

Abstract
Fast N-type inactivation of voltage-gated K+ (Kv) channels is important in fine-tuning of cellular excitability. To serve diverse cellular needs, N-type inactivation is regulated by numerous mechanisms. Here we address how reactive sulfur species – the gaseous messenger H2S and polysulfides – affect N-type inactivation of the mammalian Kv channels Kv1.4 and Kv3.4. In both channels the H2S donor NaHS slowed down inactivation with varying potency depending on the “aging” of NaHS solution. Polysulfides were >1000-times more effective than NaHS with the potency increasing with the number of sulfur atoms (Na2S2 < Na2S3 < Na2S4). In Kv1.4, C13 in the N-terminal ball domain mediates the slowing of inactivation. In recombinant protein exposed to NaHS or Na2S4, a sulfur atom is incorporated at C13 in the protein. In Kv3.4, the N terminus harbors two cysteine residues (C6, C24), and C6 is of primary importance for channel regulation by H2S and polysulfides, with a minor contribution from C24. To fully eliminate the dependence of N-type inactivation on sulfhydration, both cysteine residues must be removed (C6S:C24S). Sulfhydration of a single cysteine residue in the ball-and-chain domain modulates the speed of inactivation but does not remove it entirely. In both Kv1.4 and Kv3.4, polysulfides affected the N-terminal cysteine residues when assayed in the whole-cell configuration; on-cell recordings confirmed that polysulfides also modulate K+ channel inactivation with undisturbed cytosol. These findings have collectively identified reactive sulfur species as potent modulators of N-type inactivation in mammalian Kv channels.
Keywords: Hydrogen sulfide, Sulfhydration, K+ channel inactivation, Kcna4, Kcnc4, Reactive sulfur species
Introduction
Hydrogen sulfide (H2S) is a gaseous messenger produced in mammalian cells through enzymatic pathways of cystathionine γ-lyase (CSE) in liver, cystathionine β-synthase (CBS) in the brain, pancreas and liver, and 3-mercaptopyruvate sulfurtransferase (MST) in various tissues, from L-cysteine and homocysteine and their derivatives [44,43]. Additionally, H2S may be produced through a D-cysteine-dependent pathway with prime importance in cerebellum and kidney [40] and also by bacteria in the intestinal tract [4]. H2S has a pKa about 7 [19] and thus both H2S and HS– are expected to be present under physiological conditions. H2S and HS– in aqueous solutions are in equilibrium with more oxidized sulfur species, such as hydropersulfides and polysulfides [28,8]. In particular, glutathione persulfide (GSSH) exists at substantially higher levels in mammalian tissues than H2S [20]. Together, these sulfur-containing agents are often categorized as “reactive sulfur species” (RSS), a class of cellular messengers distinct from but closely related to the well-established reactive oxygen and reactive nitrogen species (ROS and RNS) [19].
Regulation of protein functions by RSS typically involves the addition of one or more sulfur atoms to a reactive cysteine side chain (sulfhydration). This type of modification may inactivate the modified protein but in some cases augment protein functions: increased enzyme activity in sulfhydrated GAPDH [30], enhanced actin polymerization [48], or greater open probability of KATP channels upon sulfhydration of the channel subunits [31] have been reported.
H2S, like other gaseous messengers nitric oxide (NO) and carbon monoxide (CO) and select reactive species, permeates readily through biological membranes and possesses great potential to affect ion channel proteins [6,45,29,15]. In fact, the regulation of membrane ion channels is a key mechanism for the physiological effects of H2S in blood vessels, heart, and nerves [26,34]. For instance, modulation of KATP channels by H2S plays a role in myocardial protection against ischemia/reperfusion injury [49]. H2S may also alter N-methyl-D-aspartate (NMDA) glutamate receptor channels in the mammalian brain, strengthening long-term potentiation [21].
Voltage-gated potassium (Kv) channels are among the best-investigated ion channels. A subgroup of Kv channels that undergo rapid inactivation after depolarization-induced activation are often termed A-type channels. Inactivation of A-type channels and its modulation are essential for fine-tuning of neuronal excitability, affecting action potential (AP) frequency, AP width, afterhyperpolarization, Ca2+ influx, and neurotransmitter release [9,5]. Multiple inactivation mechanisms are known [25] but N-type inactivation or “ball-and-chain” inactivation is best understood. One or more of the four cytoplasmic N-terminal ends of the α subunits harbors a “ball domain” that occludes the ion permeation pore formed of four α subunits. The mammalian Kv channel α subunits capable of mediating N-type inactivation include Kv1.4 and Kv3.4 present in neurons, heart, and endocrine cells [38].
The degree and kinetics of N-type inactivation are regulated in multiple ways. For instance, inactivation of Kv1.4 and Kv3.4 is sensitive to changes in the redox conditions [36,11,38], and for Kv1.4, a cysteine residue in the cytoplasmic N-terminal segments (C13) mediates the redox sensitivity [36]. Furthermore, the same cysteine residue is important for the channel’s sensitivity to hemin [37]. Moreover, membrane lipid interactions [32], phosphorylation state [35,2], and the intracellular pH [33] can also affect N-type inactivation of Kv1.4.
Studies on the impact of H2S on cellular electrical excitability and ion channels frequently utilize the donor NaHS; when dissolved in an aqueous solution, NaHS releases H2S. Such studies have documented a wide array of changes in cellular excitability including changes in AP characteristics [12]. Application of NaHS leads to diverse changes in functional properties of multiple ion channels including voltage-gated Ca2+ channels, KATP channels, and large-conductance Ca2+- and voltage-gated Slo1 BK channels [41]. These diverse consequences of NaHS application may reflect both direct and indirect effects of H2S. For example, H2S may modify cysteine residues of target channel proteins to form persulfides [41] and/or may also alter cellular levels of various reactive species [27].
The presence of cysteine residues in the ball domains of Kv1.4 and Kv3.4 suggests that N-type inactivation of these K+ channels may be subject to regulation by sulfhydration. In this study, we found that rapid N-type inactivation of Kv1.4 and Kv3.4 is markedly impaired by application of the H2S donor NaHS and polysulfides. Our electrophysiological and mass spectrometry results collectively suggest that application of NaHS or polysulfides leads to sulfhydration of cysteine residues in the N-terminal ball domains of Kv1.4 and Kv3.4. Impaired inactivation of Kv channels is often associated with cellular hypoexcitability and, thus, reactive sulfur species may act as long-lasting modifiers of Kv1.4/Kv3.4-expressing excitable cells.
Materials and Methods
Channel constructs and mutagenesis
The expression plasmids coding for Kv1.4 from Rattus norvegicus (Kcna4, accession no. X16002) and mutants were cloned as described before [37]. Full-length Kv3.4 from Rattus norvegicus (Kcnc4, accession no. X62841.1) was cloned into pcDNA3.1 between AflII and EcoRI sites. Mutations were created using QuikChange Site-Directed Mutagenesis Kit (Stratagene) and verified by sequencing.
For protein expression in E. coli, sequence coding for amino acid residues 1–61 of rat Kv1.4 was cloned into pETM41 containing an N-terminal MBP tag followed by a His-tag and a TEV cleavage site as described previously [37].
Cell culture and transfection
Human embryonic kidney 293T cells (HEK293T, CAMR, Porton Down, Salisbury, UK) were cultured in 45% Dulbecco’s Minimal Eagles Medium (DMEM) and 45% Ham’s F12 medium, supplemented with 10% fetal calf serum in a humid 37 °C incubator with 5% CO2. When grown to 30–50% confluence, cells were transiently transfected with the respective plasmids and CD8 using the Roti-Fect transfection kit (Carl Roth, Karlsruhe, Germany) according to the supplier instructions. Anti-CD8-coated Dynabeads (Deutsche Dynal GmbH, Hamburg, Germany) were used for visual identification of transfected cells. Electrophysiological recordings were performed 1–2 days after transfection.
Electrophysiological recordings
Ionic currents were recorded using the whole-cell or on-cell configuration at room temperature (20–24 °C) using an EPC-9 patch-clamp amplifier operated with PatchMaster software (both HEKA Elektronik, Lambrecht, Germany). Patch pipettes were fabricated from borosilicate glass (BioMedical Instruments, Zöllnitz, Germany) and were coated with dental wax (Patterson Dental, Mendota Heights, MN, USA) to reduce their capacitance. After fire-polishing the pipettes, resistances of 0.9–2.0 MΩ were obtained. An agar bridge connected the bath solution and the ground electrode. Up to 85% of the series resistance was electronically compensated and all voltages have been corrected for liquid junction potential. Leak and capacitive currents were corrected using a p/6 method. Depending on the kinetics of recovery from inactivation, test pulses were typically applied every 15 s.
The pipette solution composed of (in mM): 120 N-methyl-D-glucamine (NMG), 20 KCl, 1 MgCl2, 10 ethylene glycol tetraacetic acid (EGTA), 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pH was adjusted to 6.4, 6.9, 7.4 or 7.9 with HCl. For Kv1.4 recordings, the bath solution composed of (in mM): 148 NMG, 10 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, pH was adjusted to 6.9 or 7.4 with HCl. For Kv3.4 recordings, the bath solution composed of (in mM): 148 NMG, 4 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, pH was adjusted to 6.4, 6.9, 7.4 or 7.9 with HCl.
Chemicals
Solutions were made of high-grade chemicals obtained from Sigma-Aldrich (Taufkirchen, Germany) and Carl Roth. Sodium hydrogen sulfide (NaHS) was obtained from Cayman Chemical (Ann Arbor, MI, USA). Polysulfides (sodium disulfide (Na2S2), sodium trisulfide (Na2S3), and sodium tetrasulfide (Na2S4)) were obtained from Dojindo Molecular Technologies (Kumamoto, Japan). High-concentration stocks of NaHS (500 mM) and polysulfides (100 mM) were prepared in bath solution, and then diluted to the desired concentrations right before application. Stock solutions of NaHS or polysulfides were used within one hour of preparation (except for the NaHS “aging” experiments); they were locally applied with a fine-tipped glass pipette in close proximity to the cell examined.
Biochemical analysis and mass spectrometry
Protein consisting of amino acid residues 1–61 of the N terminus of rat Kv1.4 was expressed in E. coli BL21 (DE3) pRIL as described previously [37]. The His6-tagged protein was purified in the presence of 5 mM dithiothreitol (DTT) using a HisTrap FF crude affinity column (GE Healthcare). MBP- and His6-tag was cleaved with TEV at 20 °C overnight and uncleaved protein, the TEV site together with the MBP/His6-tag was removed using an additional HisTrap FF crude affinity column (GE Healthcare). Monomeric peptide was separated from the dimeric form by Superdex Peptide (10/300 GL) (GE Healthcare).
Purified protein with two additional amino acids (Gly-Ala) resulting from the cleavage was incubated in 20 mM Tris, 150 mM NaCl, pH 7.4 with either 1 mM NaHS or 200 μM Na2S4 at room temperature for 30 min. If applicable, samples were pretreated with 100 μM 5, 5’-dithio-bis[2-nitrobenzoic acid] (DTNB) or incubated with 5 mM DTT to remove modification. NaHS or Na2S4 was removed using a 3 kDa cut-off filter and samples were treated with 15 mM iodoacetamide at room temperature for 30 min in the dark. After removal of residual iodoacetamide, protein was cleaved with trypsin (mass spectrometry grade) by incubating overnight at 25 °C. Desalting was performed using C18 stage tips (Merck Millipore). Subsequently, the total mass of the resulting peptides was measured using MALDI-MS (Bruker Daltonics Ultraflex TOF/TOF).
Data analysis
Electrophysiological results were analyzed using FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics, Lake Oswego, OR, USA) software. Data traces shown were digitally low-pass filtered at 1 kHz. Data are presented as means±SEM with n representing the number of independent measurements.
Time constants of inactivation were estimated by fitting current traces at 50 mV according to a Hodgkin-Huxley scheme. To estimate the limiting loss of inactivation after substance application (r∞) as shown in Fig. 7b, the time course of loss of inactivation was described as follows:
| [1] |
with the starting ratio r0 and a time constant τ. An empirical exponent of 1.6 was required to describe the sigmoidal onset, presumably because of the initial rate-limiting diffusion of the substances into the cytosol across the cell membrane.
Fig. 7. Kv3.4 inactivation is subject to modification by NaHS, Na2S4, and diamide.
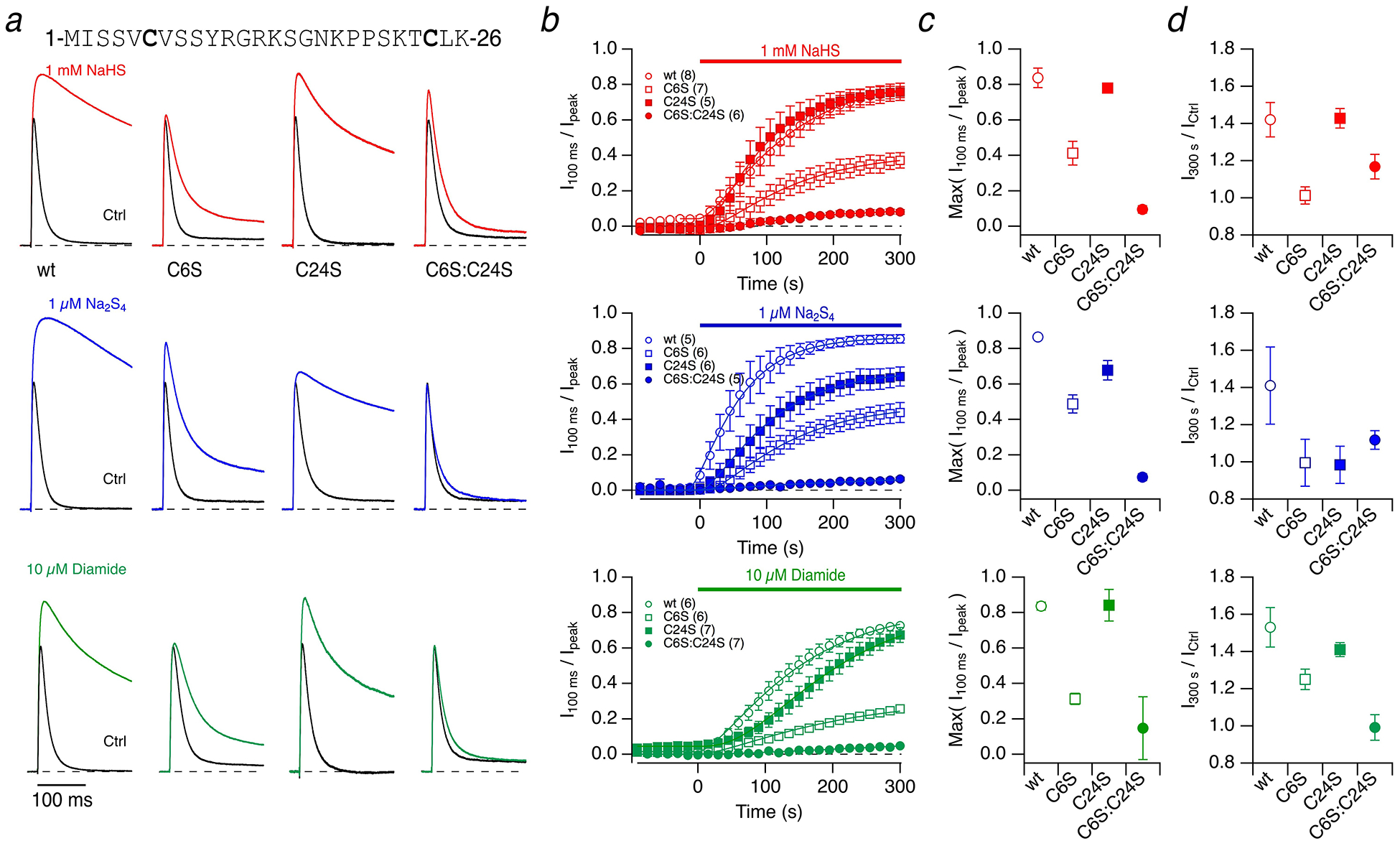
a N-terminal sequence of Kv3.4 with cysteine residues highlighted. Representative whole-cell current traces of Kv3.4 channels in response to membrane depolarization to 50 mV from a holding voltage of −100 mV before (black) and 5 minutes after application of the indicated compounds (colored): 1 mM NaHS (top), 1 μM Na2S4 (middle), 10 μM diamide (bottom) for Kv3.4 wild type (wt) and the indicated cysteine mutants. Currents are scaled to display identical control peak currents. pH 7.4. b Time course of the loss of inactivation, expressed as I100 ms / Ipeak, with compound application at time zero. Continuous curves are fits according to Equation (1), used to estimate the maximally obtained inactivation index. c Extrapolated maximal inactivation index based on the data fits shown in b). Data are means±95% confidence interval. d Fractional change in peak current after 300 s compound application (I300 s / ICtrl) for the indicated channel types. Data are means±SEM with n in parentheses in panel b)
Results
NaHS slows down N-type inactivation in Kv1.4 channels
To measure the impact of H2S on the N-type inactivation of voltage-gated K+ channels of the Kv family, rat Kcna4 was transiently expressed in HEK293T cells, and the resulting Kv1.4-mediated currents were measured in the whole-cell patch-clamp configuration. NaHS, an inorganic sulfide salt, was used as the H2S donor. With intra- and extracellular solutions adjusted to pH 7.4, channels inactivated at 50 mV with a time constant of 84.4±2.8 ms (n=7) (Fig. 1a). Extracellular application of 1 mM NaHS resulted in a progressive slowing of inactivation (Fig. 1b), increasing the time constant of inactivation more than 2-fold to 186±18 ms after 300 s (Fig. 1c). Even without any application or upon application of control bath solution, there was a slow but consistent loss of inactivation (Fig. 1b), probably because of the oxidation-sensitive C13 in the N-terminal domain. Because N-type inactivation of Kv1.4 channels depends on the intracellular pH [33], and pH is an important factor for the equilibrium between H2S and HS–, we performed similar experiments with intra- and extracellular solutions at pH 6.9 (Fig. 1a–b, right). In pH 6.9 solutions, the inactivation time constant at 50 mV was 37 ms, i.e. about twice as fast as at pH 7.4. Application of 1 mM NaHS increased the inactivation time constant by ~3-fold to 115 ms after 300 s (Fig. 1c). Compared with the results at pH 7.4, at pH 6.9 the loss of inactivation followed more closely an exponential time course and inactivation was more stable under control conditions (Fig. 1b, right). The observed instability of N-type inactivation properties at higher pH is indicative of spontaneous oxidation of C13, causing progressive loss of inactivation even under control conditions (Fig. 1b, left). For most physiological oxidants, oxidation of cysteine requires the more reactive thiolate anion as substrate [46] and lowering the pH to 6.9 favors the protonated thiol over the thiolate anion.
Fig. 1. Kv1.4 inactivation is slowed down by NaHS.
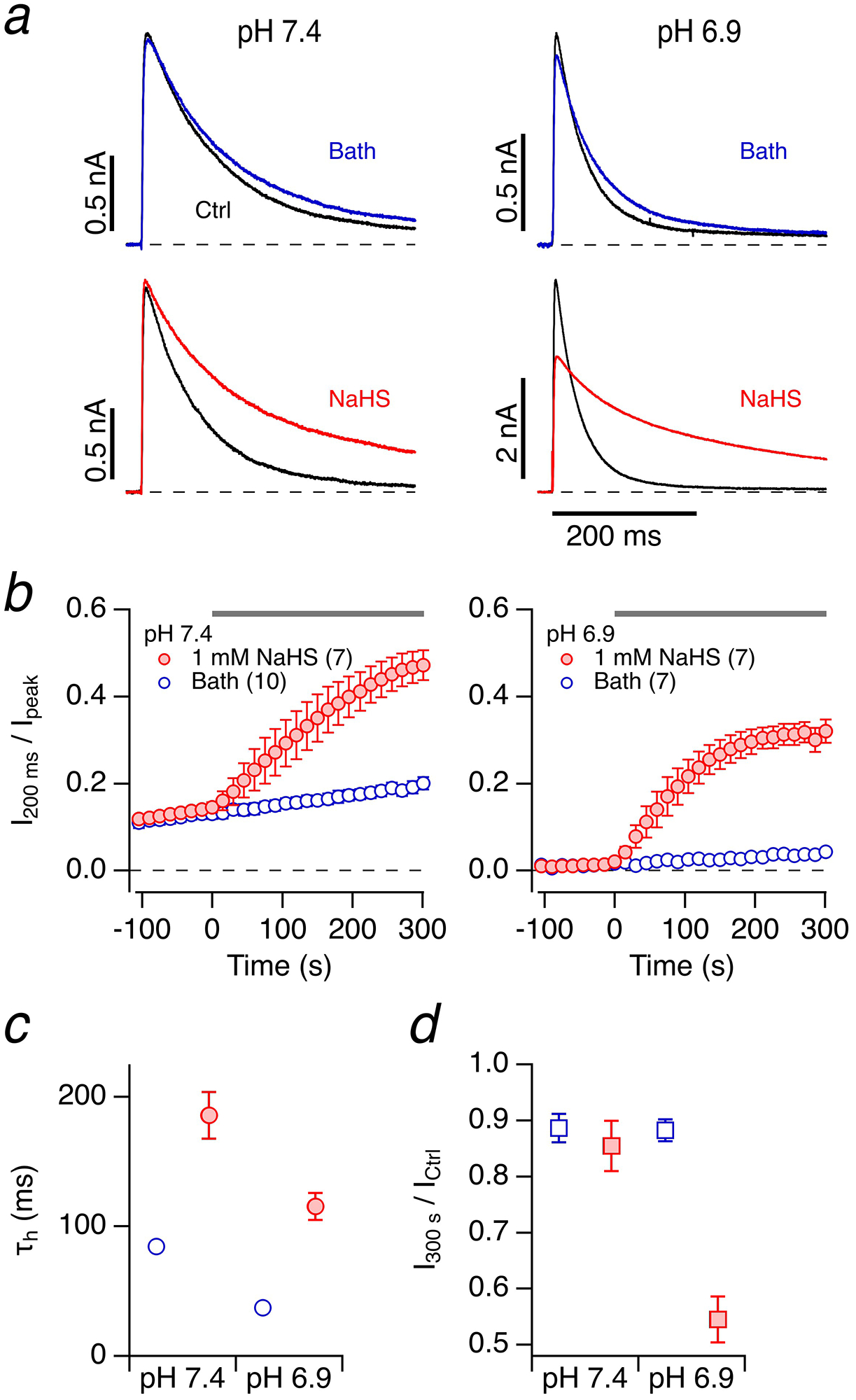
a Representative whole-cell current traces of Kv1.4 channels in HEK293T cells in response to membrane depolarization to 50 mV from a holding potential of −100 mV before (black) and 300 s after application of bath solution (top, blue) or 1 mM NaHS freshly dissolved in bath solution (bottom, red). Data are shown for pipette and bath solutions adjusted to pH 7.4 (left) and pH 6.9 (right). b Time course of the inactivation index, i.e. the current amplitude after 200 ms normalized to the peak current, for pH 7.4 (left) and pH 6.9 (right); at time zero either bath solution (open circles) or 1 mM NaHS (filled circles) were applied. Data are means±SEM (n in parentheses). c Time constants of inactivation (τh) right before (open circles) and 300 s after application of 1 mM NaHS (filled circles). d Peak current amplitude after 300 s normalized to the control before application of bath solution (open squares) or 1 mM NaHS (filled squares)
The peak current was reduced by about 10% after 300 s at both pH values when control bath solution was applied (Fig. 1d, open symbols). At pH 7.4 after application of 1 mM NaHS, the peak current was also stable, while at pH 6.9 it decreased to about 55% (Fig. 1d). This greater reduction at pH 6.9 is most likely due to a slowing of recovery from inactivation [7] because the reduction of peak current was much less pronounced in the experiments where the holding voltage was −140 mV instead of −100 mV (Suppl. Figure 1).
To investigate the concentration dependence of the impact of NaHS on Kv1.4 N-type inactivation, similar experiments were performed with extracellular application of 200 μM NaHS for 150 s; these experiments were performed at pH 6.9 because under this condition N-type inactivation is faster and more easily distinguished from C-type inactivation. The effect of 200 μM NaHS on N-type inactivation was noticeably variable, ranging from almost no effect to marked loss of inactivation (Fig. 2a). The major cause of the variability was the time elapsed between dissolving NaHS in the bath solution and its application. Therefore, we systematically assessed the impact of “aging” on the propensity of NaHS solutions to slow down N-type inactivation by analyzing the relative change in inactivation time constant at 150 s after NaHS application. NaHS was stored as 500-mM solution in a closed 1.5-ml reaction tube at room temperature for variable durations (“aging”). As shown in Fig. 2b, greater “aging” of the NaHS solution increased its potency to affect N-type inactivation.
Fig. 2. “Aging” of NaHS solutions.
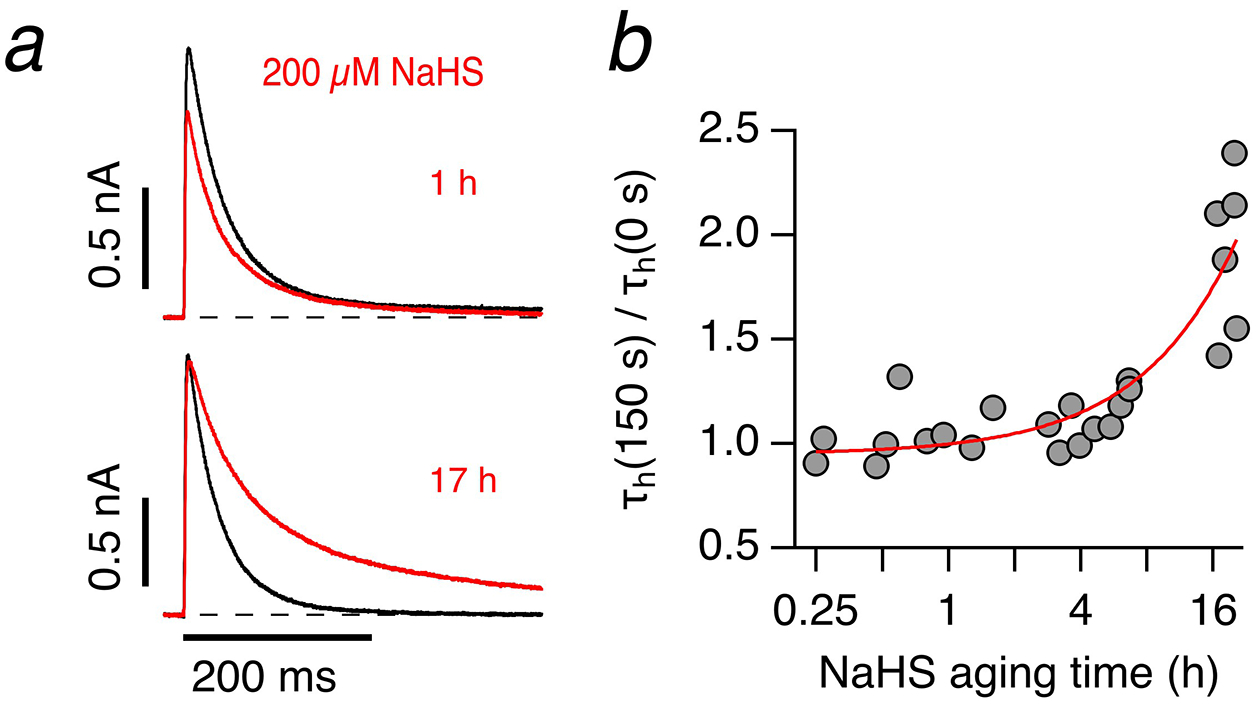
a Representative whole-cell current traces of Kv1.4 channels in HEK293T cells in response to membrane depolarization to 50 mV from a holding potential of −100 mV before (black) and 150 s after application of solution with 200 μM NaHS (red), diluted from 500 mM NaHS prepared 1 h (top, red) and 17 h (bottom, red) before the experiment. b Relative change of inactivation time constants from experiments as in a) for 200 μM NaHS, diluted from 500 mM NaHS stored in bath solution for the indicated times before application. The continuous curve indicates the increase in potency with aging time. Experiments were performed at pH 6.9
Impact of polysulfides on Kv1.4 channels
The result that “aged” NaHS solution is more potent in slowing N-type inactivation than freshly prepared NaHS solution suggests that polysulfides (Sn2–, where n ≥ 2 is the number of sulfur atoms), not H2S itself, exert the effect on the channel. We therefore tested polysulfides as sodium salts with an increasing number of sulfur atoms on the inactivation of Kv1.4 channels. As depicted in Fig. 3a, b, 1 μM Na2S2, Na2S3, and Na2S4 potently impaired N-type inactivation of Kv1.4 channels where the magnitude of the effect increased with increasing number of sulfur atoms. Because Na2S4 was most potent in interfering with N-type inactivation when tested at 1 μM, we also tested lower and higher concentrations. The inactivation index (I200 ms / Ipeak) saturated at about 0.6, i.e. a level corresponding to N-type inactivation totally removed with slower C-type inactivation intact [17], and even 100 nM Na2S4 was sufficient to noticeably impair N-type inactivation within a few minutes (Fig. 3c). Approximating the time course of the onset of loss of inactivation with single-exponential functions yielded appropriate data descriptions (Fig. 3c). The concentration dependence of the resulting time constants is shown in Fig. 3d. Loss of inactivation under bath solution application was estimated to proceed with a time constant of about 2200 s. Extrapolation of the concentration dependence of Na2S4 predicts that 10 nM Na2S4 is expected to result in a removal of inactivation with a time constant of 20 minutes, i.e. about the experimental detection limit when recording currents in the whole-cell mode.
Fig. 3. Impact of polysulfides on Kv1.4 inactivation.
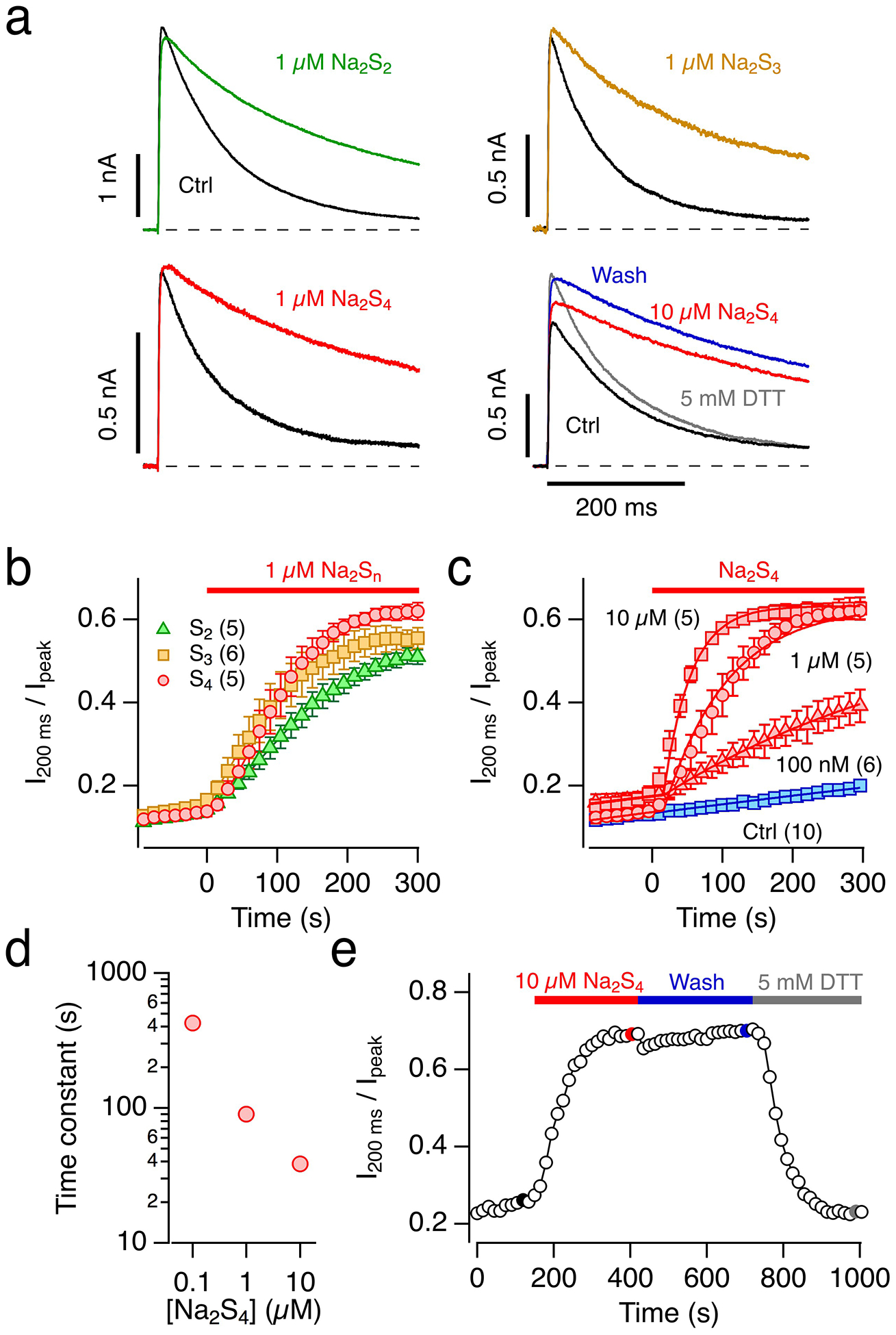
a Representative whole-cell current traces of Kv1.4 channels in HEK293T cells in response to membrane depolarization to 50 mV from a holding potential of −100 mV before (black) and 300 s after application of bath solution with 1 μM of the polysulfides Na2S2 (green), Na2S3 (orange) or Na2S4 (red), as well as 10 μM Na2S4 (red), subsequent wash with control bath solution (blue), followed by bath solution with 5 mM DTT (gray). pH 7.4. b Time course of the inactivation index, i.e. the current amplitude after 200 ms normalized to the peak current, for the indicated applications of polysulfides at time zero: Na2S2 (green triangles), Na2S3 (orange squares) or Na2S4 (red circles). Data are means±SEM (n in parentheses). c As in b) with various concentrations of Na2S4 (red) and control application of bath solution (blue). The superimposed continuous lines are single-exponential fits also taking into account the spontaneous loss of inactivation as observed in the control recordings. d Time constants of loss of inactivation as a function of Na2S4 concentration from the fits shown in panel c). e Time course of the Kv1.4 inactivation index with application of 10 μM Na2S4, subsequent wash with control bath solution, followed by bath solution with 5 mM DTT. The highlighted data points correspond to the data traces shown in a), bottom right
The inactivation, greatly impaired by Na2S4, was stable when Na2S4 was washed out (Fig. 3e). Subsequent application of the reducing agent DTT largely restored inactivation (Fig. 3a, e), further implicating a cysteine-specific modification.
Cysteine 13 in the ball domain of Kv1.4 channels
The ball domain of Kv1.4’s N terminus harbors a cysteine residue at position 13 (Fig. 4a), which is important for the channel’s sensitivity towards oxidation [36] and also for hemin binding to the channel [37]. In addition, a histidine residue at position 16 contributes to the coordination of hemin [37] and may be involved in the pH sensitivity of Kv1.4 N-type inactivation [33]. We therefore generated Kv1.4 mutants C13S and H16R in isolation and combined, and evaluated the impact of 1 mM NaHS and 10 μM Na2S4 at pH 7.4 on N-type inactivation. The mutant H16R was sensitive to 1 mM NaHS and 10 μM Na2S4; inactivation slowed down with a time course similar to that of the wild type. The inactivation remaining after a prolonged exposure to 1 mM NaHS or 10 μM Na2S4 was faster in H16R than in the wild type (Fig. 4b, c), indicating that mutant H16R probably exhibits faster N-type inactivation than the wild type when the N terminus is modified at C13. Compared with the wild type, mutant C13S inactivated more slowly under control conditions, and neither 1 mM NaHS nor 10 μM Na2S4 appreciably altered inactivation. The same treatments also failed to affect inactivation of the double mutant C13S:H16R. The diminished sensitivity of inactivation in the mutants harboring the C13S alteration to NaHS and Na2S4 suggests that H2S or polysulfides markedly impair(s) N-type inactivation of Kv1.4 channels by directly affecting C13 in the ball domain. At pH 7.4, the mutant C13S – either alone or in combination with H16R – still exhibited a low but significant spontaneous loss of inactivation after entering the whole-cell configuration under control conditions. However, application of either NaHS or Na2S4 stopped this trend and even accelerated the inactivation time course slightly (Fig. 4b, c).
Fig. 4. Kv1.4 mutants in the N terminus.
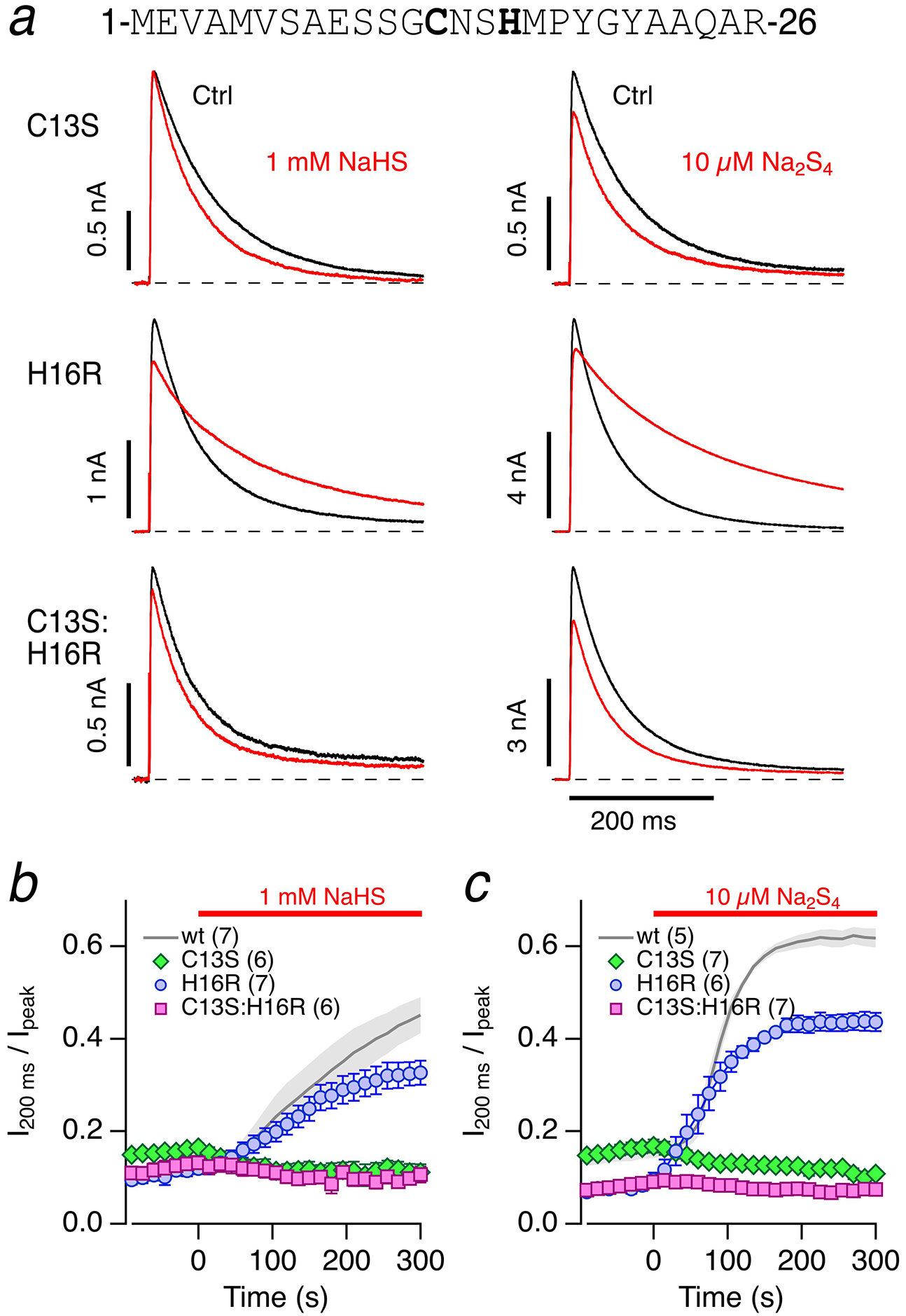
a Sequence of Kv1.4 N terminus with C13 and H16 highlighted. Representative whole-cell current traces of Kv1.4 channels in HEK293T cells in response to membrane depolarization to 50 mV from a holding potential of −100 mV before (black) and 300 s after application (red) of bath solution with 1 mM NaHS (left) or 10 μM Na2S4 (right) for Kv1.4 mutants C13S (top), H16R (middle), and C13S:H16R (bottom). pH 7.4. b Comparison of the effect of 1 mM NaHS on the inactivation indices (I200 ms / Ipeak) of the indicated Kv1.4 mutants compared with the wild type (wt) as shown in Fig. 1. c As in panel b) but with 10 μM Na2S4 application at time zero; the reference wt data were previously shown in Fig. 3). Data in b-c are means±SEM with n in parentheses
Since N-type inactivation of Kv1.4 mutant C13S:H16R was nearly insensitive to NaHS and Na2S4, this mutant is suited to infer the effects of H2S and polysulfide effects on other functional aspects of Kv1.4 channels. After incubation of cells for 7.5 minutes in 10 μM Na2S4, we failed to detect a noticeable change in the reversal potential or the channel’s voltage dependence of activation (Suppl. Figure 2).
To determine the type of protein modification that might have occurred in the channel’s N-terminal domain, we produced and purified a recombinant protein encompassing residues 1–61 and subjected it to mass spectroscopy. To stabilize persulfides, protein was alkylated with iodoacetamide. After treatment with NaHS (1 mM), only a minor peak corresponding to a mass increase of +32 Da, indicative of the presence of a persufide, was observed (Fig. 5a). Because NaHS cannot react with free cysteines [20], we further preactivated it with DTNB. NaHS cleaved this activated disulfide and resulted in a persulfide with a much higher signal. This additional peak was removed by DTT. These mass spectroscopy findings are consistent with the results of our electrophysiological measurements, which show that the reaction is reversible. In addition, they exclude the possibility of irreversible oxidation by two oxygens (+32 Da).
Fig. 5. Mass spectrometry of Kv1.4 N terminus.
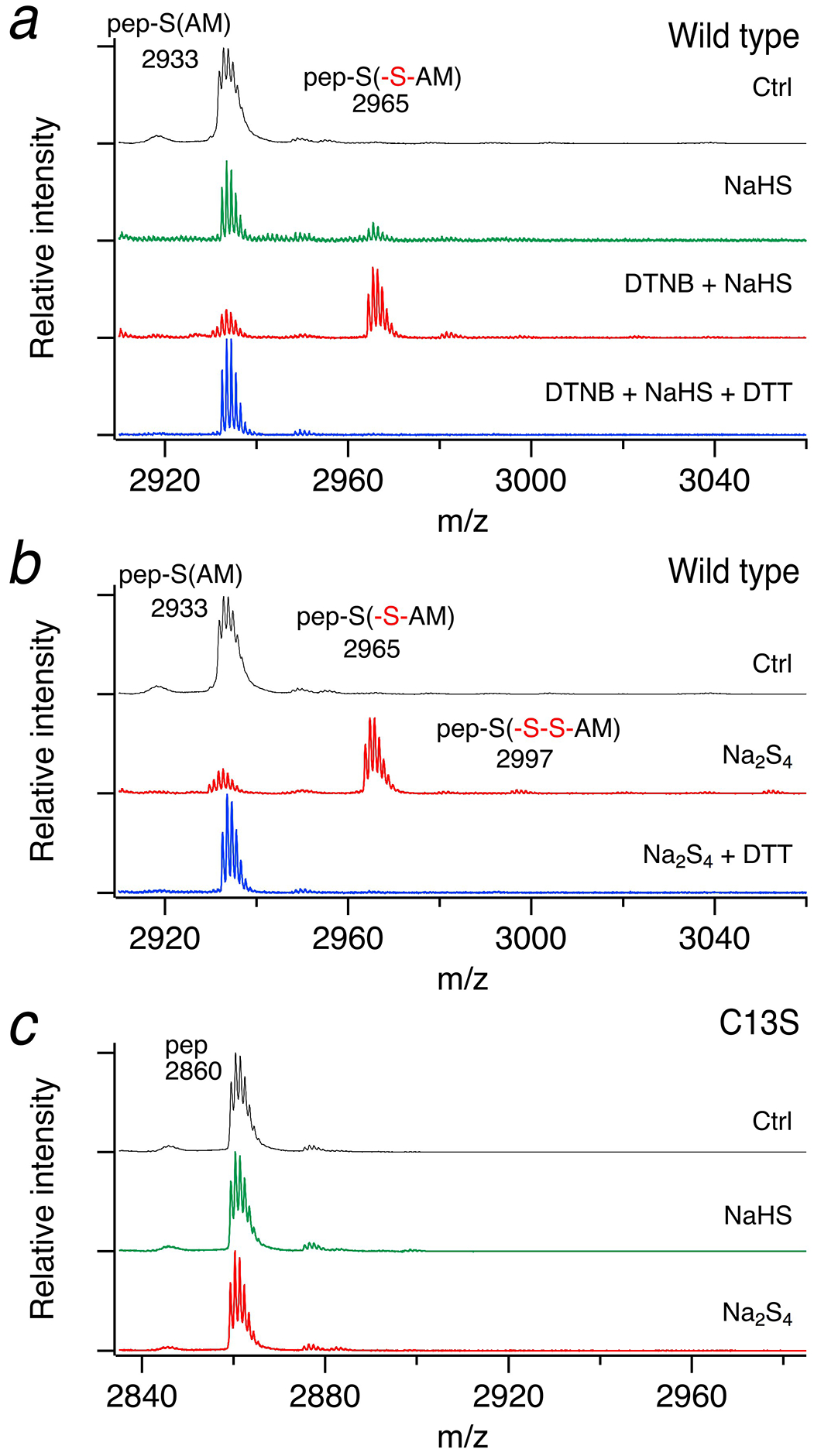
Representative MALDI MS spectra for recombinantly produced proteins encompassing residues 1–61 of the Kv1.4 N terminus (a and b) and the corresponding mutant C13S (c) after incubation with 1 mM NaHS, 100 μM DTNB and 1 mM NaHS, or 200 μM Na2S4, as indicated. Wild-type protein is sulfhydrated (+32 Da and some indication of +64 Da in panel b) in contrast to the C13S mutant. Application of 5 mM DTT removed the persulfide. Proteins were alkylated with iodoacetamid (IAM) and proteolytically digested with trypsin to yield control cysteine-containing peptide (pep-S(AM)) or pep-S(-S-AM) when previously sulfhydrated. The peptide carrying the mutation C13S does not contain the AM moiety. “m/z” refers to the ratio of mass (in Dalton) to unitary charge
The electrophysiological results showed that polysulfides were more potent than NaHS (Fig. 3). Therefore, we also incubated the recombinant protein with 200 μM Na2S4 and observed a strong signal with +32 Da (Fig. 5b). This main peak corresponds to only one additional sulfur atom. Peaks with +64 Da, +96 Da or +128 Da, indicating the presence of additional sulfur atoms, were very weak (+64 Da) or could not be clearly verified. To analyze whether the modification occurs at C13, we generated a mutant protein with C13S. Incubation with either NaHS or Na2S4 had no effect (Fig. 5c), showing clearly that C13 in the wild-type protein is sulfhydrated.
Kv1.4 is the main component of ROS-sensitive A-type K+ current found in small dorsal root ganglion (DRG) neurons, and plays a role in the generation and propagation of APs in DRG neurons [18]. In rat trigeminal ganglion neurons the H2S donor NaHS modifies AP characteristics [12], but the mechanism has remained unclear. We therefore examined the impact of Na2S4, which showed the most marked effect on the inactivation of Kv1.4 channels, on evoked APs of mouse DRG neurons. Na2S4 (50 μM) resulted in a small broadening of the APs, about 15% after 5 minutes (Suppl. Figure 3).
Impact of NaHS and polysulfides on Kv3.4 channels
The Kv3.4 channel, encoded by the Kcnc4 gene, is another mammalian N-type inactivating K+ channel exhibiting a redox-dependent inactivation time course. Similar to Kv1.4, the channel loses rapid inactivation once the cytosolic face of a membrane is exposed to oxidizing solutions, as in the inside-out configuration of the patch-clamp method [36]. Although not yet experimentally demonstrated, it is likely that the first cysteine in the N terminus at position 6 might be involved in this regulation.
We expressed rat Kcnc4 in HEK293T cells and measured voltage-dependent K+ currents in the whole-cell configuration. Kv3.4 channels show more rapid inactivation than Kv1.4 channels. At pH 7.4 (intra- and extracellular), the time course of inactivation at 50 mV was described with a single-exponential function with a time constant of 16.0±1.3 ms (n=8) (Fig. 6a, c). For comparison, the inactivation time constant of Kv1.4 channels under identical conditions was 84.4 ms (Fig. 1a, c). As noted earlier (Fig. 1), lowering the intracellular pH to 6.9 substantially increased the speed of inactivation of Kv1.4 (37.1±2.6 ms, n=7). For Kv3.4, however, there was only weak pH dependence and the direction was the opposite of what was observed for Kv1.4: for pH 6.9 we determined an inactivation time constant of 19.1±2.2 ms (n=8). Even lower pH (6.4) slightly slowed down whereas pH 7.9 further accelerated inactivation (Fig. 6a, c). Overall, the change in pH by 1.5 units had an impact on the speed of inactivation by less than a factor of 2. Unlike Kv1.4, Kv3.4 does not harbor histidine residues in its N-terminal domain, which may explain the lack of inactivation acceleration with acidification.
Fig. 6. Kv3.4 inactivation, pH and NaHS.
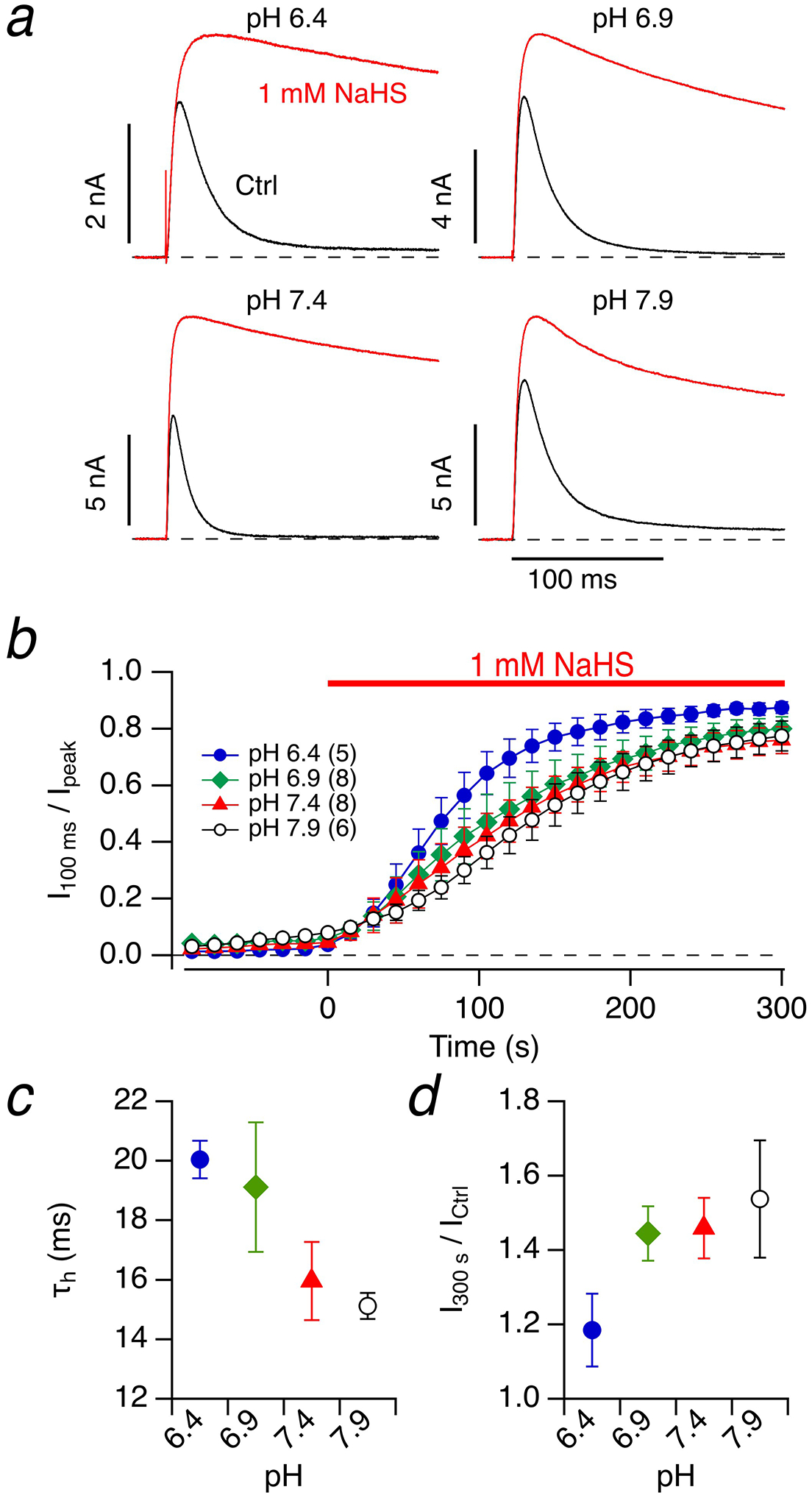
a Representative whole-cell current traces of Kv3.4 channels in HEK293T cells in response to membrane depolarization to 50 mV from a holding potential of −100 mV before (black) and 300 s after application of bath solution with 1 mM NaHS (red) at the indicated intracellular and extracellular pH values. b Time course of the inactivation index for the indicated pH values. c Time constant of inactivation under control conditions as a function of pH. d Alteration of peak current after 1 mM NaHS application for 300 s as a function of pH. Data in b-d) are means±SEM with n in parentheses in panel b)
This weak pH dependence was a practical advantage for our experiments because it allowed a quantitative assessment of the impact of the H2S donor NaHS on N-type inactivation at various pH values. For pH between 6.4 and 7.9, 1 mM NaHS resulted in a prominent loss of inactivation within 300 s yielding inactivation indices (I100 ms / Ipeak) approaching about 0.8 (Fig. 6a, b). This inactivation index is much greater than the index for Kv1.4 at 200 ms, mostly because the kinetics of N-type inactivation relative to that of C-type inactivation in Kv3.4 is much more rapid than that in Kv1.4. The time courses of NaHS-induced loss of inactivation were essentially indistinguishable between pH 6.9–7.9 (Fig. 6b).
Besides the nearly complete loss of inactivation, the peak current at 50 mV substantially increased with NaHS application with a trend of stronger current increases at higher pH (Fig. 6d), while under identical conditions the peak current of Kv1.4 channels was constant or slightly reduced (Fig. 1d). The increase in Kv3.4 peak current observed after NaHS application is consistent with the idea that N-type inactivation of Kv3.4 channels is rate-limiting in determination of ionic current time course [1].
The sensitivity of N-type inactivation to H2S is likely due to cysteine residues in the N-terminal ball-and-chain domain of Kv3.4. As depicted in Fig. 7a, there is one cysteine at position 6 and another one at position 24. We generated mutants with single and combined substitutions of serine for cysteine (C6S, C24S, C6S:C24S) and subjected these channels to 1 mM NaHS. Within 300 s of application, NaHS substantially removed N-type inactivation in the wild type and also in the mutant C24S with I100 ms / Ipeak converging to about 0.8 (Fig. 7b, top). For mutant C6S the loss of inactivation followed a similar time course but leveled off at a ratio of about 0.4 (Fig. 7b, top). Only the combination of both mutations, C6S:C24S, almost entirely removed the sensitivity of N-type inactivation to NaHS. The changes in inactivation index (Fig. 7b) were extrapolated using Equation (1) to estimate the saturating inactivation indices in the wild type and the mutant channels (Fig. 7c). Although the mutation C24S alone altered neither the impact of NaHS on the peak current (Fig. 7d) nor on the degree of inactivation loss (Fig. 7c), the mutation markedly diminished the channel’s sensitivity towards NaHS when inserted in the background of mutation C6S (C6S:C24S). C6S itself diminishes both the effects of NaHS on the peak current and inactivation, yet it still leaves the channel susceptible to NaHS-induced modification.
Similar experiments were performed with 1 μM Na2S4 (Fig. 7, middle). The mutations C6S and C24S noticeably but only partly diminished the effect of the polysulfide on the channel. The combination of both mutations made the channel virtually insensitive to Na2S4 (Fig. 7). There was no marked change in the voltage dependence of channel activation (Suppl. Figure 4). The impact of Na2S4 on the inactivation of Kv3.4 channels persisted when Na2S4 was washed out but was completely reversible when cells were exposed to solutions containing DTT (Suppl. Figure 5).
The impact of cysteine-specific modifiers on inactivation of Kv1.4 channels is well established (e.g., [36,18]). The involvement of residues C6 and C24 in N-type inactivation of Kv3.4 channels, however, has not been shown previously. We thus tested the impact of 10 μM diamide (1,1’-azobis[N,N-dimethylformamide]) because diamide is expected to induce the glutathionylation of cysteine residues in the presence of intracellular glutathione, which should be abundantly present even in the whole-cell configuration. Diamide (10 μM) readily removed inactivation in Kv3.4 channels and increased the peak current at 50 mV (Fig. 7). As found for 1 μM Na2S4, the cysteine mutations diminished these effects in a graded manner with C24S < C6S < C6S:C24S.
Because the impact of diamide on the channel may involve one or more intermediate reactions, we also measured the effect of 2 μM DTNP (2,2’-dithiobis[5-nitropyridine]), a membrane permeable cysteine-specific modifier, on N-type inactivation of Kv3.4 (Suppl. Figure 6). As found for NaHS, Na2S4, and diamide, C6 appears to be of major importance for the channel’s sensitivity to DTNP, but C24 also contributes such that only the combination of both (C6S:C24S) renders the inactivation largely resistant to the agents applied.
Polysulfide signaling in intact cells
The experiments shown thus far were performed in the whole-cell patch-clamp configuration; the intracellular milieu of the cell is dominated by the pipette solution, which does not contain any reducing compounds and, therefore, constitutes a potentially oxidizing condition. Therefore, the question arises if the observed impact of Na2S4 on the inactivation of Kv3.4 channels also occurs in intact cells or whether it relies on the oxidizing condition imposed by the patch-pipette solution. We performed experiments in which HEK293T cells expressing Kv3.4 channels were incubated for various durations in 1 μM Na2S4 before a seal was established; currents were then measured at 50 mV about 20–30 s after establishing the whole-cell mode, i.e. in a time window that is expected to allow only a minor impact on the inactivation time course based on the data shown in Fig. 7. As demonstrated in Fig. 8a–c, the initial degree of inactivation scattered substantially among the cells measured, but there was a clear trend that longer pre-incubation progressively slows down inactivation, with an estimated time constant of about 10 minutes. Experiments using pre-incubation with 10 μM diamide yielded similar results (Fig. 8a–c, bottom). Since the expression level of Kv3.4 channels in HEK293T cells is high, it was feasible to record currents in the on-cell configuration as to minimize perturbation of the intracellular milieu. Current inactivation at 50 mV slowed down considerably depending on the duration of cell pre-incubation with Na2S4 or diamide (Fig. 8d, e). A similar slowing of inactivation on preincubation of cells with Na2S4 was measured in on-cell patches of DRG neurons (Supplementary Figure 7), possibly explaining the impact of Na2S4 on the action potential shape as mentioned above.
Fig. 8. Impact of polysulfide and diamide on Kv3.4 in intact cells.
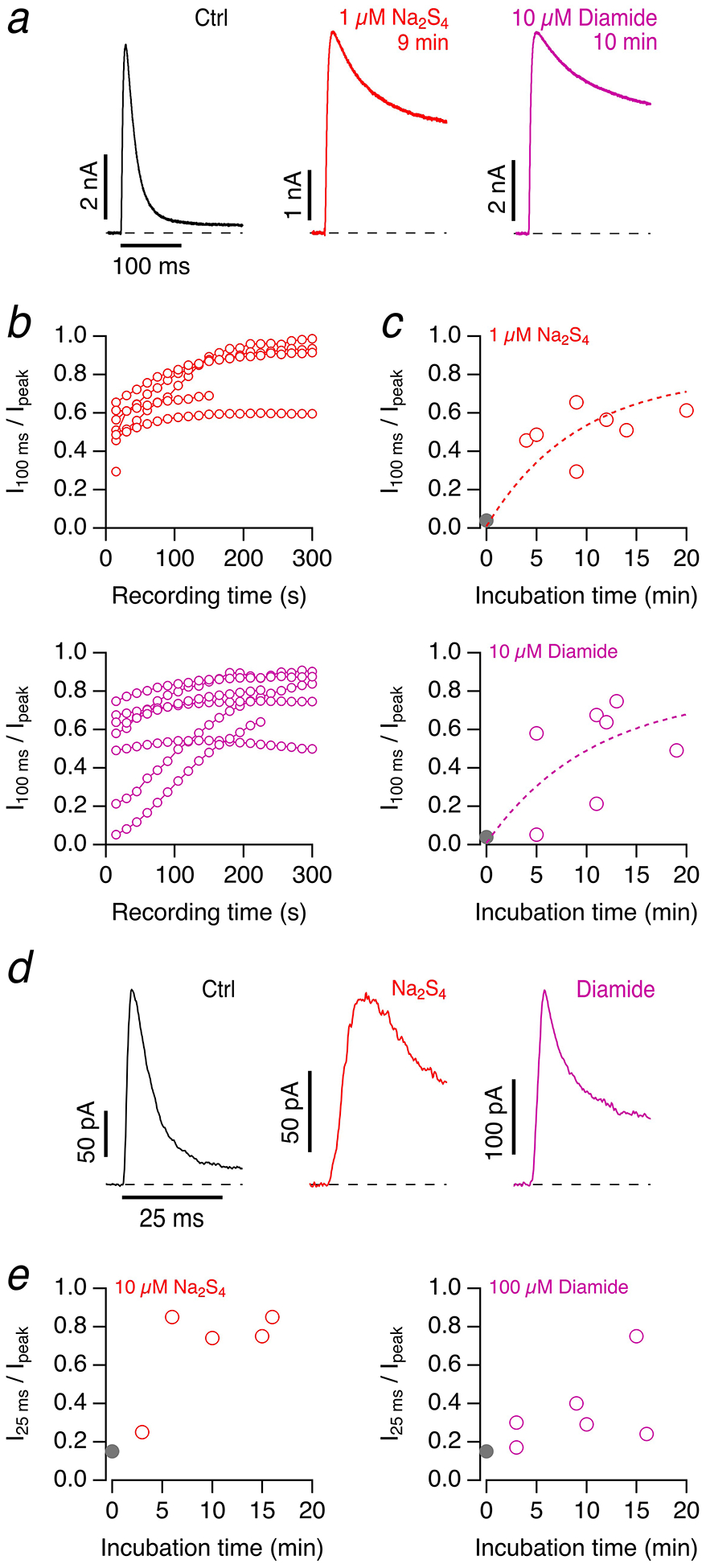
a Current traces for depolarizations to 50 mV about 20–30 s after entering the whole-cell mode for HEK293T cells expressing Kv3.4 under control conditions (left) and with preincubation with 1 μM Na2S4 (9 minutes, middle) and 10 μM diamide (10 minutes, right). b Time courses after establishing the whole-cell configuration of the inactivation index of Kv3.4 currents for 1 μM Na2S4 (top) and 10 μM diamide (bottom) at various preincubation times. c Initial inactivation indices for independent experiments in which the preincubation times for Na2S4 (top) and diamide (bottom) were varied. The gray data point at time zero indicates the inactivation index under control conditions, mean±SEM, n=7. The superimposed dashed curves are single-exponential functions approaching an estimated maximal inactivation index of 0.8 and leading time constants of 9.1±2.0 min (Na2S4) and 10.7±3.7 min (diamide); mean±95% confidence interval. d Sample on-cell current traces with depolarizations to 50 mV under control conditions (black) and after 10-min incubation of the cells with 10 μM Na2S4 (red) or 100 μM diamide (magenta). The pipette contained the same solution as the bath. e Inactivation index after 25 ms for on-cell current traces as a function of incubation time for 10 μM Na2S4 (left) or 100 μM diamide (right). The gray data points denote mean control values ±SEM (n=17)
Measurements with the redox-sensitive fluorescent protein roGFP2 [10] demonstrated that both Na2S4 and diamide in the concentration range used in this study affect the cysteine state of these cytosolic proteins (Suppl. Figure 8) thus supporting that polysulfide signaling takes places in intact cells.
Discussion
N-type inactivation of K+ channels harboring cysteine residues in their N-terminal ball-and-chain inactivation motif is subject to modulation by altered intracellular redox conditions. For example, in Kv1.4 channels expressed in a variety of neurons in the central and peripheral nervous system, the importance of C13 in such a redox-dependent modulation has been documented [36]. The exact molecular mechanism, however, has remained elusive because it is not clear if oxidation of C13 induces the formation of a disulfide bridge with another cysteine or glutathione (mixed disulfide) or another form of posttranslational modification of this residue. Here we have shown that C13 in Kv1.4 is a target for channel modification through reactive sulfur species. Exposure of Kv1.4 channels in the whole-cell recording configuration to the H2S donor NaHS results in a progressive loss of N-type inactivation, leaving the slower C-type inactivation unaffected.
Use of NaHS as H2S donor is a common experimental approach, but the chemistry of NaHS in water is complex and does not yield stable products. In water, the donor dissociates into Na+ and the hydrosulfide ion HS–. At neutral pH, water-dissolved H2S gas splits into ≈ 80% HS– and ≈ 20% of undissociated H2S in addition to an extremely small concentration of S2– [19]. Regardless of the source of HS–, ready autoxidation of this hydrosulfide always produces sulfane sulfur in form of soluble polysulfides [19]. The oxidation state (S0) of this sulfane sulfur allows direct sulfhydration of thiol groups in cysteines, whereas H2S or HS– (oxidation state −2) cannot oxidize the reduced thiol but instead require pre-oxidized substrates, such as sulfenic acid or disulfides [42,23]. Therefore, our finding that “aged” NaHS solution had increased potency in modulating Kv1.4 channel inactivation strongly suggests that spontaneously occurring sulfane sulfur (polysulfide) was the active component in our experiments with the H2S donor NaHS. In line with this idea, direct application of polysulfides was >1000-times more effective in removing inactivation than NaHS, with the greater number of sulfur atoms increasing the potency. In fact, even 100 nM Na2S4 efficiently removed inactivation from Kv1.4 channels within a couple of minutes, suggesting that endogenous polysulfides in the lower nanomolar range are capable of modulating the channels.
The findings described above are consistent with recent reports arguing that sulfane sulfur (polysulfide) is likely of greater physiological relevance than the gaseous H2S itself or the hydrosulfide ion [42,19,23]. As in the case of increased Ca2+ influx caused by activation of transient receptor potential (TRP)A1 channels [24], intermediate formation of polysulfides may often be an essential prerequisite for the regulatory function of RSS. This view is not only based on chemical properties and oxidation states but also supported by recent efforts to determine endogenous RSS concentrations in tissues. Using a novel mass spectrometric method, Ida and coworkers estimated that the glutathione persulfide GSSH exists in concentrations >100 μM in mouse brain and about 50 μM in other major organs including the heart [20]. Concentrations of H2S in tissues and plasma are controversial; however, while earlier reports indicated concentrations up to 100 μM, more recent studies suggested only nanomolar concentrations in tissues [14] and up to 1 μM in plasma [39,47]. Irrespective of the precise concentrations, a relative excess of polysulfides over H2S appears probable, given the higher toxicity of hydrogen sulfide when compared to sulfane compounds. Due to poisoning of the respiratory chain, high micromolar H2S concentrations are expected to cause tissue death, as the brain cytochrome c oxidase is 80% inhibited at 1 μM H2S [14].
The NaHS-induced slowdown of channel inactivation was reversible when DTT was applied, suggesting a reversible modification of a thiol group in the protein. This conclusion is supported by the mutation C13S largely eliminating the channel’s sensitivity toward NaHS and also polysulfides. Furthermore, a recombinant protein encompassing Kv1.4’s ball-and-chain domain gained a mass corresponding to one sulfur atom, while no such mass increase was seen for a mutant protein with C13S. Polysulfides with more than two sulfur atoms are unstable and detection using mass spectrometry is difficult [24]. Therefore, the existence of additional sulfur atoms after incubation with polysulfides cannot be excluded; in fact, a weak signal for proteins with two sulfurs was detected (Fig. 5). Nevertheless, mass spectrometry and electrophysiology measurements show that the underlying mechanism of inactivation slowing is not based on formation or breakage of a disulfide bridge with other parts of the channel protein but rather on sulfhydration at C13. An involvement of H16 can be also excluded.
H2S and polysulfides do not have noticeable effects on other aspects of Kv1.4 channels such as ion permeation or activation gating. Since N-type inactivation of Kv1.4 channels is relatively slow and overlaps with C-type inactivation in kinetics, polysulfide-induced loss of N-type inactivation only marginally alters the peak current obtained at strong depolarizations (e.g., 50 mV). Depending on the stimulation frequency, NaHS may also inhibit peak Kv1.4 current because of a slowing of recovery from N-type inactivation, which is particularly prominent at low pH. Therefore, depending on the exact conditions, NaHS-induced slowing of the recovery from N-type inactivation may actually compensate the slowing of inactivation such that it is not straightforward to predict the physiological outcome. To limit variability, we measured evoked APs under slow-voltage clamp conditions and found a small but noticeable increase of AP width with application of Na2S4 (Suppl. Figure 3), which may be one physiological consequence of Kv1.4 modulation by sulfhydration. In addition, experiments by Feng et al. [12] in rat trigeminal ganglion neurons identified a decrease in K+ outward current and an increase in AP frequency on NaHS application, which is compatible with a decreased availability of Kv1.4 channels.
Thus far, the involvement of cysteine residues in the redox sensitivity of Kv3.4 has not been directly demonstrated. N-type inactivation of Kv3.4 channels is much faster than that of Kv1.4 channels, and Kv3.4 channels only display very slow C-type inactivation. Therefore, application of NaHS and Na2S4 not only removed inactivation almost completely but also resulted in a marked increase of the peak current, suggesting that sulfhydration will result in a stronger increase in K+ current mediated by Kv3.4 compared with Kv1.4. Another marked difference resides in the pH sensitivity of N-type inactivation: while the speed of N-type inactivation of Kv1.4 decreases with increasing pH, the opposite is true for Kv3.4 channels.
The N-terminal ball-and-chain motif of Kv3.4 harbors no histidine residue but two cysteine residues (C6, C24). The cysteines are located in close proximity in a stable four-leaf clover-like conformation [3,2] and are able to form a disulfide bridge under oxidizing conditions [3]. Polysulfides not only sulfhydrate cysteines but also promote formation of disulfide bonds [22,16], both of which could impair N-type inactivation. An expected consequence of the disulfide postulate is that mutagenesis of either C6 and C24 in isolation should result in similar functional effects. However, data shown in Fig. 7 revealed that mutation C24S had a considerably smaller impact on N-type inactivation than mutation C6S. Complete insensitivity of N-type inactivation for NaHS or polysulfides was only achieved when both cysteine residues were removed (C6S:C24S) indicating a posttranslational modification different from the generation of an intra-subunit disulfide bridge. Similar results were obtained with the cysteine-specific modifier DTNP and diamide, which is expected to promote cysteine glutathionylation. Although modification of C6 contributes much more to removal of inactivation than C24, only combination of both makes the channel resistant to cysteine modifying agents. In general, modification of C24 appears to be a partially tolerated event because it does not completely eliminate but only slows down inactivation. In Kv3.3 channels only C6 exists, so we may hypothesize that inactivation of this channel is also subject to oxidative modulation, possible explaining some variability among different host cells as observed earlier [13].
As stated above, the oxidation states of H2S and HS– are incompatible with direct sulfhydration of thiol groups in proteins and require pre-oxidized substrates. Therefore, the results obtained in the whole-cell patch-clamp configuration, where the potentially oxidizing pipette solution diffuses into the cytosol, could be misleading. However, we have multiple lines of evidence to illustrate that sulfhydration of thiol groups by polysulfides occurs under physiological conditions. First, application of Na2S4 prior to establishing the whole-cell configuration slows N-type inactivation before the pipette solution diffuses into the cell. Second, application of Na2S4 is effective in the cell-attached configuration (HEK293 cells Fig. 8; DRG neurons, Suppl. Figure 7) where the cytoplasmic milieu remains largely undisturbed. Third, our imaging measurements with roGFP2 provide independent evidence. roGFP2 is a green fluorescent protein variant with two engineered cysteine residues (C147, C204) that yields ratiometric fluorescence signals when a disulfide bridge between the cysteines forms [10]. Application of Na2S4 and also of diamide to roGFP2-expressing HEK293T cells markedly changed the fluorescence signals (Suppl. Figure 8), confirming that C147 and C204 in the cytosol are affected in intact cells when exposed to polysulfides.
These experiments, together with the potency of polysulfides to sulfhydrate cysteines in the N termini of K1.4 and Kv3.4 channels in the nanomolar range, identify reactive sulfur species as potent modulators of N-type inactivation in mammalian Kv channels.
Supplementary Material
Acknowledgments:
We thank Dr. Ilka Wittig (Frankfurt, Germany) for helpful comments on the mass spectrometry experiment.
Funding: This work was supported by grants of the German Research Foundation RTG 1715 and RTG 2155 (ProMoAge) (S.H.H.). T.H. was supported in part by the National Institutes of Health grant GM121375.
Abbreviations
- AP
Action potential
- DRG
Dorsal root ganglion
- DTT
Dithiothreitol
- H2S
Hydrogen sulfide
- Kv
channel Voltage-gated potassium channel
- NaHS
Sodium hydrogen sulfide
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- roGFP2
reduction-oxidation sensitive green fluorescent protein 2
- RSS
Reactive sulfur species
- wt
Wild type
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
Ethical approval: All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
Informed consent: Not applicable.
References
- 1.Aldrich RW, Corey DP, Stevens CF (1983) A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature 306:436–441. doi: 10.1038/306436a0 [DOI] [PubMed] [Google Scholar]
- 2.Antz C, Bauer T, Kalbacher H, Frank R, Covarrubias M, Kalbitzer HR, Ruppersberg JP, Baukrowitz T, Fakler B (1999) Control of K+ channel gating by protein phosphorylation: structural switches of the inactivation gate. Nat Struct Biol 6:146–150. doi: 10.1038/5833 [DOI] [PubMed] [Google Scholar]
- 3.Antz C, Geyer M, Fakler B, Schott MK, Guy HR, Frank R, Ruppersberg JP, Kalbitzer HR (1997) NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature 385:272–275. doi: 10.1038/385272a0 [DOI] [PubMed] [Google Scholar]
- 4.Carbonero F, Benefiel AC, Alizadeh-Ghamsari AH, Gaskins HR (2012) Microbial pathways in colonic sulfur metabolism and links with health and disease. Front Physiol 3:448. doi: 10.3389/fphys.2012.00448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carrasquillo Y, Nerbonne JM (2014) IA channels: diverse regulatory mechanisms. Neuroscientist 20:104–111. doi: 10.1177/1073858413504003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen J, Daggett H, De Waard M, Heinemann SH, Hoshi T (2002) Nitric oxide augments voltage-gated P/Q-type Ca(2+) channels constituting a putative positive feedback loop. Free Radic Biol Med 32:638–649 [DOI] [PubMed] [Google Scholar]
- 7.Claydon TW, Boyett MR, Sivaprasadarao A, Ishii K, Owen JM, O’Beirne HA, Leach R, Komukai K, Orchard CH (2000) Inhibition of the K+ channel kv1.4 by acidosis: protonation of an extracellular histidine slows the recovery from N-type inactivation. J Physiol 526 Pt 2:253–264. doi: 10.1111/j.1469-7793.2000.00253.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuevasanta E, Moller MN, Alvarez B (2017) Biological chemistry of hydrogen sulfide and persulfides. Arch Biochem Biophys 617:9–25. doi: 10.1016/j.abb.2016.09.018 [DOI] [PubMed] [Google Scholar]
- 9.Debanne D, Guerineau NC, Gahwiler BH, Thompson SM (1997) Action-potential propagation gated by an axonal I(A)-like K+ conductance in hippocampus. Nature 389:286–289. doi: 10.1038/38502 [DOI] [PubMed] [Google Scholar]
- 10.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY (2004) Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem 279:22284–22293. doi: 10.1074/jbc.M312847200 [DOI] [PubMed] [Google Scholar]
- 11.Duprat F, Guillemare E, Romey G, Fink M, Lesage F, Lazdunski M, Honore E (1995) Susceptibility of cloned K+ channels to reactive oxygen species. Proc Natl Acad Sci U S A 92:11796–11800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng X, Zhou YL, Meng X, Qi FH, Chen W, Jiang X, Xu GY (2013) Hydrogen sulfide increases excitability through suppression of sustained potassium channel currents of rat trigeminal ganglion neurons. Molecular pain 9:4. doi: 10.1186/1744-8069-9-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez FR, Morales E, Rashid AJ, Dunn RJ, Turner RW (2003) Inactivation of Kv3.3 Potassium Channels in Heterologous Expression Systems. Journal of Biological Chemistry 278:40890–40898 [DOI] [PubMed] [Google Scholar]
- 14.Furne J, Saeed A, Levitt MD (2008) Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am J Physiol Regul Integr Comp Physiol 295:R1479–1485. doi: 10.1152/ajpregu.90566.2008 [DOI] [PubMed] [Google Scholar]
- 15.Gamper N, Ooi L (2015) Redox and nitric oxide-mediated regulation of sensory neuron ion channel function. Antioxid Redox Signal 22:486–504. doi: 10.1089/ars.2014.5884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greiner R, Palinkas Z, Basell K, Becher D, Antelmann H, Nagy P, Dick TP (2013) Polysulfides link H2S to protein thiol oxidation. Antioxid Redox Signal 19:1749–1765. doi: 10.1089/ars.2012.5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hollerer-Beitz G, Schonherr R, Koenen M, Heinemann SH (1999) N-Terminal deletions of rKv1.4 channels affect the voltage dependence of channel availability. Pflugers Arch 438:141–146. doi: 10.1007/s004240050891 [DOI] [PubMed] [Google Scholar]
- 18.Hsieh CP (2008) Redox modulation of A-type K+ currents in pain-sensing dorsal root ganglion neurons. Biochemical and biophysical research communications 370:445–449. doi: 10.1016/j.bbrc.2008.03.097 [DOI] [PubMed] [Google Scholar]
- 19.Iciek M, Kowalczyk-Pachel D, Bilska-Wilkosz A, Kwiecien I, Gorny M, Wlodek L (2015) S-sulfhydration as a cellular redox regulation. Biosci Rep 36. doi: 10.1042/BSR20150147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T (2014) Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A 111:7606–7611. doi: 10.1073/pnas.1321232111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura H (2000) Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochemical and biophysical research communications 267:129–133. doi: 10.1006/bbrc.1999.1915 [DOI] [PubMed] [Google Scholar]
- 22.Kimura H (2015) Hydrogen sulfide and polysulfides as signaling molecules. Proc Jpn Acad Ser B Phys Biol Sci 91:131–159. doi: 10.2183/pjab.91.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura H (2017) Hydrogen Sulfide and Polysulfide Signaling. Antioxid Redox Signal 27:619–621. doi: 10.1089/ars.2017.7076 [DOI] [PubMed] [Google Scholar]
- 24.Kimura Y, Mikami Y, Osumi K, Tsugane M, Oka J, Kimura H (2013) Polysulfides are possible H2S-derived signaling molecules in rat brain. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 27:2451–2457. doi: 10.1096/fj.12-226415 [DOI] [PubMed] [Google Scholar]
- 25.Kurata HT, Fedida D (2006) A structural interpretation of voltage-gated potassium channel inactivation. Prog Biophys Mol Biol 92:185–208. doi: 10.1016/j.pbiomolbio.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 26.Li L, Rose P, Moore PK (2011) Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol 51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505 [DOI] [PubMed] [Google Scholar]
- 27.Luc R, Vergely C (2008) Forgotten radicals in biology. Int J Biomed Sci 4:255–259 [PMC free article] [PubMed] [Google Scholar]
- 28.Miranda KM, Wink DA (2014) Persulfides and the cellular thiol landscape. Proc Natl Acad Sci U S A 111:7505–7506. doi: 10.1073/pnas.1405665111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moustafa A, Habara Y (2014) Hydrogen sulfide regulates Ca(2+) homeostasis mediated by concomitantly produced nitric oxide via a novel synergistic pathway in exocrine pancreas. Antioxid Redox Signal 20:747–758. doi: 10.1089/ars.2012.5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH (2009) H2S signals through protein S-sulfhydration. Sci Signal 2:ra72. doi: 10.1126/scisignal.2000464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH (2011) Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circulation research 109:1259–1268. doi: 10.1161/CIRCRESAHA.111.240242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B (2004) Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304:265–270. doi: 10.1126/science.1094113 [DOI] [PubMed] [Google Scholar]
- 33.Padanilam BJ, Lu T, Hoshi T, Padanilam BA, Shibata EF, Lee HC (2002) Molecular determinants of intracellular pH modulation of human Kv1.4 N-type inactivation. Mol Pharmacol 62:127–134 [DOI] [PubMed] [Google Scholar]
- 34.Peers C, Bauer CC, Boyle JP, Scragg JL, Dallas ML (2012) Modulation of ion channels by hydrogen sulfide. Antioxid Redox Signal 17:95–105. doi: 10.1089/ars.2011.4359 [DOI] [PubMed] [Google Scholar]
- 35.Roeper J, Lorra C, Pongs O (1997) Frequency-dependent inactivation of mammalian A-type K+ channel KV1.4 regulated by Ca2+/calmodulin-dependent protein kinase. J Neurosci 17:3379–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M (1991) Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature 352:711–714. doi: 10.1038/352711a0 [DOI] [PubMed] [Google Scholar]
- 37.Sahoo N, Goradia N, Ohlenschlager O, Schonherr R, Friedrich M, Plass W, Kappl R, Hoshi T, Heinemann SH (2013) Heme impairs the ball-and-chain inactivation of potassium channels. Proc Natl Acad Sci U S A 110:E4036–4044. doi: 10.1073/pnas.1313247110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahoo N, Hoshi T, Heinemann SH (2014) Oxidative modulation of voltage-gated potassium channels. Antioxid Redox Signal 21:933–952. doi: 10.1089/ars.2013.5614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen X, Peter EA, Bir S, Wang R, Kevil CG (2012) Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic Biol Med 52:2276–2283. doi: 10.1016/j.freeradbiomed.2012.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, Fukui K, Nagahara N, Kimura H (2013) A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nature communications 4:1366. doi: 10.1038/ncomms2371 [DOI] [PubMed] [Google Scholar]
- 41.Tang G, Wu L, Wang R (2010) Interaction of hydrogen sulfide with ion channels. Clinical and experimental pharmacology & physiology 37:753–763. doi: 10.1111/j.1440-1681.2010.05351.x [DOI] [PubMed] [Google Scholar]
- 42.Toohey JI (2011) Sulfur signaling: is the agent sulfide or sulfane? Anal Biochem 413:1–7. doi: 10.1016/j.ab.2011.01.044 [DOI] [PubMed] [Google Scholar]
- 43.Wallace JL, Wang R (2015) Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov 14:329–345. doi: 10.1038/nrd4433 [DOI] [PubMed] [Google Scholar]
- 44.Wang R (2012) Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92:791–896. doi: 10.1152/physrev.00017.2011 [DOI] [PubMed] [Google Scholar]
- 45.Wilkinson WJ, Kemp PJ (2011) Carbon monoxide: an emerging regulator of ion channels. J Physiol 589:3055–3062. doi: 10.1113/jphysiol.2011.206706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winterbourn CC, Hampton MB (2008) Thiol chemistry and specificity in redox signaling. Free Radic Biol Med 45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004 [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Li S, Hong M, Xu Y, Wang S, Liu Y, Qian Y, Zhao J (2014) A colorimetric and ratiometric fluorescent probe for the imaging of endogenous hydrogen sulphide in living cells and sulphide determination in mouse hippocampus. Org Biomol Chem 12:5115–5125. doi: 10.1039/c4ob00285g [DOI] [PubMed] [Google Scholar]
- 48.Zhang LJ, Tao BB, Wang MJ, Jin HM, Zhu YC (2012) PI3K p110alpha isoform-dependent Rho GTPase Rac1 activation mediates H2S-promoted endothelial cell migration via actin cytoskeleton reorganization. PloS one 7:e44590. doi: 10.1371/journal.pone.0044590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Z, Huang H, Liu P, Tang C, Wang J (2007) Hydrogen sulfide contributes to cardioprotection during ischemia-reperfusion injury by opening K ATP channels. Can J Physiol Pharmacol 85:1248–1253. doi: 10.1139/Y07-120 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.