

Key Points
A single-chain pHLA phage library was used to profile TCR specificities for pHLA.
TCRs that bind to a common peptide can target different features of the peptide.
Engaging different peptide features results in distinct peptide repertoires.
Visual Abstract
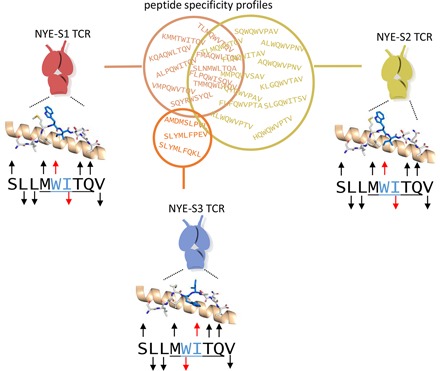
Abstract
The molecular rules driving TCR cross-reactivity are poorly understood and, consequently, it is unclear the extent to which TCRs targeting the same Ag recognize the same off-target peptides. We determined TCR–peptide–HLA crystal structures and, using a single-chain peptide–HLA phage library, we generated peptide specificity profiles for three newly identified human TCRs specific for the cancer testis Ag NY-ESO-1157–165–HLA-A2. Two TCRs engaged the same central peptide feature, although were more permissive at peripheral peptide positions and, accordingly, possessed partially overlapping peptide specificity profiles. The third TCR engaged a flipped peptide conformation, leading to the recognition of off-target peptides sharing little similarity with the cognate peptide. These data show that TCRs specific for a cognate peptide recognize discrete peptide repertoires and reconciles how an individual’s limited TCR repertoire following negative selection in the thymus is able to recognize a vastly larger antigenic pool.
Introduction
The presentation of intracellularly processed peptides by class I HLA molecules on the surface of cancer or virally infected cells enables their direct recognition and elimination by CD8+ αβ T cells (1–3). The specificity of TCRs toward a given peptide–HLA (pHLA) complex ensures appropriately targeted natural T cell responses. However, for the ∼1 × 107−8 unique TCRs present in the body to recognize the entire peptide repertoire, any given TCR must be capable of recognizing up to a million distinct theoretical peptides (4, 5). Thus, TCR cross-reactivity or polyspecificity is thought to be essential for adequate recognition of the potential pathogenic repertoire. TCRs must also simultaneously display remarkable specificity for distinguishing foreign Ag from the more limited HLA-specific self-peptide repertoire. To achieve this, TCRs typically engage a small number of exposed antigenic features or “hotspots,” whereas other peptide positions at the periphery of the TCR–pHLA interface show greater amino acid permissivity (6).
TCR specificity is driven by six CDR loops within the α- and β-chains of the receptor (2). The CDR1 and CDR2 sequences are germline encoded, whereas the CDR3 loops (ImMunoGeneTics database [IMGT] positions 105–117) have a much higher degree of sequence diversity because of somatic rearrangement, including the deletion and/or insertion of nucleotides as a consequence of V(D)J recombination (1–3). Although contacts between the germline-encoded CDRs and peptide are often observed, peptide specificity is most commonly associated with the central CDR3 residues (IMGT positions 108–113), which lie at the more flexible tip of the CDR loop, with flanking regions encoded by the germline V- and J-genes (7, 8).
TCRs that bind the same peptide have been reported to have restricted V-gene usage (9) and/or shared CDR3 sequences (7, 8). However, structural studies of multiple TCRs recognizing a common pHLA show that recognition motifs divergent in sequence but with common structural features can be used to target the same peptide hotspot. Examples include TCRs that recognize Tax–HLA-A2, HIV nef–HLA-A24 or human CMV–HLA-A2, and the MART-1–HLA-A2–specific TCRs DMF5 and Mel5 (10–15). Alternatively, TCRs can recognize different antigenic features on a given peptide, for example, on EBV–HLA-B*08:01 (16, 17). This appears to be particularly common for TCRs recognizing longmer class I peptides that are typically 11–14 aa, such as HIV–HLA-B*35:08 and NY-ESO-1–HLA-B*07:02, due to significant peptide flexibility (18, 19).
Sequence analysis of the Vα repertoires of both natural and vaccine-induced TCRs recognizing the well-studied 9-mer NY-ESO-1157–165–HLA-A2 demonstrates that, in general, they are relatively diverse in both V- and J-gene usage and CDR3α length (20, 21). Recognition of this peptide by the wild-type TCR (1G4), as well as its affinity-enhanced variants, is dominated by a single hotspot consisting of two highly hydrophobic residues, methionine and tryptophan (MW), in positions 4 and 5, respectively, which protrude from the pHLA surface (MW-peg) (22–24).
To maintain an effective functional adaptive response, multiple TCRs should not engage the exact same pool of peptides, as this would severely limit the total peptide landscape recognized by that individual’s T cells. In this study, we sought to investigate whether TCRs with a shared binding footprint, engaging the same positions of a peptide, also share overlapping cross-reactivity profiles. TCR–pHLA crystal structures and peptide variant specificity profiles were generated for three newly isolated TCRs against NY-ESO-1157–165–HLA-A2 (NYE_S1, NYE_S2, and NYE_S3). NYE_S1 and NYE_S2 TCRs have an α-chain–centric binding pocket that accommodates the MW-peg, and accordingly, these TCRs have partially overlapping peptide specificity profiles. In contrast, the NYE_S3 TCR recognizes a different conformation of the peptide leading to an unrelated peptide specificity profile.
Materials and Methods
Cell lines
PBMCs used in this study were obtained from healthy volunteers. The Oxford A Research Ethics Committee approved protocol 13/SC/0226 (Immunocore study protocol number IMCres02) was used to obtain written consent for all blood donations and was fully approved by the National Research Ethics Committee South Central. T2 APCs (LCL721 × CEM-C7) were obtained from American Type Culture Collection and cultured in R10 media.
Identification of NY-ESO-1157–165–HLA-A2–specific T cell clones
NYE_S2 and NYE_S3 TCRs were generated from T cells isolated from HLA-A*02:01+ healthy donors stimulated with NY-ESO-1157–165(9V) peptide–loaded APCs (either monocyte-derived dendritic cells or CD40-L–activated B cells). Following multiple rounds of stimulation, polyclonal T cell lines were screened for peptide specificity by IFN-γ ELISpot (BD Biosciences). T cell clones were established from polyclonal T cell lines by sorting activated T cells following peptide stimulation or tetramer binding using a BD FACSAria II. NYE_S1 TCR was identified by phage display using libraries derived from T cells isolated from HLA-A*02:01+ healthy donors (25).
X-scan mutagenesis IFN-γ ELISpot assays
X-scanning mutagenesis IFN-γ ELISpot assays were conducted according to the manufacturer’s instructions (BD Biosciences) and have been described previously (26). Briefly, TCR-transduced T cells were incubated at 5 × 104 cells per well with HLA-A*02:01+ T2 cells pulsed at 5 × 104 cells per well with 10 μM NY-ESO-1157–165–native peptide or with peptides substituted at each position with any of the 19 alternative naturally occurring amino acids (X-scanning). Plates were incubated overnight at 37°C/5% CO2 and quantified after development using an automated ELISpot reader (ImmunoSpot Series 5 Analyzer; Cellular Technology).
Protein production and biophysical measurements
Soluble disulphide-linked heterodimeric TCRs and biotinylated NY-ESO-1157–165 (9V)–HLA-A2 were cloned into pGMT7 vector encoding a C-terminal AviTag on the β-chain or H chain, respectively (27). Protein was expressed in the BL21 (DE3) Rosetta pLysS strain, refolded from inclusion bodies and purified as previously described (28, 29). In vitro site-specific biotinylation of the biotin ligase (BirA) tag was carried out prior to size exclusion chromatography using a ratio of 1 μg BirA per 100 μg TCR/pHLA.
Surface plasmon resonance equilibrium-binding analysis was performed at 25°C using a Biacore T200 instrument equipped with a CM5 sensor chip as previously reported (30). Approximately 900 response units of each pMHC were immobilized on a single CM5 sensor chip surface before all chip surfaces, including the negative control, were blocked with biotin. Increasing concentrations of each TCR were sequentially injected over the immobilized pHLAs at 20 μl/min, and each TCR concentration series was repeated three times. Binding plots (equilibrium-binding response versus TCR concentration) were plotted in GraphPad Prism 8, and curves were fitted assuming 1:1 Langmuir binding [AB = B × ABMAX/(KD + B)] to obtain KD estimates for each interaction.
Generation of single-chain HLA libraries
Single-chain HLA (scHLA) libraries were displayed on the surface of phage as disulphide trapped single-chain trimers peptide-GCGAS-(G4S)3-β2m-(G4S)3–HLA-A2 Y84C (31). Briefly, to ensure the pHLA-recognition surface was unchanged by the addition of a C-terminal linker, a Y84C mutation was introduced into the HLA-A2 H chain. This mutation opens up the F-pocket, creating a groove where the linker can sit. The insertion of cysteine into the linker immediately following the peptide allows a disulphide bond to form with the introduced Y84C mutation, anchoring the linker to the HLA surface and additionally compensating for binding affinity lost by removal of HLA contacts to the peptide C terminus.
The scHLA construct was cloned into the phagemid pim672 (32), based on the pEX922 vector (33), using Sfi1-restriction enzyme. This inserts scHLA into an open reading frame additionally encoding an N-terminal pelB leader sequence and C-terminal coat protein pIII. Diversity was encoded at the peptide level by introducing a flat distribution of 19 aa (excluding cysteine to avoid cyclic peptide formation). All 19 aa were represented at the MHC primary anchors, Pos2 and Pos9; however, to maximize the functionality of the library with peptide correctly bound in the Ag-binding groove, the amino acid distribution was biased toward known preferences for this allele, making the theoretical diversity of this library 3.22 × 1011. The library sizes were determined postelectroporation by colony counting after limiting dilutions.
The phagemid library was introduced by electroporation into Escherichia coli TG1 cells and grown in 2 × YT media containing 2% glucose, 100 μg/ml ampicillin to OD600 = 0.4, and Hyperphage (PROGEN) added at an infection ratio of ∼20:1 phage to E. coli. The cultures were then mixed by inverting, incubated for 30 min at 37°C, pelleted and resuspended in 2 × YT media containing 100 μg/ml ampicillin and 50 μg/ml kanamycin and subsequently incubated at 26°C for 16 h with shaking. Phage particles were isolated by polyethylene glycol (PEG) precipitation and 0.45 μM filtration. The use of Hyperphage provided high-valence display such that each phage displayed ∼5 copies of scHLA, enabling avidity-driven recognition of WT TCRs (34).
Panning
Two hundred nanomolar biotinylated TCRs NYE_S1, S2, and S3 were captured on streptavidin-coated paramagnetic beads (Thermo Fisher Scientific) and incubated at room temperature with gentle rotation with ∼1 × 1011 phage particles per selection, preblocked in debiotinylated 3% milk–PBS. After 1 h, beads were pulled down with a magnet and washed three times with 3% milk–PBS and twice with PBS containing 0.1% Tween. Phage particles were eluted with 10 μg/ml trypsin and used to infect early log phase TG1 E. coli cells and plated onto 2 × YT (plus 2% glucose and 100 μg/ml ampicillin) plates at 30°C for 16 h. Colonies were resuspended in 2 × YT containing 2% glucose, 100 μg/ml ampicillin, and 20% glycerol; frozen on dry ice; and stored at −80°C for the next round of panning. Three rounds of selection were performed.
Deep sequencing and cluster analysis of pHLA libraries
DNA was isolated from each glycerol stock by Miniprep (27104; QIAGEN). Sequencing libraries were prepared with molecular indexing based on a method described in Turchaninova et al. (35). Briefly, phagemid DNA was amplified with primers containing unique molecular indexes. Purified PCR products (AMPure XP beads; Beckman Coulter) were used for sequencing library preparation using NEBNext Ultra II DNA Library Prep Kit (E7645S; New England Biolabs) according to manufacturer’s instructions. Libraries were sequenced using Illumina V3 SBS chemistry on the MiSeq sequencer. FASTQ files were merged using BBMerge (36); molecular index and peptide sequence were extracted from each read, and sequence logos were generated using iceLogo (37). Sampling 0.25% of the library indicated that 98.7% of the encoded peptides were unique.
Unique peptide sequences from scHLA libraries were clustered using t-distributed stochastic neighbor embedding (tSNE) (38) on the basis of pairwise biochemical distance using the BLOSUM45 substitution matrix. The tSNE analysis was implemented using the Rtsne package in R version 3.4.4. tSNE hyperparameters were set to ensure cluster convergence, and consistency of peptide cluster identity between runs of the tSNE was ensured.
Crystallization
For NYE_S1, TCR–pHLA complex was prepared by mixing purified TCR and pHLA at equimolar ratios. Crystallization trials, using 100 nl protein solution plus 100 nl reservoir solution in sitting-drop vapor diffusion format, were set up in two-well MRC Crystallization plates using a mosquito (TTP Labtech) robot. Plates were maintained at 20°C in a Rock Imager 1000 (Formulatrix) storage system. Initial crystals grew in the PEGs II Crystallization Screen (QIAGEN), condition F6. Larger crystals were grown by cross-seeding into a grid of 10–25% PEG 4000, 0.1 M sodium citrate (pH 5–6), 0.2 M ammonium sulfate using a Seed Bead–generated seed solution and by increasing the drop size to 1 μl protein plus 1 μl well solution. Crystals were cryoprotected by addition of 15% glycerol directly to the drop and then flash-cooled at 100 K. X-ray diffraction data were collected at Diamond Light Source on beamline I02. Diffraction images were indexed, integrated, scaled, and merged using DIALS (39) and Aimless (40) through the xia2 processing suite (41).
For NYE_S2 and NYE_S3 TCR–pHLA complexes, crystallization trials using 150 nl protein solution plus 150 nl reservoir solution in sitting-drop vapor diffusion format were set up in two-well MRC Crystallization plates using a Crystal Gryphon (Art Robbins) robot. Plates were maintained at 20°C in a Rock Imager 1000 (Formulatrix) storage system. Diffraction quality crystals were grown in the following conditions: 0.02 M sodium/potassium phosphate, 20% PEG 3350 (NYE_S2 TCR–pHLA) and 0.2 M ammonium sulfate, 15% PEG 8000, and 0.1 M Tris (pH 7.5) (NYE_S3 TCR–pHLA). Crystals were cryoprotected using a 30% solution of ethylene glycol and then flash-cooled at 100 K. X-ray diffraction data were collected at Diamond Light Source (Oxfordshire, U.K.) at the I04-1 beamline at wavelength 0.91587 Å. Diffraction images were indexed, integrated, scaled, and merged using XDS and XSCALE (42, 43) through the xia2 data-processing suite (41). CC1/2 (44), Rpim, and I/σI statistics in the highest-resolution shell (criteria CC1/2 > 0.5, Rpim < 100%, and I/σI > 1) were used to determine high-resolution cutoffs (Table I).
Table I. NYE_S1, NYE_S2, and NYE_S3 TCR–pHLA data collection and refinement statistics.
| NYE_S1 Complex | NYE_S2 Complex | NYE_S3 Complex | |
|---|---|---|---|
| pdb Accession code | 6RPB | 6RPA | 6RP9 |
| Data collection | |||
| Space group | P1 | C2 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 75.3, 77.1, 184.6 | 173.1, 84.3, 111.5 | 106.6, 85.6, 171.9 |
| α, β, γ (°) | 93.0, 93.5, 118.2 | 90.0, 129.8, 90.0 | 90.0, 91.8, 90.0 |
| Resolution (Å) | 73.11-2.50 (2.56-2.50) | 66.51-2.56 (2.60-2.56) | 85.61-3.12 (3.17-3.12) |
| Rmerge (%) | 9.4 (41) | 6.4 (207) | 11.7 (99.9) |
| Rpim (%) | 9.4 (41) | 2.5 (77.5) | 4.7 (56.7) |
| CC1/2 | 0.971 (0.767) | 0.999 (0.637) | 0.999 (0.627) |
| I/σI | 2.7 (1.1) | 17.9 (0.96) | 10.99 (1.25) |
| Completeness (%) | 96.9 (96.0) | 99.3 (99.7) | 100.0 (100.0) |
| Multiplicity | 1.7 (1.7) | 7.66 (7.94) | 6.76 (3.97) |
| Refinement | |||
| Resolution (Å) | 73.09-2.50 (2.56-2.50)a | 66.51-2.56 (2.63-2.56) | 68.01-3.12 (3.20-3.12) |
| No. reflections | 125234 (9382) | 37579 (1945) | 52792 (2717) |
| Rwork/Rfree | 24.9 (34.9)/27.3 (35.6) | 25.0/29.3 (42.9/43.5) | 22.4/24.4 (36.7/41.1) |
| No. atoms | |||
| Protein | 25965 | 6277 | 16633 |
| Water | 229 | 49 | — |
| B factors | |||
| Protein | 51.1 | 83.1 | 116.8 |
| Water | 32.5 | 48.1 | — |
| RMSD | |||
| Bond lengths (Å) | 0.007 | 0.008 | 0.006 |
| Bond angles (°) | 1.098 | 1.155 | 0.996 |
Rpim (the precision-indicating merging R-value) = 1/(N−1) × Rmerge, where N is the redundancy. CC1/2 is the mean intensity correlation coefficient of half-data sets. Dashes (—) indicate that no waters were modelled for the NYE_S3 structure.
Values in parentheses are for highest resolution shell.
Structure determination and refinement
Molecular replacement was used to phase all crystal structures, using Protein Data Bank (pdb) entry 5e00 chains A and B (for HLA-A2 and β2m), pdb 3REV chain A (NYE_S2 TCRα), pdb 4DZB chain B (NYE_S2 TCRβ), 3QDJ chain D (NYE_S3 TCRα), and 5D2N chain E (NYE_S3 TCRβ) as search models in Phaser (45). For NYE_S1 TCR, a higher resolution structure of a related complex was used as the search model (data not shown), which had been solved using 4FTV as a starting model. Scoring functions after placing the final nonpeptide containing molecule were as follows: NYE_S1, TFZ 98.3, and LLG 15048; NYE_S2, TFZ 49.9, and LLG 3039; and NYE_S3, TFZ 42.1, and LLG 2218. Subsequently, 100 cycles of jelly-body refinement (σ 0.05, weighting term 0.001 and global NCS restraints) in Refmac (46) yielded R-factors as follows: NYE_S1, Rwork 31.2, and Rfree 32.1; NYE_S2, Rwork 32.7, and Rfree 33.7; and NYE_S3, Rwork 38.0, and Rfree 37.6.
Manual model adjustment was performed in Coot (47), and Refmac was used for further refinement. Translation–libration–screw-motion restraints were applied in final refinement rounds for the NYE_S1 and NYE_S3 TCR–pHLA complex structures; however, translation–libration–screw-restrained refinement was unstable for the NYE_S2 TCR–pHLA complex and was therefore not used. For refinement of the NYE_S1 TCR–pHLA complex structure, initial global noncrystallographic symmetry restraints were relaxed to automatically generated local restraints in final refinement rounds. For refinement of the NYE_S3 TCR–pHLA complex structure, initial global noncrystallographic symmetry restraints were relaxed to medium restraints in final refinement rounds (excluding the following regions in NCS domain definitions: chains D, I, and K residues 108–115 and chains E, J, and L residues 65–71). Stereochemical properties of all models were assessed using the PDB Validation Suite (48). Ramachandran statistics are the following: NYE_S1 TCR–pHLA complex, 98% favored, 2% allowed, and no rotamer outliers; NYE_S2 TCR–pHLA complex, 97% favored, 3% allowed, and no rotamer outliers; NYE_3 TCR–pHLA complex, 94% most favored, 6% additionally allowed, and a single rotamer outlier. Full data collection and refinement statistics are given in Table I.
Crystallographic figures were created using PyMOL (Schrödinger). All structural alignments were performed using Superpose (49). Buried surface area and TCR docking geometry statistics based on those described previously (2) were generated using Molecular Operating Environment (Chemical Computing Group) (50). Briefly, TCR crossing angles are calculated from the eigenvector relating the vector through the midpoints of the Vα and Vβ domains' disulphide bonds to the vector along the length of the MHC helices that defines the Ag-binding groove (V1). The tilt angle relates the pseudo–2-fold TCR symmetry axis (relating the α to the β-chain) to pHLA V1 such that a negative value reflects a tilt toward the peptide N terminus and a positive value toward the C terminus. The roll angle relates the pseudo–2-fold TCR symmetry axis to the vector perpendicular to V1 lying in the plane of the pHLA Ag-binding groove, where a negative value reflects a roll toward the HLA α1 helix, whereas a positive value reflects a roll toward the HLA α2 helix.
Crystal structure composition
The NYE_S1 TCR–pHLA crystal structure contains four copies of the TCR–pHLA complex per asymmetric unit: HLA, chain A aa 1–276 (lacking residues 221–225), chain F aa 1–276 (lacking residues 17–18), chain K aa 1–274 (lacking residues 195–198, 221–227, and 250–253), chain P aa 1–275 (lacking residues 225–227); β2m, chains B, G, L, and Q aa 0–99; peptide, chains C, H, M, and R aa 1–9; TCRα, chain D aa 3–219 (lacking residues 141–146), chain I aa 3–219 (lacking residues 141–146), chain N aa 3–217 (lacking residues 139–146 and 181–182), chain S aa 3–217 (lacking residues 140–146 and 181); and TCRβ, chain E aa 3–256, chain J aa 3–256, chain O aa 3–254, and chain T aa 2–256. TCR residue numbering was assigned according to IMGT conventions (51); despite sequence gaps between TCRα residues 29–36, 58–63, 68–74, and 110–113 and TCRβ residues 29–37, 58–63, 72–74, 81–83, and 110–112, the TCRα- and TCRβ-chains are continuous in the variable domains.
The NYE_S2 TCR–pHLA crystal structure contains one copy of the TCR–pHLA complex per asymmetric unit: HLA, chain A aa 1–276; β2m, chain B aa 1–99, peptide, chain C aa 1–9; TCRα, chain D aa 1–192 (lacking residues 145–147, 160–166, 180–184, and 193–222, which lie in disordered regions of the constant domain); and TCRβ, chain E aa 1–252 (lacking residues 236–240 and 253–257, which lie in disordered regions of the constant domain). TCR residue numbering was assigned according to IMGT conventions (51); despite sequence gaps between chain D residues 29–36, 59–62, and 68–74 and chain E residues 29–37, 59–63, 81–83, and 110–112 plus a 2 aa insertion between chain D residues 111–112, the TCRα- and TCRβ-chains are continuous in the variable domains.
The NYE_S3 TCR–pHLA crystal structure contains two copies of the TCR–pHLA complex and one apo TCR copy per asymmetric unit: HLA, chain A aa 1–275 and chain F aa 1–275; β2m, chain B aa 0–99 and chain G aa 0–99; peptide, chain C aa 1–9 and chain H aa 1–9; TCRα, chain D aa 4–217 (lacking residues 1–3 and 218–223), chain I aa 4–218 (lacking residues 1–3 and 219–223), and chain K aa 8–218 (lacking residues 1–7, 142–148, and 219–223); and TCRβ, chain E aa 1–256 (lacking residue 257), chain J aa 1–256 (lacking residue 257), and chain L aa 2–256 (lacking residues 1 and 257). TCR residue numbering was assigned according to IMGT conventions (51); despite sequence gaps between chains D, I, and K residues 29–36, 58–63, 68–74, and 110–113 and chains E, J, and L residues 29–37, 58–63, and 81–83, the TCRα- and TCRβ-chains are continuous in the variable domains.
Results
Characterization of three novel NY-ESO-1157–165–HLA-A2–specific TCRs
Three wild-type TCRs (NYE_S1, NYE_S2, and NYE_S3) recognizing NY-ESO-1157–165(9V)–HLA-A2, a heteroclytic peptide variant of NY-ESO-1157–165–HLA-A2 (52), were identified and then expressed, purified, and tested to determine their biophysical characteristics (Table I). TCR sequences are reported alongside those of previously identified NY-ESO-1157–165-specific TCRs from both natural and vaccine-induced immunodominant responses (Table II) (20, 21). NYE_S1, NYE_S2, and NYE_S3 bound to NY-ESO-1157–165(9V)–HLA-A2 with dissociation constants (KD) of ∼7.0, 7.1, and >82 μM (range of estimated KD 82–186 μM), respectively, as determined by surface plasmon resonance equilibrium–binding measurements (Supplemental Fig. 1). These KD values are consistent with other immunologically productive TCRs, which have affinities in the 0.1–500-μM range (3).
Table II. CDR sequence alignments based on IMGT numbering for known NY-ESO-1157–165–HLA-A2–specific TCRs.
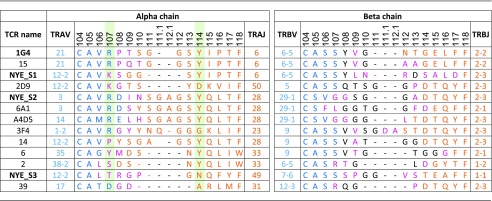 |
CDR3 sequence alignments based on IMGT numbering. TCRs for which structures have been obtained are in bold. Where several TCRs have been reported with common V-gene usage, only a single example has been randomly chosen (20); residues in the CDR3 are colored according to whether they are encoded by the V-gene (blue), D-gene (black), and J-gene (orange) or are at the junction of gene segments where the greatest amino acid diversity is obtained (pink). Residues involved in recognition of MW-peg in the NYE_S1, NYE_S2, and 1G4 TCRs, IMGT positions R/K107 and Y114, are highlighted in green.
NYE_S1 and NYE_S2 TCRs adopt an overall canonical binding geometry toward NY-ESO-1157–165(9V)–HLA-A2
We determined crystal structures of NYE_S1 and NYE_S2 TCR-NY-ESO-1157–165(9V)–HLA-A2 complexes to resolutions of 2.50 and 2.56 Å, respectively (Fig. 1A, 1D, Table I). The NYE_S1 structure contains four TCR–pHLA copies in the asymmetric unit, which are all in close overall structural agreement (root-mean-square deviation [RMSD] < 0.35 Å between any two copies, comparing Cα positions for HLA residues 1–181, peptide 1–9, Vα 3–128, and Vβ 3–128; copy 1 used for all further structural comparisons), whereas the NYE_S2 structure contains a single TCR–pHLA copy. For both structures, there is well-defined electron density at the TCR–pHLA interface (Fig. 1B, 1E). Both NYE_S1 and NYE_S2 TCRs display similar canonical pHLA-binding footprints in which the Vα and Vβ domains and, in particular, the germline-encoded CDR1α and CDR1β lay over HLA helices α2 and α1, respectively (Fig. 1C, 1F, Supplemental Table I). The V(D)J recombination–derived CDRs, CDR3α and CDR3β, sit centrally above the HLA peptide-binding groove and dominate TCR contacts with the peptide (Fig. 1C, 1F).
FIGURE 1.

NYE_S1 and NYE_S2 TCR–binding footprints on NY-ESO-1157–165(9V)–HLA-A2 and NY-ESO-1157–165 peptide conformations. Overall cartoon representation of TCR–pHLA complexes for NYE_S1 (A) and NYE_S2 (D) TCRs. NY-ESO-1157–165(9V)–HLA-A2 peptide conformation within NYE_S1 (B) and NYE_S2 (E) TCR containing structures. CDR positions above pHLA surface for NYE_S1 (C) and NYE_S2 (F) TCRs. HLA H chain, wheat; β2m, brown; TCR CDR1α, orange; TCR CDR2α, yellow; TCR CDR3α, maroon; TCR CDR1β, dark blue; TCR CDR2β, cyan; TCR CDR3β, green; NY-ESO-1 peptide, pink. 2Fo–Fc maps contoured at 1σ and carved within 2 Å of the peptide are shown in light purple. Arrows above or below the peptide sequence indicate if each side chain is either exposed or buried respectively, relative to the HLA-peptide–binding groove.
The peptide conformations in both structures are very similar (RMSD, 0.306 Å across 9 Cα positions) and show relatively minor deviations from the conformations described for previously determined NY-ESO-1157–165–HLA-A2 structures (Fig. 1B, 1E, Supplemental Fig. 2). The large, hydrophobic side chains at positions Met4 and Trp5 point upwards, away from the peptide-binding groove. Peptide positions Leu2 and Val9 are the primary anchor residues, whereas Ile6 adopts a secondary anchor position, securing the peptide in the peptide-binding groove of the HLA.
Specificity toward the peptide MW-peg motif is primarily driven by two germline CDR3α residues
For the 1G4 and NYE_S1 TCRs, the α- and β-chains both contribute to the overall peptide-binding interface, whereas for NYE_S2, the α-chain dominates the interface (Supplemental Table I). These three TCRs predominantly use the CDR3α loop to engage the MW-peg motif, with the backbone of the central residues stacking around the hydrophobic Met4 and Trp5 side chains (Fig. 2A–C). NYE_S1 makes additional contacts to Trp5 via Gln37 in the CDR1α, whereas the longer CDR3α for NYE_S2 versus NYE_S1 is able to wrap more closely around Met4 (Fig. 2D, 2F). β-Chain–mediated peptide contacts are dominated by the CDR3β loop that makes contacts to the exposed Ile6 backbone and Thr7, whereas NYE_S1 also makes additional contacts to side-chain Gln8, mediated by the CDR1β (Fig. 2G–I).
FIGURE 2.

Structural characterization of NYE_S1, 1G4, and NYE_S2 TCRs binding to NY-ESO-1157–165(9V)–HLA-A2. CDR3α–mediated interactions with the MW-peg for NYE_S1 (A), 1G4 (B), and NYE_S2 (C) TCRs, respectively. CDR3α and CDR3β stacking against the MW-peg motif for NYE_S1 (D), 1G4 (E), and NYE_S2 (F) TCRs. The areas depicted by dashed boxes are expanded in panels (G–I) to highlight CDR3β-mediated interactions. TCR CDR1α, orange; TCR CDR3α, maroon; TCR CDR3β, green; peptide, gray sticks; MW-peg, blue sticks; HLA helix-α1, wheat cartoon; potential H-bonds, yellow dashed lines.
Aligning the NYE_S1 and NYE_S2 TCR sequences with those of previously identified NY-ESO-1157–165-specific TCRs demonstrated significant diversity in CDR3α length, amino acid sequence, and variable chain usage (Table II). However, of all residues contacting the peptide, the CDR3α Arg107/Lys107 and Tyr114 residues were the most highly conserved across multiple TRAV/TRBV chain pairings. From the crystal structures, we observed that the Arg107/Lys107 side-chain carbon backbone stacks against the Trp5 side chain, whereas the Tyr114 side-chain hydrophobic ring stacks against both Met4 and Trp5 side chains, with its hydroxyl group making a putative hydrogen bond to the peptide backbone (to the Met4 carbonyl group) (Fig. 2G–I, Table II. This observation suggested that the α-centric specificity displayed by the 1G4, NYE_S1, and NYE_S2 TCRs is driven by a selective pressure for these two CDR3α amino acid positions, which, together, help to pin the loop around the MW-peg, highlighting the importance of this feature in shaping the TCR repertoire.
NYE_S3 TCR binds to a flipped NY-ESO-1157–165(9V)–HLA-A2 peptide conformation lacking an exposed MW-peg motif
A crystal structure of the NYE_S3 TCR–pHLA complex was determined to a resolution of 3.12 Å (Fig. 3, Table I). This structure contains two TCR–pHLA copies, which are in close overall structural agreement (RMSD, 0.75 Å between the two copies, comparing Cα positions for HLA residues 1–181, peptide 1–9, Vα 3–128, and Vβ 3–128; copy 1 used for all further structural comparisons) and one apo TCR copy per asymmetric unit. The most striking observation from our NYE_S3 TCR–pHLA crystal structure was a novel peptide conformation with a pronounced rearrangement of the central peptide residues, Met4, Trp5, and Ile6, relative to the conformations observed for pHLA alone (Fig. 3B, Supplemental Fig. 2G, 2H). In this structure, Ile6 no longer acts as a secondary anchor as the orientations of Trp5 and Ile6 side chains are flipped to a down (Trp5)–up (Ile6) arrangement relative to the peptide-binding groove, rather than the usual up (Trp5)–down (Ile6) configuration observed in all previous structures. This disruption of the conserved MW-peg epitope results in a very distinct TCR–pHLA-binding interface for NYE_S3 versus the NYE_S1 and NYE_S2 TCRs (Fig. 3A, 3C, Supplemental Table I).
FIGURE 3.

Structural characterization of NYE_S3 TCR binding to NY-ESO-1157–165(9V)–HLA-A2. (A) Overall cartoon representation of NYE_S3 TCR–pHLA complex. (B) NY-ESO-1157–165(9V)–HLA-A2 peptide conformation within the NYE_S3 TCR–containing structure. (C) CDR positions above pHLA surface for NYE_S3. (D) CDR3α and CDR3β stacking against the peptide Ile6 side chain. The area depicted by the dashed box is expanded in (E) to highlight specific interactions. (F) Contribution of additional NYE_S3 TCR CDRs to peptide binding. Transparent CDR3α and CDR3β lie in the foreground of this panel.
The NYE_S3 TCR binds with a typical TCR binding footprint, whereby TCRα and TCRβ-mediated contacts are directed primarily to HLA helix-α2 and HLA helix-α1, respectively (Supplemental Table I). Despite shared Vα-chain usage between NYE_S1 and NYE_S3, the TRAV12-2 germline–encoded CDRs do not bind to the same features of the pHLA. CDR1α engages peptide in NYE_S1, whereas it engages the α2 helix of the HLA in NYE_S3. In contrast to the NYE_S1 and NYE_S2 TCRs, for NYE_S3, β-chain contacts dominate the peptide-binding interface (Fig. 3D–F, Supplemental Table I). Both the CDR3α and CDR3β loops of NYE_S3 contain central Pro–Gly motifs, which form rigid type II β–hairpin turns, potentially limiting the conformational flexibility of these CDRs (Fig. 3E). For CDR3α, this tight turn sits directly above the central core of the peptide, stacking against exposed Met4 and Ile6 side chains. Further contacts to Ile6 come from the CDR3β Ser108 and Gly111, CDR1β Val37, and the CDR2β Asn57 and Tyr58 residues (Fig. 3E, 3F). In addition, contacts to the Thr7 side chain and Gln8 backbone NH group as well as the Gln8 side chain are mediated by the CDR3β backbone and CDR2β Tyr58, respectively (Fig. 3E, 3F).
Comparison of pHLA-bound and apo TCR copies within the NYE_S3 TCR–pHLA structure reveals possible conformational changes in the TCR upon engagement of pHLA (Supplemental Fig. 3). Alignment of either the Vα or Vβ domains of NYE_S3 apo TCR (unbound) with the equivalent domain in NYE_S3 TCR–pHLA (bound) reveals close structural agreement, apart from the CDR3α loop (Supplemental Fig. 3A, 3B). In its unbound form, the CDR3α loop of the NYE_S3 TCR adopts an elongated conformation that is stabilized by an extension of the flanking β-strands. However, when bound to pHLA, flexion at Arg108 and Gln115 enables CDR3α to bend, without which the tip of the loop would clash with peptide Met4 (Supplemental Fig. 3C). This CDR3α conformation stacks between exposed peptide Met4 and Ile6 side chains. To accommodate the bent CDR3α conformation, a small twist in the TCRβ-chain relative to TCRα also occurred in the NYE_S3 TCR–pHLA, compared with the NYE_S3 apo TCR (Supplemental Fig. 3D).
All three TCRs display unique peptide specificity profiles, enabling recognition of distinct off-target peptides
We performed peptide specificity profiling on the three identified TCRs using two complementary approaches. In the first cellular X-scan approach, each of the three NY-ESO-1157–165-specific TCRs were transduced into T cells. HLA-A*02:01–positive target cells were pulsed with NY-ESO-1157–165 peptide in which each amino acid position was sequentially replaced with all 19 alternative naturally occurring amino acids (171 peptides in total), and IFN-γ release was measured relative to pulsing with cognate peptide by ELISpot (Fig. 4). Using this assay, we found Trp5 to be absolutely required for recognition by all three TCRs, as no IFN-γ release was observed when this residue was replaced (Fig. 4D–F). However, Met4 could be substituted more readily for NYE_S1, as compared with NYE_S2 or NYE_S3. Surprisingly, both NYE_S1 (Fig. 4D) and NYE_S2 (Fig. 4E) could also accommodate a Gln at position 4, potentially because of the similar length of the side chains and introduction of additional hydrogen bonding potential to the exposed CDR3α backbone. Consistent with its role as a secondary anchor, Ile6 displayed limited selectivity for NYE_S1 and NYE_S2, tolerating substitutions to other hydrophobic amino acid side chains. However, the upward facing conformation of this residue in the NYE_S3 structure resulted in this TCR tolerating Arg in this position, a large charged residue that is not able to act as a downward-facing secondary anchor (Fig. 4F). In line with the structural observations, the NYE_S1 and NYE_S3 TCRs also showed restricted specificity for Gln at position 8, whereas this position was more permissive in NYE_S2 (Fig. 4D–F).
FIGURE 4.

Cellular potency and X-scan TCR specificity profiles. Cellular potency was assessed using T cells transduced with NYE_S1 (A), NYE_S2 (B), and NYE_S3 (C) TCRs, exposed to HLA-A2+ T2 cells pulsed with increasing concentrations of NY-ESO-1157–165 peptide. T cell activation was measured using an IFN-γ ELISpot assay at 24 h, and EC50 values were determined (solid triangles). No response was seen at the highest concentration of peptide used for pulsing a nontransduced T cell control (open triangles) or irrelevant TAX peptide (open squares). (D–F) X-scanning mutagenesis of all NY-ESO-1157–165 single amino acid variants. Data shown are from a representative donor (n = 2). An average of triplicate data points is presented for each of the 171 separate experiments normalized to the NY-ESO-1157–165 peptide control highlighted in (A)–(C). Each amino acid is shown in one-letter code and colored according to functional similarity: positive (blue), aromatic uncharged (green), aliphatic (black), small nonpolar (orange), polar (pink), and negative (red). Outlined boxes highlight the cognate residues.
Because of the biased nature of X-scans toward the cognate peptide, we generated a more extensive, high throughput, and unbiased approach by profiling TCR specificity using scHLA phage libraries in which peptide diversity was encoded at all positions, including position 2 and position 9 (which were biased toward HLA-A2–specific anchor residues), and presented in a disulphide-linked single-chain trimer format (Fig. 5A). This method is a significant advancement for TCR specificity profiling over previous methods, as it enables the ability to simultaneously monitor the enrichment of millions of individual sequences through the use of next-generation DNA sequencing (6, 53, 54). Phage libraries encoding 5 × 108 variants were generated, and following three cycles of avidity-driven phage display panning (Fig. 5B), a peptide specificity profile was generated for each TCR (Fig. 5C).
FIGURE 5.

Molecular analysis of TCR specificity for NY-ESO-1157–165 peptide. (A) Schematic of multivalent disulphide trapped single-chain trimers (dsSCT) displayed on HLA-A2 phage libraries. (B) Schematic of phage display panning protocol. (C) Heatmaps and sequence logos displaying amino acid permissivity generated from next-generation DNA sequencing (NGS) workflow showing initial peptide diversity in which positions 2 and 9 are biased toward known HLA-A2 preferences, and Cys is excluded prior to TCR selection (left panel). The following three panels show peptide diversity following three cycles of panning using each of the three NY-ESO-1157–165 specific TCRs. Outlined boxes highlight the cognate residues.
Analysis of the sequence landscape revealed that, when given free choice of peptide, the NYE_S1 and NYE_S2 TCRs both recognized peptides shaped by the MW-peg: for both TCRs, more than 99% of the peptides (n > 2 sequence counts) contained a tryptophan at position 5 (Fig. 5C). Cluster analysis showed that more than 99% of the high-confidence peptides enriched in response to the NYE_S1 TCR were found in the same sequence cluster as the NY-ESO-1157–165 peptide (Fig. 6). Approximately 40% of peptides enriched in response to the NYE_S2 TCR were also found in this cluster. The NYE_S2 TCR displayed a preference for peptides with Gln at position 4 and was relatively permissive at position 8, whereas the NYE_S1 TCR displayed a preference for Gln at position 8 and was relatively permissive at position 4 (Figs. 5C, 6). Within the cluster containing the NY-ESO-1157–165 peptide, it was possible to identify a subcluster with restricted specificity at Q4 and Q8. This overlapping region of the NYE_S1 and NYE_S2 specificity profiles accounted for 35.6 and 8.2% of the total TCR-specific peptides, respectively (Fig. 6C). Approximately 59% of peptides enriched by the NYE_S2 TCR recognized peptides belonging to a sequence cluster related to the NY-ESO-1157–165 peptide but displayed a preference for peptides with Pro at position 6 and an aromatic residue at position 3 (Fig. 6). Recognition of this cluster was restricted to the NYE_S2 TCR. Neither the NYE_S1 or NYE_S3 TCR bind to the consensus sequence YQWQWVPAV HLA-A2 (Supplemental Fig. 1). The specificity profiles for the NY-ESO-1157–165-containing cluster were predicted by the cellular X-scan, but the NYE_S2-specific subcluster could not be predicted with single amino acid substitutions alone.
FIGURE 6.

Sequence cluster analysis of the most abundant peptides identified in response to the three NY-ESO-1157–165–specific TCRs. (A) The 500 most enriched peptides for each NY-ESO-1157–165-specific TCR were clustered by calculating BLOSUM 45 distance between all peptides, and unique sequences plotted with a two-dimensional tSNE analysis. Peptides are colored according to the TCR, to which binding has been observed. TCR NYE_S1 (orange), NYE_S2 (blue), and NYE_S3 (pink) and peptides recognized by both TCR NYE_S1 and NYE_S2 (green). The NY-ESO-1157–165 peptide is shown in black. (B) Sequence logos for each of the clusters, including a cluster that has no convergence and is presumed to be noise, are shown. (C) The number of unique peptide sequences and the percentage of peptides recognized by each TCR from each cluster are reported.
By contrast, the NYE_S3 TCR appeared to recognize a nonoverlapping peptide repertoire that did not share sequence or physiochemical properties with the NY-ESO-1157–165 peptide. The peptides enriched in response to NYE_S3 formed a single sequence cluster with the consensus sequence SLYMLFPEV that contained 48 unique peptides and accounted for 93% of the high-confidence peptides analyzed. Only 0.02% of peptides enriched in response to the NYE_S3 TCR were found in the same cluster as the NY-ESO-1157–165 peptide.
NYE_S3 bound to SLYMLFPEV–HLA-A2 with a dissociation constant (KD) of ∼110 μM, whereas neither NYE_S1 nor NYE_S2 TCR bound (Supplemental Fig. 1). Further an accurate KD could not be determined for the NYE_S3 TCR binding to NY-ESO157–165-like peptides that are the consensus of peptides enriched by the NYE_S1 (estimated KD > 350 μM) or NYE_S2 TCR. Assessment of pHLA-A2 stability at 25°C was comparable for NY-ESO-1157–165, the heteroclytic 9V variant and SLYMLFPEV (data not shown).
Discussion
In this study, we show that TCRs binding the same peptide hotspot have partially overlapping specificity profiles, whereas those binding different hotspots have nonoverlapping specificity profiles. We observed that two TCRs (NYE_S1 and NYE_S2) bound peptide in which the MW-peg is a prominent feature (24). This binding configuration has previously been reported for the 1G4 TCR (22–24) (55) and, taken together, this provides further evidence that an antigenic feature within a peptide is able to shape the TCR repertoire.
The MW-peg is likely a dominant epitope because MW are the two least abundant amino acids in human proteins (only a single codon exists for each) and are thus rarely found in the self-peptidome. This premise is supported by the observation that only 0.69% of all HLA-A2 9-mer peptides have a tryptophan at position 5 (analysis of data from 24 publicly available large scale HLA-A2 peptidomic datasets) (56). Rarely occurring sequence hotspots that distinguish the peptide from self are likely to enable the avoidance of T cell negative selection in the thymus and generate a TCR repertoire with a low potential for cross-reactivity.
Two different TCR binding modes to NY-ESO-1157–165 were observed. NYE_S1 and NYE_S2 TCR recognition of the MW-peg involves an extensive interface with the CDR3α even though the NY-ESO-1157–165–HLA-A2–specific TCR repertoire is diverse in Vα and Jα sequences and CDR3α length (20, 21). Despite this diversity, we have identified two germline-encoded CDR3α positions (IMGT107 and 114) that have conserved amino acid usage that helps to form an α-chain–centric binding pocket to accommodate the MW-peg. In contrast, the NYE_S3 TCR does not bind to the MW-peg but, instead, engages a different conformational epitope, an upward-facing Ile6 residue. This epitope, as with the MW-peg, is unlikely to be a commonly exposed epitope in the self-peptidome for two additional reasons: first, a novel backbone conformation is adopted, and second, an exposed hydrophobic residue at position 6 is unusual, as this residue tends to serve as a secondary anchor residue.
We have developed a TCR specificity screening tool able to assess up to 5 × 108 peptides simultaneously. Using this tool, the NYE_S1 and NYE_S2 TCRs enriched for peptides that share many features with the NY-ESO-1157–165 peptide and were consistent with the TCR-specific cellular X-scan. Using the scHLA approach, we were unable to identify molecular mimics of the NY-ESO-1157–165 peptide for the NYE_S3 TCR. Consistent with a requirement to engage an unusual peptide conformation, the NYE_S3 cellular X-scan has an extremely restricted permissivity at positions 4–8. Despite interrogating up to 5 × 108 variants in the scHLA libraries, it is possible that none shared the necessary combination of residues at positions 4–8 for the peptide to adopt the more unusual NY-ESO-1157–165 conformation and so allow NYE_S3 TCR binding.
The dynamic nature of peptides, especially those that are longer than 9 aa, in the HLA-binding groove is well established (57, 58), as conformational plasticity plays an important role in enabling shape complementarity between TCR, peptide, and HLA. It is perhaps unsurprising that for highly dynamic peptides, multiple TCRs can recognize and bind the peptide in distinct conformations (19). However, 9-mer peptides, such as the NY-ESO-1157–165 peptide, are usually more constrained, with different conformations typically being observed only as small backbone and side-chain rotamer movements (57–60).
Previous studies have demonstrated that off-target peptides do not need to share sequence, physiochemical, or backbone geometry with the cognate peptide (61, 62), and, in addition, multiple TCRs can bind distinct peptide hotspots on the same pHLA (17). In one instance, this was driven by conformational plasticity in the peptide (19). In this study, we build on these observations to demonstrate that TCRs that recognize distinct peptide hotspots in the same peptide can display nonoverlapping specificity profiles, thereby providing a mechanism by which TCRs could potentially evade thymic deletion caused by a specific self-peptide. This has implications for the clinic, as new strategies are currently being developed that exploit the T cell–based targeting of tumor cells by engineering highly specific TCRs against pHLA (63). Understanding a TCRs potential off-target repertoire forms an important part of an immunotherapeutic TCR safety profile (26, 64–66). With sufficient screening at the TCR discovery stage, it may be possible to identify TCRs that recognize distinct antigenic features of the peptide such that their peptide specificity profiles are different, and in consequence, a specific off-target peptide is no longer limiting.
The T cell repertoire must maintain the ability to avoid recognizing the self-peptide repertoire. TCR specificity profiles do not typically involve all peptide positions, and restricted amino acid preferences are limited to a subset of the peptide (6). Therefore, targeting a limited number of peptide positions, particularly in which these antigenic features allow discrimination from the self-repertoire, enables T cells to survive negative selection in the thymus. Equally, the permissive nature of several peptide positions enables TCR recognition of a significant number of theoretical peptides, thereby providing the coverage required by the TCR repertoire to recognize a diverse and evolving pathogenic sequence landscape (13). However, Ag-driven expansion of TCR clonotypes skews the TCR repertoire such that an individual may have many TCRs that recognize a single Ag. In this study, we show that even when multiple TCRs have the capacity to engage a common Ag, they can, nevertheless, recognize distinct and, in some instances, almost completely nonoverlapping peptide repertoires.
Supplementary Material
Acknowledgments
We thank David Cole and Ita O’Kelly for critically reading the manuscript. The authors thank Jacob Robinson, Miriam Hock, Penio Todorov, Luise Weigand, and Vanessa Clark for generating technical reagents. We also thank Diamond Light Source for beamtime and the staff of beamlines I02 and I04-1 for assistance with crystal testing and data collection.
The coordinates and structure factors presented in this article have been submitted to Protein Data Bank (http://www.rcsb.org/pdb/) under accession numbers 6RPB, 6RPA, and 6RP9.
The online version of this article contains supplemental material.
- IMGT
- ImMunoGeneTics database
- MW
- methionine and tryptophan
- MW-peg
- MW in positions 4 and 5, respectively, which protrude from the pHLA surface
- pdb
- Protein Data Bank
- pHLA
- peptide–HLA complex
- RMSD
- root-mean-square deviation
- scHLA
- single-chain HLA
- tSNE
- t-distributed stochastic neighbor embedding.
Disclosures
K.J.S. and S.G. are employees of GlaxoSmithKline. All other authors are or were employees of Immunocore, Ltd. The study was entirely funded by these organizations.
References
- 1.Turner S. J., Doherty P. C., McCluskey J., Rossjohn J. 2006. Structural determinants of T-cell receptor bias in immunity. Nat. Rev. Immunol. 6: 883–894. [DOI] [PubMed] [Google Scholar]
- 2.Rudolph M. G., Stanfield R. L., Wilson I. A. 2006. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 24: 419–466. [DOI] [PubMed] [Google Scholar]
- 3.Rossjohn J., Gras S., Miles J. J., Turner S. J., Godfrey D. I., McCluskey J. 2015. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33: 169–200. [DOI] [PubMed] [Google Scholar]
- 4.Wooldridge L., Ekeruche-Makinde J., van den Berg H. A., Skowera A., Miles J. J., Tan M. P., Dolton G., Clement M., Llewellyn-Lacey S., Price D. A, et al. 2012. A single autoimmune T cell receptor recognizes more than a million different peptides. J. Biol. Chem. 287: 1168–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arstila T. P., Casrouge A., Baron V., Even J., Kanellopoulos J., Kourilsky P. 1999. A direct estimate of the human alphabeta T cell receptor diversity. Science 286: 958–961. [DOI] [PubMed] [Google Scholar]
- 6.Adams J. J., Narayanan S., Birnbaum M. E., Sidhu S. S., Blevins S. J., Gee M. H., Sibener L. V., Baker B. M., Kranz D. M., Garcia K. C. 2016. Structural interplay between germline interactions and adaptive recognition determines the bandwidth of TCR-peptide-MHC cross-reactivity. Nat. Immunol. 17: 87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dash P., Fiore-Gartland A. J., Hertz T., Wang G. C., Sharma S., Souquette A., Crawford J. C., Clemens E. B., Nguyen T. H. O., Kedzierska K., et al. 2017. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glanville J., Huang H., Nau A., Hatton O., Wagar L. E., Rubelt F., Ji X., Han A., Krams S. M., Pettus C., et al. 2017. Identifying specificity groups in the T cell receptor repertoire. Nature 547: 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen G., Yang X., Ko A., Sun X., Gao M., Zhang Y., Shi A., Mariuzza R. A., Weng N. P. 2017. Sequence and structural analyses reveal distinct and highly diverse human CD8+ TCR repertoires to immunodominant viral antigens. Cell Rep. 19: 569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garboczi D. N., Ghosh P., Utz U., Fan Q. R., Biddison W. E., Wiley D. C. 1996. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature 384: 134–141. [DOI] [PubMed] [Google Scholar]
- 11.Ding Y.-H., Smith K. J., Garboczi D. N., Utz U., Biddison W. E., Wiley D. C. 1998. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity 8: 403–411. [DOI] [PubMed] [Google Scholar]
- 12.Shimizu A., Kawana-Tachikawa A., Yamagata A., Han C., Zhu D., Sato Y., Nakamura H., Koibuchi T., Carlson J., Martin E., et al. 2013. Structure of TCR and antigen complexes at an immunodominant CTL epitope in HIV-1 infection. Sci. Rep. 3: 3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh N. K., Riley T. P., Baker S. C. B., Borrman T., Weng Z., Baker B. M. 2017. Emerging concepts in TCR specificity: rationalizing and (maybe) predicting outcomes. J. Immunol. 199: 2203–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borbulevych O. Y., Santhanagopolan S. M., Hossain M., Baker B. M. 2011. TCRs used in cancer gene therapy cross-react with MART-1/Melan-A tumor antigens via distinct mechanisms. J. Immunol. 187: 2453–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cole D. K., Yuan F., Rizkallah P. J., Miles J. J., Gostick E., Price D. A., Gao G. F., Jakobsen B. K., Sewell A. K. 2009. Germ line-governed recognition of a cancer epitope by an immunodominant human T-cell receptor. J. Biol. Chem. 284: 27281–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kjer-Nielsen L., Clements C. S., Purcell A. W., Brooks A. G., Whisstock J. C., Burrows S. R., McCluskey J., Rossjohn J. 2003. A structural basis for the selection of dominant alphabeta T cell receptors in antiviral immunity. Immunity 18: 53–64. [DOI] [PubMed] [Google Scholar]
- 17.Gras S., Saulquin X., Reiser J. B., Debeaupuis E., Echasserieau K., Kissenpfennig A., Legoux F., Chouquet A., Le Gorrec M., Machillot P., et al. 2009. Structural bases for the affinity-driven selection of a public TCR against a dominant human cytomegalovirus epitope. J. Immunol. 183: 430–437. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y. C., Miles J. J., Neller M. A., Gostick E., Price D. A., Purcell A. W., McCluskey J., Burrows S. R., Rossjohn J., Gras S. 2013. Highly divergent T-cell receptor binding modes underlie specific recognition of a bulged viral peptide bound to a human leukocyte antigen class I molecule. J. Biol. Chem. 288: 15442–15454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan K. F., Gully B. S., Gras S., Beringer D. X., Kjer-Nielsen L., Cebon J., McCluskey J., Chen W., Rossjohn J. 2018. Divergent T-cell receptor recognition modes of a HLA-I restricted extended tumour-associated peptide. Nat. Commun. 9: 1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Derré L., Bruyninx M., Baumgaertner P., Ferber M., Schmid D., Leimgruber A., Zoete V., Romero P., Michielin O., Speiser D. E., Rufer N. 2008. Distinct sets of alphabeta TCRs confer similar recognition of tumor antigen NY-ESO-1157-165 by interacting with its central Met/Trp residues. Proc. Natl. Acad. Sci. USA 105: 15010–15015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le Gal F.-A., Ayyoub M., Dutoit V., Widmer V., Jäger E., Cerottini J.-C., Dietrich P.-Y., Valmori D. 2005. Distinct structural TCR repertoires in naturally occurring versus vaccine-induced CD8+ T-cell responses to the tumor-specific antigen NY-ESO-1. J. Immunother. 28: 252–257. [DOI] [PubMed] [Google Scholar]
- 22.Chen J. L., Stewart-Jones G., Bossi G., Lissin N. M., Wooldridge L., Choi E. M., Held G., Dunbar P. R., Esnouf R. M., Sami M., et al. 2005. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med. 201: 1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sami M., Rizkallah P. J., Dunn S., Molloy P., Moysey R., Vuidepot A., Baston E., Todorov P., Li Y., Gao F., et al. 2007. Crystal structures of high affinity human T-cell receptors bound to peptide major histocompatibility complex reveal native diagonal binding geometry. Protein Eng. Des. Sel. 20: 397–403. [DOI] [PubMed] [Google Scholar]
- 24.Webb A. I., Dunstone M. A., Chen W., Aguilar M. I., Chen Q., Jackson H., Chang L., Kjer-Nielsen L., Beddoe T., McCluskey J., et al. 2004. Functional and structural characteristics of NY-ESO-1-related HLA A2-restricted epitopes and the design of a novel immunogenic analogue. J. Biol. Chem. 279: 23438–23446. [DOI] [PubMed] [Google Scholar]
- 25.Li Y., Moysey R., Molloy P. E., Vuidepot A.-L., Mahon T., Baston E., Dunn S., Liddy N., Jacob J., Jakobsen B. K., Boulter J. M. 2005. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotechnol. 23: 349–354. [DOI] [PubMed] [Google Scholar]
- 26.Harper J., Adams K. J., Bossi G., Wright D. E., Stacey A. R., Bedke N., Martinez-Hague R., Blat D., Humbert L., Buchanan H., et al. 2018. An approved in vitro approach to preclinical safety and efficacy evaluation of engineered T cell receptor anti-CD3 bispecific (ImmTAC) molecules. PLoS One 13: e0205491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fairhead M., Howarth M. 2015. Site-specific biotinylation of purified proteins using BirA. Methods Mol. Biol. 1266: 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boulter J. M., Glick M., Todorov P. T., Baston E., Sami M., Rizkallah P., Jakobsen B. K. 2003. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 16: 707–711. [DOI] [PubMed] [Google Scholar]
- 29.Garboczi D. N., Hung D. T., Wiley D. C. 1992. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. USA 89: 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liddy N., Bossi G., Adams K. J., Lissina A., Mahon T. M., Hassan N. J., Gavarret J., Bianchi F. C., Pumphrey N. J., Ladell K., et al. 2012. Monoclonal TCR-redirected tumor cell killing. Nat. Med. 18: 980–987. [DOI] [PubMed] [Google Scholar]
- 31.Mitaksov V., Truscott S. M., Lybarger L., Connolly J. M., Hansen T. H., Fremont D. H. 2007. Structural engineering of pMHC reagents for T cell vaccines and diagnostics. Chem. Biol. 14: 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jakobsen B. K., Molloy P. E., Vuidepot A. B., Liddy N. R., inventors; Immunocore, Ltd. and Adaptimmune Ltd., assignees. TCR libraries. United States patent application 15/262,160, Publication No. US20170051036A1. 2017 Feb 23. [Google Scholar]
- 33.Zhao Y., Bennett A. D., Zheng Z., Wang Q. J., Robbins P. F., Yu L. Y. L., Li Y., Molloy P. E., Dunn S. M., Jakobsen B. K., et al. 2007. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J. Immunol. 179: 5845–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rondot S., Koch J., Breitling F., Dübel S. 2001. A helper phage to improve single-chain antibody presentation in phage display. Nat. Biotechnol. 19: 75–78. [DOI] [PubMed] [Google Scholar]
- 35.Turchaninova M. A., Davydov A., Britanova O. V., Shugay M., Bikos V., Egorov E. S., Kirgizova V. I., Merzlyak E. M., Staroverov D. B., Bolotin D. A., et al. 2016. High-quality full-length immunoglobulin profiling with unique molecular barcoding. Nat. Protoc. 11: 1599–1616. [DOI] [PubMed] [Google Scholar]
- 36.Bushnell B., Rood J., Singer E. 2017. BBMerge - accurate paired shotgun read merging via overlap. PLoS One 12: e0185056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. 2009. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6: 786–787. [DOI] [PubMed] [Google Scholar]
- 38.van der Maaten L., Hinton G. 2008. Visualizing data using t-SNE. J. Mach. Learn. Res. 9: 2579–2605. [Google Scholar]
- 39.Waterman D. G., Winter G., Gildea R. J., Parkhurst J. M., Brewster A. S., Sauter N. K., Evans G. 2016. Diffraction-geometry refinement in the DIALS framework. Acta Crystallogr. D Struct. Biol. 72: 558–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans P. 2006. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62: 72–82. [DOI] [PubMed] [Google Scholar]
- 41.Winter G. 2010. xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 43: 186–190. [Google Scholar]
- 42.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66: 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kabsch W. 2010. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66: 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karplus P. A., Diederichs K. 2012. Linking crystallographic model and data quality. Science 336: 1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. 2007. Phaser crystallographic software. J. Appl. Cryst. 40: 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murshudov G. N., Vagin A. A., Dodson E. J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53: 240–255. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60: 2126–2132. [DOI] [PubMed] [Google Scholar]
- 48.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. 2000. The protein data bank. Nucleic Acids Res. 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krissinel E., Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60: 2256–2268. [DOI] [PubMed] [Google Scholar]
- 50.Vilar S., Cozza G., Moro S. 2008. Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 8: 1555–1572. [DOI] [PubMed] [Google Scholar]
- 51.Ehrenmann F., Kaas Q., Lefranc M. P. 2010. IMGT/3Dstructure-DB and IMGT/DomainGapAlign: a database and a tool for immunoglobulins or antibodies, T cell receptors, MHC, IgSF and MhcSF. Nucleic Acids Res. 38(Database issue): D301–D307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen J. L., Dunbar P. R., Gileadi U., Jäger E., Gnjatic S., Nagata Y., Stockert E., Panicali D. L., Chen Y. T., Knuth A., et al. 2000. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J. Immunol. 165: 948–955. [DOI] [PubMed] [Google Scholar]
- 53.Birnbaum M. E., Mendoza J. L., Sethi D. K., Dong S., Glanville J., Dobbins J., Ozkan E., Davis M. M., Wucherpfennig K. W., Garcia K. C. 2014. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 157: 1073–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams J. J., Narayanan S., Liu B., Birnbaum M. E., Kruse A. C., Bowerman N. A., Chen W., Levin A. M., Connolly J. M., Zhu C., et al. 2011. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 35: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang H., Lim H. S., Knapp B., Deane C. M., Aleksic M., Dushek O., van der Merwe P. A. 2016. The contribution of major histocompatibility complex contacts to the affinity and kinetics of T cell receptor binding. Sci. Rep. 6: 35326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boehm K. M., Bhinder B., Raja V. J., Dephoure N., Elemento O. 2019. Predicting peptide presentation by major histocompatibility complex class I: an improved machine learning approach to the immunopeptidome. BMC Bioinformatics 20: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ayres C. M., Corcelli S. A., Baker B. M. 2017. Peptide and peptide-dependent motions in MHC proteins: immunological implications and biophysical underpinnings. Front. Immunol. 8: 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cole D. K., Bulek A. M., Dolton G., Schauenberg A. J., Szomolay B., Rittase W., Trimby A., Jothikumar P., Fuller A., Skowera A., et al. 2016. Hotspot autoimmune T cell receptor binding underlies pathogen and insulin peptide cross-reactivity. J. Clin. Invest. 126: 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Macdonald W. A., Chen Z., Gras S., Archbold J. K., Tynan F. E., Clements C. S., Bharadwaj M., Kjer-Nielsen L., Saunders P. M., Wilce M. C., et al. 2009. T cell allorecognition via molecular mimicry. Immunity 31: 897–908. [DOI] [PubMed] [Google Scholar]
- 60.Hawse W. F., De S., Greenwood A. I., Nicholson L. K., Zajicek J., Kovrigin E. L., Kranz D. M., Garcia K. C., Baker B. M. 2014. TCR scanning of peptide/MHC through complementary matching of receptor and ligand molecular flexibility. J. Immunol. 192: 2885–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riley T. P., Hellman L. M., Gee M. H., Mendoza J. L., Alonso J. A., Foley K. C., Nishimura M. I., Vander Kooi C. W., Garcia K. C., Baker B. M. 2018. T cell receptor cross-reactivity expanded by dramatic peptide-MHC adaptability. Nat. Chem. Biol. 14: 934–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yin Y., Mariuzza R. A. 2009. The multiple mechanisms of T cell receptor cross-reactivity. Immunity 31: 849–851. [DOI] [PubMed] [Google Scholar]
- 63.Lowe K. L., Cole D., Kenefeck R., OKelly I., Lepore M., Jakobsen B. K. 2019. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat. Rev. 77: 35–43. [DOI] [PubMed] [Google Scholar]
- 64.Cameron B. J., Gerry A. B., Dukes J., Harper J. V., Kannan V., Bianchi F. C., Grand F., Brewer J. E., Gupta M., Plesa G., et al. 2013. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 5: 197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raman M. C. C., Rizkallah P. J., Simmons R., Donnellan Z., Dukes J., Bossi G., Le Provost G. S., Todorov P., Baston E., Hickman E., et al. 2016. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci. Rep. 6: 18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Antunes D. A., Rigo M. M., Freitas M. V., Mendes M. F. A., Sinigaglia M., Lizée G., Kavraki L. E., Selin L. K., Cornberg M., Vieira G. F. 2017. Interpreting T-cell cross-reactivity through structure: implications for TCR-based cancer immunotherapy. Front. Immunol. 8: 1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.