using a SYBR Green Real-Time PCR kit (Thermo Fisher) with an Applied Biosystems QuantStudio 6 Flex Real-Time PCR System thermocycler (Thermo Fisher). Using GAPDH mRNA levels as an internal loading control, variation was determined using the DDCT method (Livak and Schmittgen, 2001). The following primers were used: Human MYC forward 5′-GGCTCCTGGCAAAAGGTCA, human MYC reverse 5′- CTGCGTAGTTGTGCTGATGT, mouse MYC forward 5′- ATGCCCCTCAACGTGAACTTC, mouse MYC reverse 5′- GTCGCAGATGAAATAGGGCTG, mouse GAPDH forward 5′-AGGTCGGTGTGAACGGATTTG and mouse GAPDH reverse 5′-TGTAGACCATGTAGTTGAGGTCA.
Immunostaining
Mitotic spindle staining and quantification of spindle defects
Cells were grown on glass coverslips (Fisherbrand), fixed with 100% methanol at −20°C for 3 min, rehydrated with Tris-buffered sa-line supplemented with 0.1% Tween 20 (TBS-T), washed three times with TBS-T for 5 minutes, blocked with antibody diluting solution (Abdil; 2% BSA, 0.1% sodium azide in TBS-T) for 20 minutes and incubated with primary antibodies diluted in Abdil for one hour. Then, cells were washed three times with TBS-T, incubated with secondary antibodies in Abdil for 30 minutes, washed three times with TBS-T, incubated with 4’,6-diamidino-2-phenylindole (DAPI, 1:10,000) and mounted with ProLong Gold reagent (Invitrogen). The following antibodies were used as indicated: mouse anti-α-tubulin DM1 (1:1,000, T6199, Sigma), human anti-centromere (CREST, 1:50, 15–234 Antibodies Incorporated), rabbit anti-γ-tubulin (1:1,000, T3559, Sigma), rabbit anti-TPX2 (1:500, HPA005487, Sigma), mouse anti-centrin 20H5 (1:300, 04–1624, Millipore). Mouse and human secondary antibodies conjugated to Alexa Fluor 488 and 594, respectively (1:1,000, A11029 and A11014, Thermo Fisher) and a rabbit secondary antibody conjugated to Alexa Fluor 647 (1:1,000, ab150079, Abcam) were used.
For quantification of mitotic spindle defects, mitotic cells with CREST-positive (kinetochore containing) lagging and misaligned chromosomes were counted and the percentage of total mitotic cells calculated. For MTB-TOM cells, lagging chromosomes were quantified from time-lapse microscopy experiments of H2B-mCherry expressing cells.
Mad1 staining
Cells were permeabilized in PHEM (120 mM PIPES, 50 mM HEPES, 20mM EGTA, 4 mM Magnesium Acetate) supplemented with 1% Triton-100 for 15 s, fixed with 3% paraformaldehyde in PHEM for 15 minutes at 37°C, washed four times with PHEM supplemented with 0.05% Triton-100, incubated with PHEM supplemented with 1% Triton-100 for 10 minutes, washed twice with PHEM supplemented with 0.05% Triton-100, blocked with PHEM supplemented with 0.05% Triton-100 and 2% BSA for one hour and incubated with mouse anti-MAD1 BB3–8 (1:300, MABE867, Millipore) and human anti-centromere (CREST, 1:50, 15–234 Antibodies Incorporated) diluted in PHEM supplemented with 0.05% Triton-100 and 2% BSA for one hour. Then, cells were washed four times with PHEM supplemented with 0.05% Triton-100 and incubated with mouse and human secondary antibodies conjugated to Alexa Fluor 488 and 594, respectively (1:1,000, A11029 and A11014, Thermo Fisher) for one hour, washed four times and mounted with ProLong Gold reagent (Thermo Fisher).
Microtubule regrowth assay
Cells were seeded onto 12 mm coverslips into 24-well plates at 100,000 cells per well. To depolymerize microtubules, cells were incubated with 0.2 μg/ml nocodazole (M1404, Sigma) for 5 to 7 hours. Coverslips were drained on tissue and washed by placing into 5 mL prewarmed media without nocodazole. Cells were fixed at indicated time points after nocodazole wash-out and stained with anti-α-tubulin (α-tub), anti-γ-tubulin (γ-tub, centrosome), anti-CREST (kinetochores) and DAPI (DNA). α-tubulin positive microtubule foci and asters were counted manually. Co-localization with γ-tubulin indicated centrosomal localization. Centrosomal distance was manually measured using Fiji if positioned in the same z-plane. For quantifying cells with aligned chromosomes, all cells that started to align their chromosomes plus cells with metaphase plates were included. Monopolar spindles were not included in the quantification. Microtubule aster length was manually determined from confocal images using Fiji.
Microscopy
Time-lapse microscopy
For live-cell analyses, cells were seeded into 12 well plates at 50,000 cells/well and followed by time-lapse microscopy at 37°C and 5% CO2. Images were acquired on an inverted microscope (Nikon Eclipse Ti), operated by NIS-Elements software and equipped with a CoolSNAP HQ2 CCD camera (Photometrics). Cells were imaged with phase contrast (20–100 ms exposure), 488 nm or 594 nm laser light (75–200 ms exposure) through a 20× 0.45 Ph1 objective using perfect focus every 7–10 min, in a stage-top incubation chamber (Okolab) maintained at 37°C and 5% CO2. The time from nuclear envelope breakdown (loss of nuclear FUCCI) or chromosome condensation (H2B-mCherry/bright field) until the beginning of anaphase (start of chromosome movement to the poles) or cell death was determined. Graphs were generated using the Prism software package, version 7 (GraphPad Software).
Fluorescence microscopy
Fixed cells were imaged with exposure times of 5–200ms with DAPI, GFP, TRITC, and CY5 filter cubes and a mercury arc lamp on a Zeiss AxioPlan2 epifluorescence microscope (operated by MicroManager 1.4.13) with a 40× 1.3 DIC oil objective and a QIClick camera (QImaging). Images were recorded with a Z-optical spacing of 0.2 μm and analyzed using the Fiji software (Schindelin et al., 2012).
Confocal microscopy
TPX2 immunofluorescence was imaged at 37°C with a 100× NA 1.49 objective lens (CFI APO TIRF; Nikon) on an inverted microscope system (TE2000 Perfect Focus System; Nikon) equipped with a Borealis modified spinning disk confocal unit (CSU10; Yokogawa) with 200-mW, 405 nm, 488 nm, 561 nm and 643 nm solid-state lasers (LMM5; Spectral Applied Research), electronic shutters, a Clara cooled scientific-grade interline CCD camera (Andor), and controlled by NIS-Elements software (Nikon).
Imaging of multipolar spindles and nocodazole wash-out was performed on an inverted (Eclipse Ti-E; Nikon, Tokyo, JPN), spinning disk confocal (CSU- X1; Yokogawa Electric Corporation, Tokyo, JPN) microscope. 4-color fixed imaging was performed with a Di01-T405/488/568/647–13×15×0.5 (Semrock, Lake Forest, IL) head dichroic along with four diode laser lines [405 nm (100mW), 488 nm (150 mW), 561 nm (100 mW), and 642 nm (100 mW)], different emission filters [ET460/50 m, ET525/50M, ET630/75M, and ET690/50M (Chroma, 323 Bellows Falls, VT)], and a Zyla camera (Andor Technology, Belfast, UK; bin = 1, 65.7 nm/pixel). Fluorescent cell images were acquired in z-planes spaced 200–500 nm apart, and laser powers and exposures optimized (but acquisition settings were kept constant across all coverslips in a single experiment) with a 100 × 1.45 Ph3 oil objective through a 1.5 × lens (Metamorph 7.7.8.0; Molecular Devices, San Jose, CA).
Western blotting
Cells were lysed in Laemmli buffer (60 mM Tris-HCl, pH 6.8, 1 μM DTT, 2% (w/v) SDS) supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Roche). Protein concentration was determined using the DC Protein Assay (BioRad). 30 μg Protein extracts were resolved using 4%–12% SDS-PAGE gels (Life Technologies) and transferred to nitrocellulose membranes using iBlot (Life Technologies). Membranes were probed with primary antibodies overnight on a 4°C shaker, then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies, and signals were visualized with ECL (Bio-Rad). The following primary antibodies were used: Anti-β-actin (actin) (1:10,000, sc-47778 HRP, Santa Cruz Biotechnology), as loading control, Anti-c-MYC (MYC) (1:1,000, ab32072, Abcam), Anti-TPX2 (1:1,000, HPA005487, Sigma), Anti-cleaved PARP (1:1000, 9542, Cell signaling), Anti-Cyclin A (1:1,000, sc-596, Santa Cruz Biotechnology), Anti-Cylin B1 (1:1,000, ab2949, abcam). Protein levels were quantified by normalizing to b-actin using ImageJ.
Quantification of TPX2 and MYC protein levels (Figure 4B)
To quantify TPX2 and MYC protein level, band intensities were measured using Fiji, and normalized to β-actin from at least three independent experiments. To compare between different blots, TPX2 and MYC levels were normalized to RPE-NEO. The correlation coefficient was computed using Pearson correlation.
Cell viability assays
To screen spindle genes in RPE-NEO and RPE-MYC, cells were seeded in 12-well plates at 50,000 cells/well and transfected with 50 nM siRNA as described above or treated with 10 μM of the CDK1 inhibitor Purvalanol A (P4484, Sigma) (Figure 4A). Cells were harvested after 72 h and cell viability was assessed by performing the flow cytometry-based Guava ViaCount Viability Assay (4000–0040, Millipore) according to the manufacturer’s instructions. For further validation of the effects of siTPX2 treatment, cells were seeded in 6-well plates at 100,000 cells per well and transfected with 1.7 nM siRNA as described above. Cells were harvested at 72 hours and cell viability determined using the PrestoBlue Cell Viability Reagent (A13261, Thermo Fisher) (Figure 5E) or the Countess Automated Cell Counter and Trypan Blue Stain (0.4%) (Invitrogen) (Figure 5D) according to the manufacturer’s instructions.
Cell cycle profiles
To determine cell cycle profiles after siTPX2 treatment RPE-NEO and RPE-MYC cells were seeded in 6-well plates at 100,000 cells per well and transfected with 1.7 nM siRNA as described above. Cells were harvested after 48 hours, trypsinized, washed with PBS and fixed in 95% ethanol overnight at 4°C. Fixed cells were collected by centrifugation at 5000 rcf. at 4°C. The pellet was washed twice with PBS, then incubated in staining solution (80 μg/ml propidium iodide, 150 μg/ml and RNaseA, 1% Tween 20 in PBS) for one hour. Cells were centrifuged at 5000 rcf. at 4°C and the pellet was resuspended in 300 μL PBS. Flow cytometry was performed on a BD LSRII flow cytometer (Becton Dickinson) and at least 20,000 events were counted. Data were analyzed using FlowJo 2 (FlowJo, LLC).
ChIP-qPCR
RPE-NEO and RPE-MYC cells were grown to 80% confluency on 15 cm2 dishes. On the day of harvest, cells were fixed on the dish with 1% formaldehyde in PBS for 8 minutes at room temperature. The reactions were quenched with 0.125 M glycine for 5 minutes. The cells were then washed with 1X PBS, twice, and lifted off in cell lysis buffer (10 mM Tris-HCl pH 8.1, 10 mM NaCl, 1.5 mM MgCl2,0.5% NP-40), supplemented with protease inhibitors (Roche). The cell lysate was then incubated for 15 minutes on ice and crude nuclear extracts were collected by centrifugation at 600 rcf. for 5 min at 4°C. The pellet was resuspended in 0.5 mL of nuclear lysis buffer (50 mM Tris-HCl pH 8.1, 5 mM EDTA, 1% SDS) supplemented with protease inhibitors. The chromatin was fragmented with a Diagenode Bioruptor (20 cycles of 30 s on followed by 30 s off, at 4°C). To remove insoluble components, the samples were centrifuged at 13,000 rcf. for 15 min at 4°C and the supernatant was recovered. The supernatant was diluted 1:1 with dilution buffer (16.7 mM Tris-HCl pH 8.1, 1.1% Triton X-100, 0.01% SDS, 167 mM NaCl, 1.2 mM EDTA) supplemented with protease inhibitors and quantified by DC protein assay (Biorad). For each sample, 20 uL of protein A Dynabead magnetic beads (Invitrogen) was added to 500 ug of protein in 500 uL of dilution buffer and incubated with 3 ug of MYC [Y69] antibody (Abcam, ab32072) or no antibody control overnight at 4°C. The next day, samples were washed with each of the following buffers, once, in the order of: low salt (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris HCl pH 8.1, 150 mM NaCl), high salt (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris HCl pH 8.1, 500 mM NaCl), LiCl (1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris-HCl pH 8.1, 250 mM LiCl), and TE (10 mM Tris-HCl pH 8.1, 1 mM EDTA). ChIP DNA was eluted off the beads by incubating beads in 100 uL elution buffer (10 mM Tris-HCl pH 8.1, 1 mM EDTA, 1% SDS, 150 mM NaCl), supplemented with DTT at a final concentration of 5 mM, for 10 minutes at 65°C. The supernatant was removed, and the ChIP DNA was eluted a second time in the same fashion. The combined supernatant was then incubated overnight at 65°C to reverse crosslinks and proteinase K treated for 1 hour the next morning. Samples were purified using a ChIP DNA purification kit (Active Motif) per manufacturer’s protocol.
ChIP samples were diluted 1:10 and used as template in the Power Up Sybr Master Mix (Invitrogen) and DNA was amplified using the QuantStudio 6 & 7 Flex Real-Time PCR system. The data was analyzed using the fold enrichment method. The following primer sequences were used: negative control LDHA Forward: 5′-GGGCCACCGAATGCTC-3′ and Reverse: 5′-AATGGCCCTGGCTGCAG-3′; positive control LDHA Forward: 5′-TCCTGACTCAGGCTCATGGC-3′ and Reverse: 5′-AGACAACCGACCGGCAGA-3′ (Kim et al., 2004). For TPX2, primer was designed around the transcription start site. Forward primer 5′-GAGC ACTCCGGTCTTTGTAATA-3′ and Reverse primer 5′-CAATACCGGAAGTCAGAAGGAG-3′.
RNA sequencing and gene expression analysis
MTB-TOM tumors from 10 mice that were fed doxycycline (MYC ON), mammary glands from three mice that were never fed doxycycline and MTB-TOM tumors from five mice that were taken off doxycycline for 72 hours (MYC OFF) were flash-frozen in liquid nitrogen. The Eμ-tTX/TRE-MYC lymphoma cell line was grown in the absence of doxycycline (MYC ON) and in the presence of doxycycline for three days (MYC OFF) as previously described and flash frozen using liquid nitrogen (Felsher and Bishop, 1999b; Goga et al., 2007). Three biological replicates were performed. RNA was isolated using the RNAeasy kit (QIAGEN). Library preparation and Illumina RNA sequencing was performed by Q2 Solutions (https://www.q2labsolutions.com). Sequencing reads were mapped to the mouse transcriptome (mm9 for lymphoma cells, GRC38/mm10 for MTBTOM) using STAR aligner (Dobin et al., 2013). The read count was used for differential gene expression (DGE). DGE analysis of Eμ-tTX/TRE-MYC cells MYC OFF compared to MYC ON was processed using the edgeR package (Robinson et al., 2010). DGE analysis of MTB-TOM tumors MYC ON compared to normal mammary gland, and MYC OFF tumors compared to MYC ON tumors was performed using the DESeq2 package (Love et al., 2014). The human TCGA breast-invasive carcinoma dataset was sourced from the TCGA Research Network (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga), made available on the University of California, Santa Cruz (UCSC) Cancer Browser. DGE analysis of triple-negative compared to receptor positive breast cancer excluding normal tissue was calculated using the limma R package and as described in Camarda et al. (2016). DGE of MYC-driven liver mouse tumor compared normal liver was sourced from Anderton et al. (2017). A gene set was compiled containing genes associated with the Gene Ontology terms kinetochore, microtubule, mitotic spindle and mitosis with log2 fold change ≥ 1 or ≤ −1 and a false discovery rate (FDR) ≤ 0.01 (Table S2).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are shown as mean ± standard deviation (SD) or standard error of the mean (SEM) as indicated. For comparisons, unpaired two-sided Student’s t test for continous data or Fisher’s exact test on raw counts were applied using the Prism software (GraphPad) as indicated. All cell-based experiments were independently repeated at least three times. Statistical significance is indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. No statistical method was used to pre-determine sample size throughout this study. The investigators were not blinded to allocation for the in vivo experiments.
DATA AND CODE AVAILABILITY
RNA sequencing data from MTB-TOM tumors and Eμ-tTA/TRE-MYC lymphoma cells were deposited in NCBI GEO under accession number GEO: GSE130922.
Secondary accessions: the human breast cancer RNA-sequencing datasets are derived from the TCGA Research Network (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) and are available from GEO: GSE62944. Gene expression values for the MYC-driven liver tumors are available from GEO: GSE73295.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-α-tubulin, clone DM1 | Sigma | Cat# T6199; RRID:AB_477583 |
| Human anti-centromere | Antibodies Incorporated | Cat# 15–234; RRID:AB_2687472 |
| Rabbit anti-γ-tubulin | Sigma | Cat# T3559; RRID:AB_477575 |
| Mouse anti-centrin, clone 20H5 | Millipore | Cat# 04–1624; RRID:AB_10563501 |
| Rabbit anti-TPX2 | Sigma | Cat# HPA005487; RRID:AB_1858223 |
| Mouse anti-MAD1, clone BB3–8 | Millipore | Cat# MABE867; RRID:AB_831526 |
| Anti-β-actin-HRP | Santa Cruz | Cat# sc-47778; RRID:AB_2714189 |
| Anti-c-MYC, clone Y69 | Abcam | Cat# ab32072; RRID:AB_731658 |
| Anti-cleaved PARP | Cell Signaling Technology | Cat# 9542; RRID:AB_2160739 |
| Rabbit anti-Cyclin B1 | Abcam | Cat# ab2949 |
| Rabbit anti-Cyclin A, clone C-19 | Santa Cruz Biotechnology | Cat# sc-596; RRID:AB_631330 |
| Anti-mouse conjugated to Alexa Fluor 488 | Thermo Fisher | Cat# A11029; RRID:AB_138404 |
| Anti-human conjugated to Alexa Fluor 594 | Thermo Fisher | Cat# A11014; RRID:AB_2534081 |
| Anti-rabbit conjugated to Alexa Fluor 647 | Abcam | Cat# ab150079; RRID:AB_2722623 |
| Biological Samples | ||
| Patient-derived xenogafts (PDX) | DeRose et al., 2011 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nocodazole | Sigma | Cat# M1404 |
| Purvalanol A | Sigma | Cat# P4484 |
| Propidium iodide | Sigma | Cat# P4170 |
| RNase A | Sigma | Cat# R6513 |
| Critical Commercial Assays | ||
| PrestoBlue Cell Viability Reagent | Thermo Fisher | Cat# A13261 |
| Guava ViaCount Viability Assay | Millipore | Cat# 4000–0040 |
| Trypan Blue Stain | Thermo Fisher | Cat# T10282 |
| ChIP DNA purification kit | Active Motif | Cat# 58002 |
| Power Up Sybr Master Mix | Thermo Fisher | Cat# A25741 |
| Deposited Data | ||
| RNA sequencing of Eμ-tTA/TRE-MYC lymphoma cells and MMTV-rtTA/TetO-MYC tumors | This paper | GEO: GSE130922 |
| Gene expression values of LAP-tTA × TetO-MYC (LT2-MYC) liver tumors | Anderton et al., 2017 | GEO: GSE73295 |
| The Cancer Genome Atlas | Cancer Genome Atlas Network, 2012 | GEO: GSE62944 |
| Experimental Models: Cell Lines | ||
| RPE-NEO | Goga et al., 2007 | N/A |
| RPE-MYC | Goga et al., 2007 | N/A |
| MDA-MB-231 | ATCC | HTB-26 |
| HCC1143 | ATCC | CRL-2321 |
| BT549 | ATCC | HTB-122 |
| HCC3153 | Neve et al., 2006 | N/A |
| HCC1428 | ATCC | CRL-2327 |
| T47D | ATCC | HTB-133 |
| LY2 | Neve et al., 2006 | N/A |
| ZR75B | ATCC | CRL-1500 |
| HMEC MYC ER | Horiuchi et al., 2012 | N/A |
| eμ-tTX/TRE-MYC cells | Goga et al., 2007 | N/A |
| MTB-TOM cells | This study | N/A |
| 293T | ATCC | CRL-3216 |
| Rat1a | Goga et al., 2007 | N/A |
| Rat1a myc−/−MYC | Schwinkendorf and Gallant, 2009 | N/A |
| Rat1a myc−/− MYCΔMBII | Schwinkendorf and Gallant, 2009 | N/A |
| RPE-MYC shTPX2 CDS1 | This study | N/A |
| RPE-MYC shTPX2 CDS2 | This study | N/A |
| RPE-MYC shTPX2 CDS3 | This study | N/A |
| RPE-MYC shGFP | This study | N/A |
| RPE-MYC shResTPX2-mEm | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: MMTV-rtTA/TetO-MYC | D’Cruz et al., 2001 | N/A |
| Mouse: NOD/MrkBomTac-Prkdcscid | Taconic Biosciences | N/A |
| Oligonucleotides | ||
| shRNA sequences see Table S3 | This study | N/A |
| Primer for TPX2 shRNA resistant see Table S3 | This study | N/A |
| Primer for qPCR see Table S3 | This study | N/A |
| Primer for ChIP PCR see Table S3 | This study | N/A |
| Recombinant DNA | ||
| pCDII-EF-MCS mKO2-hCdt1(30/120) | Sakaue-Sawano et al., 2008 | GenBank accession# AB370332 |
| pCDII-EF-MCS mAG-hGeminin(1/110) | Sakaue-Sawano et al., 2008 | GenBank accession# AB370333 |
| pLenti6/V5-DEST H2B-mCherry | Pemble et al., 2017 | N/A |
| Tet-PLKO-puro | Wiederschain et al., 2009 | Addgene Plasmid #21915 |
| pHR-mEmerald | Hu and Mullins, 2019 | N/A |
| Software and Algorithms | ||
| Fiji | Schindelin et al., 2012 | https://fiji.sc/ |
| STAR aligner | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| edgeR package | Robinson et al., 2010 | https://doi.org/10.18129/B9.bioc.edgeR |
| DESeq2 package | Love et al., 2014 | https://doi.org/10.18129/B9.bioc.DESeq2 |
| limma R package | Ritchie et al., 2015 | https://doi.org/10.18129/B9.bioc.limma |
Highlights.
MYC overexpression reversibly induces CIN by reprogramming mitotic gene expression
MYC impairs mitotic spindle formation
High TPX2 expression allows cells that overexpress MYC to adapt to spindle stress
TPX2 depletion is synthetic lethal with MYC overexpression
ACKNOWLEDGMENTS
We thank Dr. Torsten Wittmann and Dr. Antonius van Haren for the plasmids and help with microscopy. We thank Dr. J. Michael Bishop for the cell lines and Dr. Stephen Floor for help with Encode analysis. We thank all of the members of the Goga laboratory for critically reading the manuscript. We thank Dr. David Morgan and Ms. Hannah Klein-Connolly for useful discussions and advice. This work was supported by the Susan G. Komen postdoctoral fellowship grant PDF15331114 (to J.R.); NIH F32 CA243548-01 (to J.V.L.); a CIRM Predoctoral Award (to M.T.); the NIH F99/K00 Predoctoral to Postdoctoral Transition Award F99CA212488 (to R.C.); the Breast Cancer Research Foundation (BCRF) (to H.S.R.); NIH DP2 GM119177 and NSF 1548297 (to S.D.); and the CDMRP Breast Cancer Research Program (W81XWH-12-1-0272), Give Breast Cancer the Boot (GBCTB), NIH R01- CA170447, the Gazarian Family Chair, and the Lymphoma Scholar Award (all to A.G.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.041.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aguirre-Portolés C, Bird AW, Hyman A, Cañamero M, Pérez de Castro I, and Malumbres M (2012). Tpx2 controls spindle integrity, genome stability, and tumor development. Cancer Res. 72, 1518–1528. [DOI] [PubMed] [Google Scholar]
- Amati B, Alevizopoulos K, and Vlach J (1998). Myc and the cell cycle. Front. Biosci 3, d250–d268. [DOI] [PubMed] [Google Scholar]
- Anderton B, Camarda R, Balakrishnan S, Balakrishnan A, Kohnz RA, Lim L, Evason KJ, Momcilovic O, Kruttwig K, Huang Q, et al. (2017). MYC-driven inhibition of the glutamate-cysteine ligase promotes glutathione depletion in liver cancer. EMBO Rep. 18, 569–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Thompson SL, Manning AL, and Compton DA (2009). Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. Cell Biol 11, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss R, Sardon T, Vernos I, and Conti E (2003). Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol. Cell 12, 851–862. [DOI] [PubMed] [Google Scholar]
- Bird AW, and Hyman AA (2008). Building a spindle of the correct length in human cells requires the interaction between TPX2 and Aurora A. J. Cell Biol 182, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T (2008). Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci 121 (Suppl 1), 1–84. [DOI] [PubMed] [Google Scholar]
- Bretones G, Delgado MD, and León J (2015). Myc and cell cycle control. Biochim. Biophys. Acta 1849, 506–516. [DOI] [PubMed] [Google Scholar]
- Bush A, Mateyak M, Dugan K, Obaya A, Adachi S, Sedivy J, and Cole M (1998). c-myc null cells misregulate cad and gadd45 but not other proposed c-Myc targets. Genes Dev. 12, 3797–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, et al. (2016). Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med 22, 427–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappellen D, Schlange T, Bauer M, Maurer F, and Hynes NE (2007). Novel c-MYC target genes mediate differential effects on cell proliferation and migration. EMBO Rep. 8, 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carretero M, Ruiz-Torres M, Rodríguez-Corsino M, Barthelemy I, and Losada A (2013). Pds5B is required for cohesion establishment and Aurora B accumulation at centromeres. EMBO J. 32, 2938–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, and Szallasi Z (2006). A signature ofchromosomalinstability inferred from geneexpressionprofiles predicts clinical outcome in multiple human cancers. Nat. Genet 38, 1043–1048. [DOI] [PubMed] [Google Scholar]
- Cavazza T, Malgaretti P, and Vernos I (2016). The sequential activation of the mitotic microtubule assembly pathways favors bipolar spindle formation. Mol. Biol. Cell 27, 2935–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. (2008). Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117. [DOI] [PubMed] [Google Scholar]
- Cimini D (2008). Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim. Biophys. Acta 1786, 32–40. [DOI] [PubMed] [Google Scholar]
- Cimini D, Fioravanti D, Salmon ED, and Degrassi F (2002). Merotelic kinetochore orientation versus chromosome mono-orientation in the origin of lagging chromosomes in human primary cells. J. Cell Sci 115, 507–515. [DOI] [PubMed] [Google Scholar]
- D’Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE, Cox JD, Ha SI, Belka GK, Golant A, et al. (2001). c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat. Med 7, 235–239. [DOI] [PubMed] [Google Scholar]
- Dauch D, Rudalska R, Cossa G, Nault J-C, Kang T-W, Wuestefeld T, Hohmeyer A, Imbeaud S, Yevsa T, Hoenicke L, et al. (2016). A MYC-aurora kinase A protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med 22, 744–753. [DOI] [PubMed] [Google Scholar]
- Dechat T, Shimi T, Adam SA, Rusinol AE, Andres DA, Spielmann HP, Sinensky MS, and Goldman RD (2007). Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc. Natl. Acad. Sci. USA 104, 4955–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. (2011). BET bromodo-main inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose YS, Wang G, Lin Y-C, Bernard PS, Buys SS, Ebbert MTW, Factor R, Matsen C, Milash BA, Nelson E, et al. (2011). Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, and Mak TW (2015). Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 60, 524–536. [DOI] [PubMed] [Google Scholar]
- Eibes S, Gallisà-Suñé N, Rosas-Salvans M, Martínez-Delgado P, Vernos I, and Roig J (2018). Nek9 Phosphorylation Defines a New Role for TPX2 in Eg5-Dependent Centrosome Separation before Nuclear Envelope Breakdown. Curr. Biol 28, 121–129.e4. [DOI] [PubMed] [Google Scholar]
- Eyers PA, Erikson E, Chen LG, and Maller JL (2003). A novel mechanism for activation of the protein kinase Aurora A. Curr. Biol 13, 691–697. [DOI] [PubMed] [Google Scholar]
- Faire K, Waterman-Storer CM, Gruber D, Masson D, Salmon ED, and Bulinski JC (1999). E-MAP-115 (ensconsin) associates dynamically with microtubules in vivo and is not a physiological modulator of microtubule dynamics. J. Cell Sci 112, 4243–4255. [DOI] [PubMed] [Google Scholar]
- Felsher DW, and Bishop JM (1999a). Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc. Natl. Acad. Sci. USA 96, 3940–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsher DW, and Bishop JM (1999b). Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 4, 199–207. [DOI] [PubMed] [Google Scholar]
- Gallaud E, Caous R, Pascal A, Bazile F, Gagné J-P, Huet S, Poirier GG, Chrétien D, Richard-Parpaillon L, and Giet R (2014). Ensconsin/Map7 promotes microtubule growth and centrosome separation in Drosophila neural stem cells. J. Cell Biol 204, 1111–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett S, Auer K, Compton DA, and Kapoor TM (2002). hTPX2 is required for normal spindle morphology and centrosome integrity during vertebrate cell division. Curr. Biol 12, 2055–2059. [DOI] [PubMed] [Google Scholar]
- Goga A, Yang D, Tward AD, Morgan DO, and Bishop JM (2007). Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med 13, 820–827. [DOI] [PubMed] [Google Scholar]
- Greaves M (2015). Evolutionary determinants of cancer. Cancer Discov. 5, 806–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruss OJ, Carazo-Salas RE, Schatz CA, Guarguaglini G, Kast J, Wilm M, Le Bot N, Vernos I, Karsenti E, and Mattaj IW (2001). Ran induces spindle assembly by reversing the inhibitory effect of importin alpha on TPX2 activity. Cell 104, 83–93. [DOI] [PubMed] [Google Scholar]
- Gruss OJ, Wittmann M, Yokoyama H, Pepperkok R, Kufer T, Silljé H, Karsenti E, Mattaj IW, and Vernos I (2002). Chromosome-induced micro-tubule assembly mediated by TPX2 is required for spindle formation in HeLa cells. Nat. Cell Biol 4, 871–879. [DOI] [PubMed] [Google Scholar]
- Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, et al. (2014). Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 26, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heald R, and McKeon F (1990). Mutations of phosphorylation sites in lamin A that prevent nuclear lamina disassembly in mitosis. Cell 61, 579–589. [DOI] [PubMed] [Google Scholar]
- Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, Creasman KJ, Bazarov AV, Smyth JW, Davis SE, et al. (2012). MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med 209, 679–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Anderton B, and Goga A (2014). Taking on challenging targets: making MYC druggable. Am. Soc. Clin. Oncol. Educ. Book, e497–e502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, Corella AN, Eyob H, Kessenbrock K, Lawson DA, et al. (2016). PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nat. Med 22, 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, and Mullins RD (2019). LC3 and STRAP regulate actin filament assembly by JMY during autophagosome formation. J Cell Biol. 218 (1), 251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Wu G, Rusch M, Lukes L, Buetow KH, Zhang J, and Hunter KW (2012). Integrated cross-species transcriptional network analysis of metastatic susceptibility. Proc. Natl. Acad. Sci. USA 109, 3184–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer J, and Tsai M-Y (2012). A novel role for TPX2 as a scaffold and co-activator protein of the Chromosomal Passenger Complex. Cell. Signal 24, 1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar AP (2016). A Cell Biological Perspective on Past, Present and Future Investigations of the Spindle Assembly Checkpoint. Biology (Basel) 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A, Deb-Basu D, Cherry A, Turner S, Ford J, and Felsher DW (2003). Defective double-strand DNA break repair and chromosomal translocations by MYC overexpression. Proc. Natl. Acad. Sci. USA 100, 9974–9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler JD, Kahle KT, Sun T, Meerbrey KL, Schlabach MR, Schmitt EM, Skinner SO, Xu Q, Li MZ, Hartman ZC, et al. (2012). A SUMOylation-dependent transcriptional subprogram is required for Myc-driven tumorigenesis. Science 335, 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Zeller KI, Wang Y, Jegga AG, Aronow BJ, O’Donnell KA, and Dang CV (2004). Evaluation of myc E-box phylogenetic footprints in glycolytic genes by chromatin immunoprecipitation assays. Mol. Cell. Biol 24, 5923–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knouse KA, Lopez KE, Bachofner M, and Amon A (2018). Chromosome Segregation Fidelity in Epithelia Requires Tissue Architecture. Cell 175, 200–211.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufer TA, Silljé HHW, Körner R, Gruss OJ, Meraldi P, and Nigg EA (2002). Human TPX2 is required for targeting Aurora-A kinase to the spindle. J. Cell Biol 158, 617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn J, and Dumont S (2017). Spindle assembly checkpoint satisfaction occurs via end-on but not lateral attachments under tension. J. Cell Biol 216, 1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang C-Y, et al. (2015). Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526, 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann FM, Feicht S, Helm F, Maurberger A, Ladinig C, Zimber-Strobl U, Kühn R, Mautner J, Gerbitz A, and Bornkamm GW (2012). Humanized c-Myc mouse. PLoS One 7, e42021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, and Vogelstein B (1997). Genetic instability in colorectal cancers. Nature 386, 623–627. [DOI] [PubMed] [Google Scholar]
- Lin CY, Lovén J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, and Young RA (2012). Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151, 56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler S, Sloss O, Geary B, Pierce A, Whetton AD, and Taylor SS (2019). Oncogenic MYC amplifies mitotic perturbations. Open Biol. 9, 190136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma N, Titus J, Gable A, Ross JL, and Wadsworth P (2011). TPX2 regulates the localization and activity of Eg5 in the mammalian mitotic spindle. J. Cell Biol 195, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado M, and Kapoor TM (2011). Constitutive Mad1 targeting to kinetochores uncouples checkpoint signalling from chromosome biorientation. Nat. Cell Biol 13, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins MM, Zhou AY, Corella A, Horiuchi D, Yau C, Rakhshandehroo T, Gordan JD, Levin RS, Johnson J, Jascur J, et al. (2015). Linking tumor mutations to drug responses via a quantitative chemical-genetic interaction map. Cancer Discov. 5, 154–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack SJ, Weaver Z, Deming S, Natarajan G, Torri J, Johnson MD, Liyanage M, Ried T, and Dickson RB (1998). Myc/p53 interactions in transgenic mouse mammary development, tumorigenesis and chromosomal instability. Oncogene 16, 2755–2766. [DOI] [PubMed] [Google Scholar]
- Menssen A, Epanchintsev A, Lodygin D, Rezaei N, Jung P, Verdoodt B, Diebold J, and Hermeking H (2007). c-MYC delays prometaphase by direct transactivation of MAD2 and BubR1: identification of mechanisms underlying c-MYC-induced DNA damage and chromosomal instability. Cell Cycle 6, 339–352. [DOI] [PubMed] [Google Scholar]
- Meraldi P, Draviam VM, and Sorger PK (2004). Timing and checkpoints in the regulation of mitotic progression. Dev. Cell 7, 45–60. [DOI] [PubMed] [Google Scholar]
- Musinipally V, Howes S, Alushin GM, and Nogales E (2013). The microtubule binding properties of CENP-E’s C-terminus and CENP-F. J. Mol. Biol 425, 4427–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumayer G, Belzil C, Gruss OJ, and Nguyen MD (2014). TPX2: of spindle assembly, DNA damage response, and cancer. Cell. Mol. Life Sci 71, 3027–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe LP, Tong F, et al. (2006). A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 10, 515–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. (2012). c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda M (2002). The role of nucleophosmin in centrosome duplication. Oncogene 21, 6170–6174. [DOI] [PubMed] [Google Scholar]
- Owens B (2013). Kinesin inhibitor marches toward first-in-class pivotal trial. Nat. Med 19, 1550. [DOI] [PubMed] [Google Scholar]
- Pemble H, Kumar P, van Haren J, and Wittmann T (2017). GSK3-mediated CLASP2 phosphorylation modulates kinetochore dynamics. J. Cell Sci 130, 1404–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CBL, Leal MF, Abdelhay ESFW, Demachki S, Assumpção PP, de Souza MC, Moreira-Nunes CA, Tanaka AMDS, Smith MC, and Burbano RR (2017). MYC Amplification as a Predictive Factor of Complete Pathologic Response to Docetaxel-based Neoadjuvant Chemotherapy for Breast Cancer. Clin. Breast Cancer 17, 188–194. [DOI] [PubMed] [Google Scholar]
- Petry S (2016). Mechanisms of Mitotic Spindle Assembly. Annu. Rev. Biochem 85, 659–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petry S, and Vale RD (2015). Microtubule nucleation at the centrosome and beyond. Nat. Cell Biol 17, 1089–1093. [DOI] [PubMed] [Google Scholar]
- Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, Torres-Arzayus MI, Brown M, Egan SE, Wahl GM, et al. (2013). Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol. 14, R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riffell JL, Zimmerman C, Khong A, McHardy LM, and Roberge M (2009). Effects of chemical manipulation of mitotic arrest and slippage on cancer cell survival and proliferation. Cell Cycle 8, 3025–3038. [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids Research 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryl T, Kuchen EE, Bell E, Shao C, Flórez AF, Mönke G, Gogolin S, Friedrich M, Lamprecht F, Westermann F, and Höfer T (2017). Cell-Cycle Position of Single MYC-Driven Cancer Cells Dictates Their Susceptibility to a Chemotherapeutic Drug. Cell Syst. 5, 237–250.e8. [DOI] [PubMed] [Google Scholar]
- Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, et al. (2014). Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 511, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwinkendorf D, and Gallant P (2009). The conserved Myc box 2 and Myc box 3 regions are important, but not essential, for Myc function in vivo. Gene 436, 90–100. [DOI] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. (2004). MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 431, 1112–1117. [DOI] [PubMed] [Google Scholar]
- Sheen J-H, and Dickson RB (2002). Overexpression of c-Myc alters G(1)/S arrest following ionizing radiation. Mol. Cell. Biol 22, 1819–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton KR, Crawford L, Tsui E, Manchester HE, Maertens O, Liu X, Liberti MV, Magpusao AN, Stein EM, Tingley JP, et al. (2017). Melanoma Therapeutic Strategies that Select against Resistance by Exploiting MYC-Driven Evolutionary Convergence. Cell Rep. 21, 2796–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, and Evan GI (2010). The ups and downs of Myc biology. Curr. Opin. Genet. Dev 20, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Ertych N, Kienitz A, Vogel C, Schneider V, Fritz B, Jacob R, Dittmar G, Weichert W, Petersen I, and Bastians H (2010). The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat. Cell Biol 12, 492–499. [DOI] [PubMed] [Google Scholar]
- Struntz NB, Chen A, Deutzmann A, Wilson RM, Stefan E, Evans HL, Ramirez MA, Liang T, Caballero F, Wildschut MHE, et al. (2019). Stabilization of the Max Homodimer with a Small Molecule Attenuates Myc-Driven Transcription. Cell Chem. Biol 26, 711–723.e14. [DOI] [PubMed] [Google Scholar]
- Sugihara E, Kanai M, Matsui A, Onodera M, Schwab M, and Miwa M (2004). Enhanced expression of MYCN leads to centrosome hyperamplification after DNA damage in neuroblastoma cells. Oncogene 23, 1005–1009. [DOI] [PubMed] [Google Scholar]
- Talapatra SK, Harker B, and Welburn JP (2015). The C-terminal region of the motor protein MCAK controls its structure and activity through a conformational switch. eLife 4, e06421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Macůrek L, Janssen A, Geers EF, Alvarez-Fernández M, and Medema RH (2009). Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr. Biol 19, 1703–1711. [DOI] [PubMed] [Google Scholar]
- Thompson SL, and Compton DA (2008). Examining the link between chromosomal instability and aneuploidy in human cells. J. Cell Biol 180, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Bakhoum SF, and Compton DA (2010). Mechanisms of chromosomal instability. Curr. Biol 20, R285–R295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, Ridgway RA, Huels D, Littler S, Schandl C, et al. (2015). MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell 28, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turajlic S, and Swanton C (2017). Implications of cancer evolution for drug development. Nat. Rev. Drug Discov 16, 441–442. [DOI] [PubMed] [Google Scholar]
- Walz S, Lorenzin F, Morton J, Wiese KE, von Eyss B, Herold S, Rycak L, Dumay-Odelot H, Karim S, Bartkuhn M, et al. (2014). Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 511, 483–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chauhan J, Hu A, Pendleton K, Yap JL, Sabato PE, Jones JW, Perri M, Yu J, Cione E, et al. (2013). Disruption of Myc-Max heterodimerization with improved cell-penetrating analogs of the small molecule 10074-G5. Oncotarget 4, 936–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao Y-M, Lengauer C, et al. (2009). Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504. [DOI] [PubMed] [Google Scholar]
- Wittmann T, Bokoch GM, and Waterman-Storer CM (2004). Regulation of microtubule destabilizing activity of Op18/stathmin downstream of Rac1. J. Biol. Chem 279, 6196–6203. [DOI] [PubMed] [Google Scholar]
- Xia F, Canovas PM, Guadagno TM, and Altieri DC (2008). A survivin-ran complex regulates spindle formation in tumor cells. Mol. Cell. Biol 28, 5299–5311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Liu H, Goga A, Kim S, Yuneva M, and Bishop JM (2010). Therapeutic potential of a synthetic lethal interaction between the MYC protooncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. USA 107, 13836–13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Kwiatkowski NP, Zhang T, Nabet B, Xu M, Liang Y, Quan C, Wang J, Hao M, Palakurthi S, et al. (2018). Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. eLife 7, e39030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Roostalu J, Surrey T, and Nogales E (2017). Structural insight into TPX2-stimulated microtubule assembly. eLife 6, e30959. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA sequencing data from MTB-TOM tumors and Eμ-tTA/TRE-MYC lymphoma cells were deposited in NCBI GEO under accession number GEO: GSE130922.
Secondary accessions: the human breast cancer RNA-sequencing datasets are derived from the TCGA Research Network (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) and are available from GEO: GSE62944. Gene expression values for the MYC-driven liver tumors are available from GEO: GSE73295.