

Abstract
Bioluminescent antibodies represent attractive detection agents in both bioanalytical assays and imaging. Currently, their preparation relies on genetic fusion of luciferases to antibodies or nonspecific chemical conjugation strategies. Here, we report a generic method to generate well-defined covalent antibody–luciferase conjugates starting from commercially available monoclonal antibodies. Our approach uses fusion proteins consisting of the bright blue light-emitting luciferase NanoLuc (NL) and an Fc-binding protein domain (Gx) that can be photo-cross-linked to the antibody using UV light illumination. Green and red color variants were constructed by tight fusion of the NanoLuc with a green fluorescent acceptor domain and introduction of Cy3, respectively. To increase the already bright NanoLuc emission, tandem fusions were successfully developed in which the Gx domain is fused to two or three copies of the NanoLuc domain. The Gx-NL fusion proteins can be efficiently photo-cross-linked to all human immunoglobulin G (IgG) isotypes and most mammalian IgG’s using 365 nm light, yielding antibodies with either one or two luciferase domains. The bioluminescent antibodies were successfully used in cell immunostaining and bioanalytical assays such as enzyme-linked immunosorbent assay (ELISA) and Western blotting.
Introduction
Luminescence represents an attractive optical detection method, both in bioanalytical assays and for (in vivo) imaging applications.1,2 Even though the photon output of luminescence is lower than that of fluorescence, luminescence detection is typically orders of magnitude more sensitive because the absence of background fluorescence and scattering provides for a very low background.1 Chemiluminescent detection has found widespread use in immunoassays such as enzyme-linked immunosorbent assay (ELISA) and Western blots, whereas bioluminescence has become an attractive detection method for in vivo optical imaging. The recent development of more efficient and stable luciferases and luciferase substrates has further expanded the application of bioluminescent detection in cell-based screening assays, point-of-care diagnostics, and in vivo imaging.1,3
A key step in the application of bioluminescence in immunoassays and immunostaining is connecting the reporter luciferase to the antibody used for molecular recognition. A classical approach is to use antibody–reporter conjugates such as horseradish peroxidase (HRP)-conjugated secondary antibodies to detect the presence of a primary antibody. While this approach allows the use of a limited number of antibody–reporter conjugates to detect a large number of primary antibodies, the approach adds an additional incubation and washing step to immunoassays and is not suitable for in vivo imaging applications. Two approaches to generate direct luciferase–antibody conjugates have been used: genetic fusion of the luciferase to an antibody (fragment) and chemical conjugation of luciferases to monoclonal antibodies. Genetic fusion has the advantage of generating homogeneous conjugates with a well-defined antibody–luciferase stoichiometry.4−11 However, genetic fusion requires cloning for each new antibody–luciferase conjugate and often involves cumbersome expression optimization and access to mammalian expression systems. A second general approach is to chemically conjugate the luciferase and antibody proteins, either covalently or noncovalently.12−14 While several approaches are available for covalent conjugation to commercially available monoclonal antibodies, these approaches do not allow precise control over the conjugation site, yielding a heterogeneous mixture of luciferase–antibody conjugates with little control over conjugation site and stoichiometry.15 The latter can be improved by fusing a luciferase to antibody-binding domains targeting the invariable part of antibodies such as protein A or protein G.16−18 However, this approach results in the formation of a noncovalent complex, which can dissociate under dilute conditions or extensive washing.
Here we report a generic method to generate antibody–luciferase conjugates that combines the best of both strategies. Our approach uses NanoLuc luciferase that is genetically fused to a protein G domain that contains the photo-cross-linkable non-natural amino acid para-benzoylphenylalanine (pBPA, Figure 1A). This protein G variant was recently developed by Hui and co-workers, who reported efficient and very specific cross-linking of the protein domain to the Fc part of antibodies upon photoactivation with 365 nm light.19 Importantly, efficient cross-linking was observed for all major human IgG subclasses and many other mammalian IgG’s. We developed a series of fusion proteins in which this photo-cross-linkable protein G is fused to blue, green, and red light-emitting variants of NanoLuc luciferase (Figure 1B). To further improve sensitivity, we also report the tandem fusion of multiple copies of NanoLuc to a single protein G domain. Following successful conjugation of the protein–luciferase fusion proteins to a variety of monoclonal antibodies, several applications of these bioluminescent antibodies are explored, including cell immunostaining, ELISA, and Western blotting.
Figure 1.

Development of bioluminescent antibodies using Gx-NL fusion proteins. (A) Gx-NL fusions contain a pBPA moiety (inset), which is able to conjugate to the backbone of the Fc domain of an antibody under 365 nm illumination. (B) Schematic representation of protein sequence of Gx-NL fusion proteins. (C) Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) analysis of purified adapter proteins. (D) Normalized luminescence spectra of different colored adapter proteins. (E) Luminescence spectra of 100 pM fusion proteins with multiple tandem repeats of NanoLuc.
Results and Discussion
Protein Expression, Purification, and Characterization
In recent years, several groups have reported the use of photo-cross-linkable Fc-binding protein domains as a generic approach to synthesize antibody conjugates with a well-defined stoichiometry.19−23 Here, we chose to use a protein G domain variant developed by Tsourkas and co-workers for LASIC, light-activated site-specific conjugation of native IgG’s.19 This small protein domain, which we here refer to as Gx, contains the photoreactive non-natural amino acid para-benzoylphenylalanine (pBPA) at position 24. Unlike protein-A-derived photo-cross-linkable domains, which are isotype-specific, Gx can be efficiently photo-cross-linked to a broad variety of IgG’s, including all major human subtypes, most mice (IgG2a,2b,2c,3), and some rat (IgG2c) and rabbit (polyclonal) subclasses. NanoLuc luciferase was chosen because of its small size, thermodynamic stability, and brightness.1,24 Oxidation of furimazine by NanoLuc produces bright blue light (460 nm) that is relatively stable over time and ∼100 times more intense than the commonly used Renilla and Firefly luciferases. In addition to simply fusing Gx to NanoLuc via a flexible linker, we also designed fusion proteins in which NanoLuc was tightly fused to the green fluorescent protein mNeonGreen or the red-emitting tdTomato fluorescent proteins (Figure 1B).25 In these fusion proteins, the energy of the product’s excited state is efficiently transferred to the fluorescent domains via bioluminescence resonance energy transfer (BRET), providing green and red light-emitting NanoLuc variants. In an effort to further increase the bioluminescent intensity, we also generated tandem fusion proteins containing two and three NanoLuc domains fused to a single Gx domain. All fusion proteins contained an N-terminal Strep-tag and a C-terminal 6xHis-tag to allow for straightforward affinity-based purification of the full-size proteins.
Expression plasmids encoding the fusion proteins were cotransformed in E. coli BL21(DE3) with the pEVOL-pBpF vector containing the tRNA/tRNA synthetase for the incorporation of the pBPA non-natural amino acid. All proteins were efficiently expressed and purified to homogeneity using a combination of nickel affinity and Strep-Tactin affinity chromatography (Figure 1C), typically yielding 30 mg of pure protein per liter of culture. Electrospray ionization quadrupole time-of-flight (ESI-Q-TOF) analysis confirmed the expected molecular weight for all fusion proteins showing incorporation of the pBPA amino acid and full maturation of the fluorescent proteins (Figure S1). All fusion proteins showed the expected bioluminescent spectra (Figures 1D and S2). The Gx-mNG-NL protein shows almost exclusively green emission, consistent with highly efficient BRET between NanoLuc and mNeonGreen. As reported before, BRET is less efficient for the Gx-tdTom-NL protein, showing residual blue luminescence at 460 nm in addition to the main red peak at 600 nm.25 When comparing the absolute intensities of the fusion proteins with multiple NanoLuc domains, the intensity of the blue luminescence clearly increased with the number of NanoLuc domains (Figure 1E). The luminescent intensities appear to not be completely proportional to the number of NLs, but it can be challenging to compare absolute luminescent intensities between different proteins because the luminescent intensity is not stable over time.
Photo-Cross-Linking
When testing optimal conditions for photo-cross-linking, we noticed that the red fluorescence of the Gx-tdTom-NL protein was slowly bleached upon illumination with the 365 nm light required for photoactivation of the pBPA group, showing almost complete bleaching after 1 h, the time typically used for photoconjugation (Figure S3A). Fortunately, the mNeonGreen protein in Gx-mNG-NL was more stable under these conditions, showing only a 10% decrease after 1 h of illumination with 365 nm light (Figure S3B). To provide an alternative red bioluminescent variant, we decided to introduce Cy3 as a red fluorescent acceptor, after establishing that Cy3 fluorescence is not affected by the illumination conditions used during photo-cross-linking (Figure S3C). To do so, the native cysteine present at position 166 in NanoLuc was mutated to serine, and another cysteine was introduced near the C-terminus of NanoLuc. Conjugation to the native cysteine is known to inactivate the enzymatic activity of NanoLuc, while conjugation at the C-terminus was previously shown to not affect the luciferase activity.26 Incubation of Gx-NL-Cys with a 15-fold molar excess of maleimide-functionalized Cy3 resulted in a labeling efficiency of 93%. The emission spectrum of the resulting Gx-NL-Cy3 protein was similar to that of Gx-tdTom-NL (Figure 1D). The bioluminescent intensity was found to be attenuated, which is partially due to Cys166Ser mutation (Figure S2).28
Optimal conditions for photo-cross-linking were established using cetuximab, a human IgG1-type therapeutic antibody that blocks the epidermal growth factor receptor. Different ratios of Gx-mNG-NL to antibody were irradiated with 365 nm for 15–180 min, while keeping the sample on ice to prevent overheating. Photo-cross-linking efficiency was monitored using nonreducing SDS-PAGE to allow the distinction between non-, once-, and twice-conjugated cetuximab (Figure 2A). The bioconjugation yield depended on both the irradiation time and the amount of Gx-mNG-NL protein used (Figure S4). On the basis of these results, we chose 60 min of irradiation with 8 equiv of protein G–luciferase fusion protein per IgG (four per Fc domain) as our standard condition for subsequent conjugations, as this allowed essentially complete conjugation of antibody with at least one copy of the bioluminescent reporter within a reasonable irradiation time. Similar conjugation efficiencies were observed for all other Gx-NL fusion proteins with cetuximab, including the fusion proteins with multiple NanoLuc domains (Figure 2B).
Figure 2.

Photoconjugation of Gx-NL fusion proteins to cetuximab. (A) Nonreducing SDS-PAGE analysis of photo-cross-linking 8 equiv of Gx-mNG-NL (3.2 μM) to cetuximab (0.4 μM) with increasing illumination times. (B) Nonreducing SDS-PAGE analysis of 1 h photoconjugation of 0.4 μM cetuximab with 3.2 μM of various protein G–luciferase fusion proteins.
Immunotargetting of Cell Surface Receptors
As a first application, we explored the performance of our bioluminescent antibodies in cellular targeting and quantification. An advantage of the Gx-mNG-NL and Gx-NL-Cy3 protein labels is that they allow for both fluorescent detection by direct excitation of the fluorophore and bioluminescent detection following addition of furimazine substrate. The fluorescent detection allowed us to use fluorescence-assisted cell sorting (FACS) to first explore targeting specificity using two human, IgG1-type therapeutic antibodies, cetuximab and trastuzumab, targeting the EGFR and HER2 cell surface receptors, respectively. Both antibodies were labeled with Gx-NL-Cy3 or Gx-mNG-NL (Figure S5). FACS analysis using the EGFR-overexpressing A431 tumor cell line shows efficient labeling by cetuximab–luciferase conjugates and no/very low labeling by trastuzumab–luciferase (Figure 3A and B). In contrast, efficient binding of bioluminescent trastuzumab was observed for SK-BR-3 cells, a tumor cell line that overexpresses the HER2 receptor, whereas 20-fold lower fluorescent intensity was observed upon incubation of SK-BR-3 cells with bioluminescent cetuximab (Figure S6).
Figure 3.
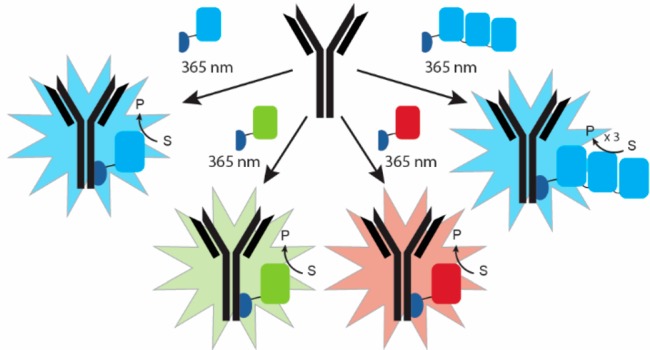
Immunostaining of cell surface receptors on A431 and SK-BR-3 cells using bioluminescent antibodies. Flow cytometry of A431 cells stained with 10 nM cetuximab (C) or trastuzumab (T) photo-cross-linked with (A) Gx-mNG-NL (mNG) or (B) Gx-NL-Cy3 (Cy3). (C, D) Plate reader read-out of stained cells (A, A431; S, SK-BR-3) with 1 nM bioluminescent antibody (C, cetuximab; T, trastuzumab) with either (C) luminescence or (D) fluorescence detected. (E) Analysis of the experiments described in (C) and (D) using a digital camera.
While fluorescent immunostaining of cells can be efficiently done at the single-cell level using FACS, fluorescent detection is much more cumbersome in plate reader-based assays, where cellular quantification and identification are hampered by background fluorescence and light scattering. The ability to use both fluorescence and bioluminescence allowed us to directly compare their performance for cellular quantification in plate reader-based assays. Bioluminescence detection allowed sensitive detection of A431 cells and SK-BR-3 cells over a wide range of cell concentrations using bioluminescent cetuximab and trastuzumab conjugates, respectively (Figure 3C and D). A linear correlation was observed between the number of cells and the bioluminescence intensity for each combination, allowing cell quantification over a broad range between 10 and 10 000 cells (Figure 3C). The higher bioluminescence intensity of cetuximab/A431 vs trastuzumab/SK-BR-3 cells is consistent with the higher labeling efficiency observed in FACS (Figure 3A and B) and probably reflects higher cell surface receptor expression levels. The intensity of green bioluminescence observed in experiments using Gx-mNG-NL-labeled antibodies is ∼5-fold higher compared to red bioluminescence using Gx-NL-Cy3-labeled antibodies, which reflects both attenuated luciferase activity and noncomplete BRET in the latter conjugate. Fluorescent detection under otherwise identical conditions was much less sensitive. A significant increase in fluorescence is only observed for the A431–cetuximab combination, and only above 5 000 cells per well. (Figure 3D). None of the other combinations showed a significant increase in fluorescence above the background under these conditions, showing that bioluminescence detection is at least 3 orders of magnitude more sensitive than fluorescence. Figure 3E shows that the luminescent signal is bright enough to be easily detected using an ordinary digital camera, making bioluminescent immunolabeling of cells an attractive, low-cost alternative to fluorescence-based cellular quantification methods such as FACS.
ELISA
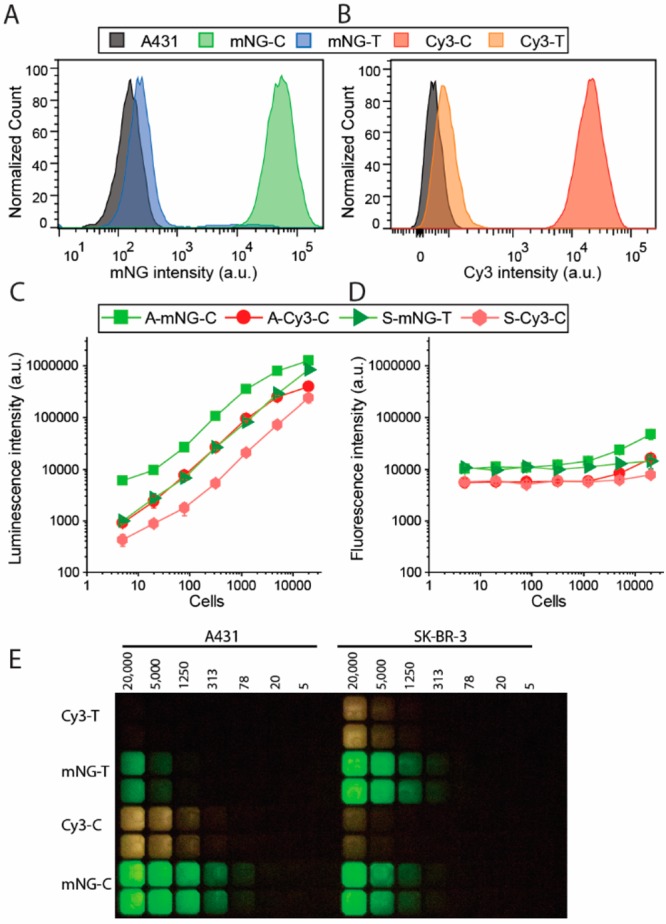
Classical ELISAs typically use a primary antibody that specifically binds to the molecular target, which is subsequently detected by incubation with a second antibody conjugated to a reporter enzyme such as alkaline phosphatase or horseradish peroxidase (HRP) that binds to the constant part of the first antibody. The ability to directly conjugate almost any monoclonal antibody with a bioluminescent reporter enzyme would simplify this procedure by requiring at least one less binding and washing step and by the ability to directly measure the NanoLuc-catalyzed bioluminescence in real time, whereas many reporter enzymes that use colorimetric detection are end-point assays that require an additional color-development step. An ELISA experiment was performed in which Gx-mNG-NL–cetuximab was used to detect the presence of anti-cetuximab antibodies (Figure 4). Detection of antidrug antibodies (ADAs) is important because the occurrence of ADAs is the main reason that treatment with therapeutic antibodies becomes ineffective in a significant number of patients.27 A 96-well plate was first coated with cetuximab overnight and blocked with a milk solution. Next, various concentrations of anti-cetuximab antibody were allowed to bind. After washing away the nonbound antibodies, the amount of bound anti-cetuximab antibody was determined by incubation with 1.3 nM of Gx-mNG-NL–cetuximab. During photoconjugation, a 2-fold excess of adapter was used. A 200-fold excess of human IgG’s was added to Gx-mNG-NL–cetuximab to capture any nonconjugated Gx-mNG-NL protein and prevent it from binding to immobilized cetuximab. The use of Gx-mNG-NL–cetuximab allowed us to directly compare the performance of fluorescent and bioluminescent detection in the same assay (Figure 4). Fluorescent detection showed a small dynamic range (50% increase in fluorescence) and allowed detection of anti-cetuximab over a limited concentration range between 500 and 5 000 ng/mL. In contrast, the dynamic range using bioluminescent detection was much larger, showing a 100-fold increase in bioluminescent signal and allowing detection of 10 ng/mL (0.1 nM) of anti-cetuximab. The bioluminescent titration curve could be fitted with a 1:1 binding model, yielding an EC50 of 1.25 ± 0.13 nM.
Figure 4.
ELISA assay for the detection of the anti-cetuximab antibody using bioluminescently labeled cetuximab. A 1 μg/mL cetuximab-coated plate was incubated with anti-cetuximab antibody (1.2–10 000 ng/μL) and subsequently with 1.33 nM Gx-mNG-NL–cetuximab antibody. Both fluorescence and luminescence were recorded using a plate reader.
Western Blot
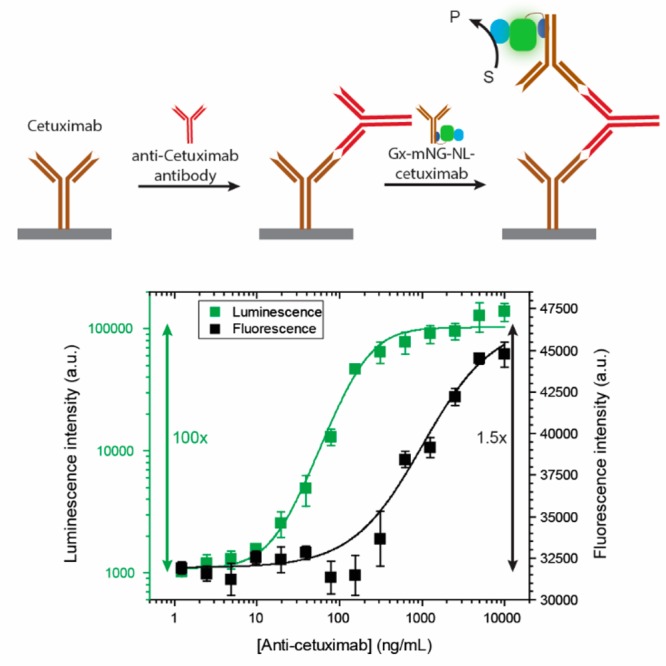
Another important bioanalytical application that uses antibody-based detection is Western blotting. Like ELISA, direct conjugation of the NanoLuc reporter enzyme to the primary antibody would avoid the need for a second antibody incubation and washing step. As proof of principle, we photoconjugated a mouse Ig2a-type anti-HA antibody using Gx conjugated to either a single NanoLuc domain (Gx-NL) or three copies of the NanoLuc domain (Gx-NL3). E. coli lysate was spiked with different concentrations of a purified 65 kDa 2·HA-tag-labeled protein (Figure 5A) and ran over a 12% SDS-PAGE gel (Figure 5B). Following transfer of the proteins from the gel to a nitrocellulose blot, the presence of HA-labeled protein was detected by incubation with 3.32 nM bioluminescent anti-HA antibody (Figure S7). In Coomassie-stained SDS-PAGE gels, the HA-labeled protein can be detected up to a 25-fold dilution (6.5 μg/mL), whereas bioluminescent detection on the Western blot clearly shows the presence of the HA protein at 125-fold lower concentrations (Figure 5C). The increased bioluminescent activity for the Gx-NL3 protein compared to Gx-NL (Figure 1E) also translates into a stronger signal in Western blot detection, making this the protein of choice for this application (Figure 5D).
Figure 5.
Bioluminescent antibody used as detection antibody in immunostaining of a Western blot. (A) Reducing SDS-PAGE gel showing the purified 65 kDa 2·HA-tag protein. (B) Coomassie-stained SDS-PAGE gel of cell lysate spiked with various dilutions (1–3 125×) of the 2·HA-tag protein. (C, D) Western blot of (B) stained with 3.32 nM Gx-NL-anti-HA (C) or Gx-NL3-anti-HA (D).
Conclusion
The photo-cross-linkable protein G–luciferase proteins reported here provide an easily accessible, efficient, and chemoselective new method for the synthesis of antibody–luciferase conjugates. A particular strength of our approach is that it does not require cloning or recombinant antibody expression and can be directly applied to almost all human and many mammalian monoclonal IgG antibodies. In addition to their application in various bioanalytical assays as demonstrated here, these bioluminescent antibodies also represent attractive reagents for in vivo imaging, in particular when using the red-shifted luciferase. The lower bioluminescence intensity of the red Gx-NL-Cy3 variant could be further improved by optimization of the BRET efficiency and the use of tandem repeats. Finally, the approach reported here could be easily extended to other red-shifted luciferases, including the H-Luc and S-Luc NanoLuc variants developed by the Johnsson group and various other luciferase–luciferin pairs optimized for in vivo imaging.28−30
Experimental Section
General Reagents
All chemicals were purchased from Merck unless stated otherwise. Therapeutic antibodies cetuximab (Erbitux, Merck) and trastuzumab (Herceptin, Roche) were obtained via the Catherina Hospital pharmacy in Eindhoven, The Netherlands. The NanoLuc substrate Nano-Glo was obtained from Promega. The non-natural amino acid pBPA was purchased from Bachem (4017646). The anti-cetuximab antibody (Clone HCA221) was ordered at Bio-Rad. The anti-HA antibody (Clone: 5BD1D10) was purchased from Invitrogen.
Protein Expression and Purification
All fusion protein constructs were cloned in pET28a vectors, and their sequences were verified using Sanger dideoxy sequencing (StarSEQ/BaseClear). The pEVOL-pBpF plasmid containing a tRNA/tRNA synthetase pair enabling incorporation of pBPA was a gift from Peter Schultz (Addgene plasmid no. 31190).31 For a detailed description of cloning procedures, please see the Supporting Information. Expression plasmids for the fusion proteins were cotransformed in Escherichia coli BL21(DE3) cells (Novagen) with the pEVOL-pBpF vector and cultured in 0.5 L 2xYT medium (2.5 g of NaCl, 5 g of yeast extract, and 8 g of peptone in 0.5 L of dH2O) supplemented with 50 μg/mL kanamycin and 25 μg/mL chloramphenicol. When the OD600 reached 0.5–0.6, expression was induced using 0.1–1 mM isopropyl β-d-1-thiogalactopyramoside, 0.02 w/v % arabinose, and 1 mM para-benzoylphenylalanine (pBPA). After overnight expression at 20 °C, cells were harvested by centrifugation at 10 000g for 10 min. Cells were then lysed using Bugbuster protein-extraction reagent (Novagen) and Benzonase endonuclease (Novagen) and centrifuged at 16 000g for 20–40 min. Protein G–luciferase fusion proteins were purified using Ni-affinity chromatography (Novagen, His-bind resin) and Strep-Tactin XT (IPA) purification according to the manufacturer’s instructions (see the Supporting Information). All proteins were stored frozen at −80 °C in Strep-Elution buffer (100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 50 mM Biotin, pH 8.0) until further use. Protein concentration was determined by measuring the absorption at 280 nm using the extinction coefficients listed in the Supporting Information (Table S1). Purity of protein was confirmed with both SDS-PAGE analysis and ESI-Q-TOF (Figure S1).
Cy3 Labeling of Gx-NL-Cys
Gx-NL-Cys (50 μM) was incubated with 1 mM tris(2-carboxyethyl)phosphine (TCEP) for 20 min at room temperature. Next, sulfo-Cy3-maleimide (LumiProbe, no. 21380) was added in a 15× molar excess and incubated overnight at 4 °C. The excess of dye was removed using a PD-10 desalting column (GE healthcare, 17-0851-01), using 2 mL of elution buffer (100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 8.0). Protein and dye concentration were determined using Nanodrop 3000 at 280 and 552 nm using the extinction coefficients of the protein (40 910 M–1 cm–1) and the dye (162 000 M–1 cm–1), respectively. The absorption at 280 nm was corrected for the contribution of the Cy3-dye by subtracting 0.06·A552nm. The degree of labeling was 0.93. Aliquots of the labeled protein were snap-frozen in liquid nitrogen and stored at −80 °C until use.
Luminescence Spectra
Luminescence spectra of nonconjugated protein G-NanoLuc fusion proteins were recorded with a plate reader (Tecan Spark). The proteins were diluted in luminescence buffer (100 mM NaCl, 50 mM Tris, 10% glycerol, 0.05% Tween-20, pH 7.4) to a final concentration of 100 pM, and NanoGlo (Promega) was added in a 2000× dilution. Full luminescence spectra were recorded with a 5 nm bandwidth and a 1 s integration time.
Photo-cross-linking
Antibodies (400 nM) were mixed with the Gx-NL fusion protein (2 or 8 equiv) in 50 mM Tris-HCl (pH 8.5). Mixtures were illuminated in a 200 μL Eppendorf tube, on ice, for 1 h with 365 nm UV light (Promed UV-lamp, 36 W). After photo-cross-linking, bioluminescent antibodies were stored at 4 °C until use.
Cell Culturing
Human A431 carcinoma cells were cultured in RPMI-1640 medium (Gibco, 21875) supplemented with 10% fetal bovine serum (Gibco, 26140) and 1% penicillin/streptomycin (Gibco, 15140) at 37 °C, 5% CO2. The human SK-BR-3 adenocarcinoma cells were cultured in the same medium supplemented with 1 mM sodium pyruvate at 37 °C, 5% CO2. Cells were passed at 80% confluency.
Cell Experiments
Cells were detached using 2 mL of trypsin (Gibco, 25300) for a T75 flask for 5 min. Once cells were detached, the trypsin was neutralized by addition of 4 equiv of full medium. Cells were counted on a Neubauer hemocytometer. Next, cells were centrifuged (A431, 10 min at 100g; SK-BR-3, 5 min at 150g) and washed once in PBS+ (PBS + 1 mg/mL BSA). Cells were resuspended in PBS+ and aliquoted in 1.5 mL Eppendorf tubes.
For FACS measurements, a final concentration of 10 nM antibody–conjugate was added to 100 000 cells and incubated for 15 min at 25 °C, 400 rpm. Cells were centrifuged for 5 min at 100g and resuspended in PBS+. All FACS measurements were done using a BD FACS Aria III equipped with a 70 μm nozzle. mNeonGreen was excited by a 488 nm laser and detected through a 530/30 bandpass filter. Cy3 was excited by a 561 nm laser and detected through a 582/15 bandpass filter. For all analyses, doublet cells were excluded by standard doublet discrimination with forward and side scatter area versus height plots. Histograms were created with FlowJo software.
For luminescence measurements, a final concentration of 1 nM antibody-conjugate was added to 100 000 cells and incubated for 15 min at 25 °C, 400 rpm. Cells were centrifuged for 5 min at 100g and washed three times in PBS+. A 4-fold dilution series of the cells was made in a white 384-well plate (Greiner) and black plate (Thermo Nunc LumiNunc). After addition of NanoGlo (4000× dilution) to the white plate, the luminescence was recorded using a digital camera (Sony DSC-RX100, 20 s, ISO 6400) and a plate reader (Tecan Safire, 250 ms). The fluorescence was recorded from the black plate (Tecan Safire; mNG, 500/10–530/10; Cy3, 550/10–580/10)
ELISA
A white or black 96-well plate (Thermo Nunc LumiNunc) was coated with 100 μL of 1 μg/mL cetuximab in phosphate-buffered saline (PBS) and incubated overnight at 4 °C. The next day, wells were washed 3 times with 250 μL of PBST (PBS + 0.05% Tween-20). Wells were blocked with 240 μL of PBSM (PBST + 2 w/v % skim milk) for 1.5 h at room temperature. After washing 3 times with PBST, the wells were incubated with 100 μL of a dilution series of anti-cetuximab (HCA221, 1.2–10 000 ng/mL in PBSM) for 1 h at room temperature. After washing 3 times, 100 μL of 1.33 nM bioluminescent antibody (Gx-mNG-NL-cetuximab with 2 equiv of Gx-mNG-NL used for photo-cross-linking) preincubated with 266 nM human serum IgG (Sigma, I8640) in PBSM was added and incubated for 1 h at room temperature. After incubation the wells were washed 3 times with PBST and once with luminescence buffer. In the black plate, fluorescence was recorded (Tecan Spark, 488/530 nm). For the white plate, after addition of 100 μL of luminescence buffer with a 4000× dilution of NanoGlo, a picture was taken (Sony DSC-RX100, ISO 6400, 30 s) and the luminescence was recorded with a plate reader (Tecan Spark, 533 nm, 1 s).
Western Blot
A β-lactamase-based antibody sensor protein containing two HA-tags (Abs-4) was expressed in E. coli BL21(DE3) and purified as described before.32 A 5-fold dilution series (2.5 μM–0.8 nM) of the protein was made in supernatant of lysed E. coli NovaBlue(DE3) cells (Novagen). Samples were loaded on two 12% SDS-PAGE gels and run for 1 h at 150 V in TGS buffer (25 mM Tris, 192 mM glycine, 0.1% SDS, pH 8.3). One gel was stained with Bio-Safe Coomassie stain (Bio-Rad, 1610787), and a picture was taken using the ImageQuant 350 (GE Healthcare). The other gel was used for the Western blot. The proteins were transferred to a nitrocellulose membrane (Whatman, GE) using a standard procedure. After blotting, the membrane was cut in two identical pieces and individually blocked using a 5 w/v % skim milk in PBST (PBS + 0.1% Tween-20) in a 50 mL tube on a tube roller. Next, the anti-HA antibody (IgG2a, Clone 5B1D10) with either the Gx-NL or Gx-NL3 photoconjugated was added in a final antibody concentration of 3.32 nM in 5 mL of 5 w/v % skim milk in PBST for 1 h. Next, the membrane was washed once in 5% w/v skim milk in PBST, 3 times in PBST, and 2 times in luminescence buffer. The blots were dried on a tissue and placed in a container. NanoGlo was added in a 1000× dilution in luminescence buffer covering the entire blot (2.5 mL), and a picture was taken in the dark using a digital camera (Sony DSC-RX100, ISO 6400, 30 s).
Acknowledgments
The research leading to these results has received funding from the Ministry of Education, Culture and Science (Gravitation program 024.001.035). We thank Ir. Eva van Aalen for measuring the kinetics of Cy3 photobleaching.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.bioconjchem.9b00804.
Additional experimental procedures (cloning, protein purification, and fluorescent spectra) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kaskova Z. M.; Tsarkova A. S.; Yampolsky I. V. (2016) 1001 Lights: luciferins, luciferases, their mechanisms of action and applications in chemical analysis, biology and medicine. Chem. Soc. Rev. 45, 6048–6077. 10.1039/C6CS00296J. [DOI] [PubMed] [Google Scholar]
- Yeh H.-W.; Ai H.-W. (2019) Development and applications of bioluminescent and chemiluminescent reporters and biosensors. Annu. Rev. Anal. Chem. 12, 129–150. 10.1146/annurev-anchem-061318-115027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England C. G.; Ehlerding E. B.; Cai W. (2016) NanoLuc: A small luciferase is brightening up the field of bioluminescence. Bioconjugate Chem. 27, 1175–1187. 10.1021/acs.bioconjchem.6b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiald P.; Mousli M.; Goyffon M.; Vaux D. (1997) Engineering of a bioluminescent antigen-binding protein. Biotechnol. Lett. 19, 1037–1041. 10.1023/A:1018463804651. [DOI] [Google Scholar]
- Venisnik K. M.; Olafsen T.; Loening A. M.; Iyer M.; Gambhir S. S.; Wu A. M. (2006) Bifunctional antibody-renilla luciferase fusion protein for in vivo optical detection of tumors. Protein Eng., Des. Sel. 19, 453–460. 10.1093/protein/gzl030. [DOI] [PubMed] [Google Scholar]
- Han C.; Ihara M.; Ueda H. (2013) Expression of an antibody-enzyme complex by the L-chain fusion method. J. Biosci. Bioeng. 116, 17–21. 10.1016/j.jbiosc.2013.01.012. [DOI] [PubMed] [Google Scholar]
- Oyama H.; Morita I.; Kiguchi Y.; Miyake S.; Moriuchi A.; Akisada T.; Niwa T.; Kobayashi N. (2015) Gaussia Luciferase as a genetic fusion partner with antibody fragments for sensitive immunoassay monitoring of clinical biomarkers. Anal. Chem. 87, 12387–12395. 10.1021/acs.analchem.5b04015. [DOI] [PubMed] [Google Scholar]
- Boute N.; Lowe P.; Berger S.; Malissard M.; Robert A.; Tesar M. (2016) NanoLuc luciferase - a multifunctional tool for high throughput antibody screening. Front. Pharmacol. 7, 27. 10.3389/fphar.2016.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath N.; Flemming R.; Godat B.; Urh M. (2017) Development of nanoluc bridging immunoassay for detection of anti-drug antibodies. J. Immunol. Methods 450, 17–26. 10.1016/j.jim.2017.07.006. [DOI] [PubMed] [Google Scholar]
- Mori A.; Ojima-Kato T.; Kojima T.; Nakano H. (2018) Zipbodyzyme: development of new antibody-enzyme fusion proteins. J. Biosci. Bioeng. 125, 637–643. 10.1016/j.jbiosc.2017.12.021. [DOI] [PubMed] [Google Scholar]
- Ren W.; Li Z.; Xu Y.; Wan D.; Barnych B.; Li Y.; Tu Z.; He Q.; Fu J.; Hammock B. D. (2019) One-step ultrasensitive bioluminescent enzyme immunoassay based on nanobody/nanoluciferase fusion for detection of aflatoxin B1 in cereal. J. Agric. Food Chem. 67, 5221–5229. 10.1021/acs.jafc.9b00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye S.; Sato J. (2008) Recombinant aequorin with a reactive cysteine residue for conjugation with maleimide-activated antibody. Anal. Biochem. 378, 105–107. 10.1016/j.ab.2008.03.044. [DOI] [PubMed] [Google Scholar]
- Lomakina G. Y.; Istrate A.; Rudenko N. V.; Ugarova N. N. (2014) Synthesis and application of firefly luciferase antibody conjugates in a bioluminescent immunoassay of Salmonella cells. Moscow Univ. Chem. Bull. 69, 49–55. 10.3103/S0027131414020047. [DOI] [Google Scholar]
- Moutsiopoulou A.; Hunt E.; Broyles D.; Pereira C. A.; Woodward K.; Dikici E.; Kaifer A.; Daunert S.; Deo S. K. (2017) Bioorthogonal protein conjugation: application to the development of a highly sensitive bioluminescent immunoassay for the detection of interferon-γ. Bioconjugate Chem. 28, 1749–1757. 10.1021/acs.bioconjchem.7b00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennler P.; Fischer E.; Schibli R. (2015) Antibody conjugates: from heterogeneous populations to defined reagents. Antibodies 4, 197–224. 10.3390/antib4030197. [DOI] [Google Scholar]
- Jablonski E. (1985) The preparation of bacterial luciferase conjugates for immunoassay and application to rubella antibody detection. Anal. Biochem. 148, 199–206. 10.1016/0003-2697(85)90646-3. [DOI] [PubMed] [Google Scholar]
- Lindbladh C.; Mosbach K.; Bülow L. (1991) Preparation of a genetically fused protein A/luciferase conjugate for use in bioluminescent immunoassays. J. Immunol. Methods 137, 199–207. 10.1016/0022-1759(91)90025-B. [DOI] [PubMed] [Google Scholar]
- Tsuboi S.; Jin T. (2018) Recombinant protein (luciferase-IgG binding domain) conjugated quantum dots for BRET-coupled near-infrared imaging of epidermal growth factor receptors. Bioconjugate Chem. 29 (4), 1466–1474. 10.1021/acs.bioconjchem.8b00149. [DOI] [PubMed] [Google Scholar]
- Hui J. Z.; Tamsen S.; Song Y.; Tsourkas A. (2015) LASIC: light activated site-specific conjugation of native IgGs. Bioconjugate Chem. 26, 1456–1460. 10.1021/acs.bioconjchem.5b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perols A.; Karlström A. E. (2014) Site-specific photoconjugation of antibodies using chemically synthesized IgG-binding domains. Bioconjugate Chem. 25, 481–488. 10.1021/bc400440u. [DOI] [PubMed] [Google Scholar]
- Lee Y.; Jeong J.; Lee G.; Moon J. H.; Lee M. K. (2016) Covalent and oriented surface immobilization of antibody using photoactivatable antibody Fc-binding protein expressed in Escherichia coli. Anal. Chem. 88, 9503–9509. 10.1021/acs.analchem.6b02071. [DOI] [PubMed] [Google Scholar]
- Yang H.-M.; Bao R.-M.; Yu C.-M.; Lv Y.-N.; Zhang W.-F.; Tang J.-B. (2017) Fc-specific biotinylation of antibody using an engineered photoactivatable Z-biotin and its biosensing application. Anal. Chim. Acta 949, 76–82. 10.1016/j.aca.2016.10.039. [DOI] [PubMed] [Google Scholar]
- Rosier B. J. H. M.; Cremers G. A. O.; Engelen W.; Merkx M.; Brunsveld L.; de Greef T. F. A. (2017) Incorporation of native antibodies and Fc-fusion proteins on DNA nanostructures via a modular conjugation strategy. Chem. Commun. 53, 7393–7396. 10.1039/C7CC04178K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M. P.; Unch J.; Binkowski B. F.; Valley M. P.; Butler B. L.; Wood M. G.; Otto P.; Zimmerman K.; Vidugiris G.; Machleidt T.; et al. (2012) Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem. Biol. 7, 1848–1857. 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K.; Kimura T.; Shinoda H.; Bai G.; Daniels M. J.; Arai Y.; Nakano M.; Nagai T. (2016) Five colour variants of bright luminescent protein for real-time multicolour bioimaging. Nat. Commun. 7, 13718 10.1038/ncomms13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen W.; van de Wiel K. M.; Meijer L. H. H.; Saha B.; Merkx M. (2017) Nucleic acid detection using BRET-beacons based on bioluminescent protein-DNA hybrids. Chem. Commun. 53, 2862–2865. 10.1039/C6CC10032E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willrich M. A. V. (2017) Therapeutic monoclonal antibodies in the clinical laboratory. J. Appl. Lab. Med. 2, 454–457. 10.1373/jalm.2016.022160. [DOI] [PubMed] [Google Scholar]
- Chu J.; Oh Y.; Sens A.; Ataie N.; Dana H.; Macklin J. J.; Laviv T.; Welf E. S.; Dean K. M.; Zhang F.; et al. (2016) A bright cyan-excitable orange fluorescent protein facilitates dual-emission microscopy and enhances bioluminescence imaging in vivo. Nat. Biotechnol. 34, 760–767. 10.1038/nbt.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh H.-W.; Karmach O.; Ji A.; Carter D.; Martins-Green M. M.; Ai H. (2017) Red-shifted luciferase-luciferin pairs for enhanced bioluminescence imaging. Nat. Methods 14, 971–974. 10.1038/nmeth.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiblot J.; Yu Q.; Sabbadini M. D. B.; Reymond L.; Xue L.; Schena A.; Sallin O.; Hill N.; Griss R.; Johnsson K. (2017) Luciferases with tunable emission wavelengths. Angew. Chem., Int. Ed. 56, 14556–14560. 10.1002/anie.201708277. [DOI] [PubMed] [Google Scholar]
- Chin J. W.; Martin A. B.; King D. S.; Wang L.; Schultz P. G. (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 99, 11020–11024. 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banala S.; Aper S. J. A.; Schalk W.; Merkx M. (2013) Switchable reporter enzymes based on mutually exclusive domain interactions allow antibody detection directly in solution. ACS Chem. Biol. 8, 2127–2132. 10.1021/cb400406x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.