

Abstract
Phylogenetic analysis of complete genomes of the avian coronaviruses avian infectious bronchitis (AIBV) and turkey coronavirus (TCoV) supported the hypothesis that numerous recombination events have occurred between these viruses. Although the two groups of viruses differed markedly in the sequence of the spike protein, the gene (S) encoding this protein showed no evidence of positive selection or of an elevated mutation rate. Rather, the data suggested that recombination events have homogenized the portions of the genome other than the S gene between the two groups of viruses, while continuing to maintain the two distinct, anciently diverged versions of the S gene. The latter hypothesis was supported by a phylogeny of S proteins from representative coronaviruses, in which S proteins of AIBV and TCoV fell in the same clade.
Electronic supplementary material
The online version of this article (doi:10.1007/s00705-011-1061-5) contains supplementary material, which is available to authorized users.
Keywords: Recombination Event, Bootstrap Support, Synonymous Substitution, Nonsynonymous Substitution, Much Recent Common Ancestor
Introduction
Avian infectious bronchitis virus (AIBV), a member of the single-stranded, positive-sense RNA virus family Coronaviridae, infects domestic chickens and causes major economic losses to the poultry industry worldwide [1–5]. AIBV causes disease in the upper respiratory tract, but it can also infect other organs such as the kidney and gonads [2]. Turkey coronavirus (TCoV), which causes acute enteric disease in turkeys and consequent economic loss, is closely related to AIBV [6]. Sequence analysis has suggested that recombination may have played a key role in the evolutionary origin of TCoV. Because the spike proteins of TCoV isolates were much more divergent from those of AIBV than were other TCoV proteins, Jackwood and colleagues [7] proposed that TCoV arose through a recombination event involving replacement of the S gene (encoding the spike or S protein) of an AIBV genome with one derived from a more distant, unidentified coronavirus.
Largely because of interest in the origin of SARS coronavirus, numerous studies have addressed the role of recombination in coronavirus evolution [8]. In the case of AIBV, there has been long-standing interest in recombination among free-living strains [7, 9–12], and the occurrence of recombination among genotypes has been demonstrated experimentally in vitro [13]. As the genomes of AIBV isolates have been sequenced, several naturally occurring genomes have been reported with novel gene sets and/or gene orders compared to those seen in classic AIBV isolates [14, 15]. The sequences of these genomes show the effects of gene deletion and rearrangement, processes to which recombination may have contributed.
Here I applied phylogenetic methods to sequences of protein-coding genes of 11 classic AIBV genomes and 6 TCoV genomes in order to reconstruct past recombination events among members of these two taxa. I examined the pattern of nucleotide substitution in the S gene in order to test the hypothesis that this gene is subject to an unusually high rate of nucleotide substitution, which might account for the high level of divergence in this region, even in the absence of recombination. In addition, I conducted a phylogenetic analysis of S proteins from a wide variety of coronaviruses in order to test the hypothesis that the S protein of TCoV is derived from a distantly related virus.
Materials and methods
I analyzed 17 representative complete genomes of avian infectious bronchitis virus (AIBV) and turkey coronavirus (TCoV; Table 1). Only genomes including open reading frames corresponding to the 11 protein-coding genes in the AIBV isolate BJ (AY319651) were used so that phylogenies of each gene could be constructed for the same set of genomes, and attenuated laboratory strains were avoided. Sequences of each protein-coding gene (Table 2) were aligned at the amino acid level with the CLUSTAL X program [16], and the alignment was imposed on the DNA sequences. In addition, additional S protein sequences from 37 representative coronaviruses were downloaded from the NCBI protein sequence database and aligned using CLUSTAL X.
Table 1.
Sequences used in analyses
| Virus | Isolate accession number | Isolate |
|---|---|---|
| AIBV | AY319651 | BJ |
| AY338732 | LX4 | |
| AY514485 | California 99 | |
| AY641576 | Peafowl/GD/KQ6/2006 | |
| AY646283 | Partridge/GD/S14/2003 | |
| AY851295 | Mass 41 | |
| DQ288927 | SAIBK | |
| DQ646405 | TW2575/98 | |
| EU637854 | CK/CH/LSD/05I | |
| EU714029 | SC021202 | |
| FN430414 | ITA/90254/2005 | |
| TCoV | EU022525 | TCoV-540 |
| EU022526 | TCoV-ATCC | |
| EU095850 | MG10 | |
| GQ427174 | TCoV/TX-GL/01 | |
| GQ427175 | TCoV/IN-517/94 | |
| GQ427176 | TCoV/TX-1038/98 |
Table 2.
Proteins encoded by avian infectious bronchitis virus (AIBV) and turkey coronavirus (TCoV)
| Protein | Gene | No. of amino acidsa | Reading framea |
|---|---|---|---|
| 1a | 1a | 3963 | 3′ end overlaps b portion of 1ab |
| 1ab | 1ab | 6641 | Reading frame change by ribosomal slippage |
| Spike | S | 1169 | 5′ end overlaps b portion of 1ab reading frame |
| 3a | 3a | 57 | First nucleotide of start is last nucleotide of stop of S; last nucleotide of stop is first nucleotide of start of 3b |
| 3b | 3b | 62 | First nucleotide of start is last nucleotide of stop of 3a; 3′ end overlaps Env reading frame |
| Envelope | Env | 109 | 5′ end overlaps 3b reading frame; 3′ end overlaps M reading frame |
| Membrane glycoprotein | M | 226 | 5’ end overlaps Env reading frame |
| 5a | 5a | 65 | 3′ end overlaps 5b reading frame |
| 5b | 5b | 81 | 5′ end overlaps 3a reading frame; 3′ end overlaps N reading frame |
| Nucleocapsid | N | 409 | 5′ end overlaps 5b reading frame |
aBased on IBV isolate BJ (AY319651)
Phylogenetic trees were constructed by the neighbor-joining (NJ) [17] and maximum-likelihood (ML) [18] methods, as implemented in the MEGA5 program [19]. NJ trees of DNA sequences from AIBV and TCoV were constructed on the basis of the maximum composite likelihood (MCL) distance [19], while ML trees were based on the Tamura-Nei model [20]. NJ trees of S protein sequences were constructed on the basis of the JTT distance, and ML trees of S protein sequences used the JTT model [19]. The reliability of clustering patterns in trees was assessed by bootstrapping [21]; 1000 bootstrap samples were used. Since the NJ and ML methods yielded very similar results, only the NJ trees are shown below; the ML trees are included as supplementary material (Supplementary Figures S1-S5). In all computations of evolutionary distances, any site at which the alignment postulated a gap in any sequence was excluded from all pairwise comparisons.
The reading frames in AIBV and TCoV genomes show considerable overlap (Table 2). The pattern of nucleotide substitution in regions of viral genes encoded by overlapping reading frames can be atypical because sites that are synonymous in one reading frame may be nonsynonymous in other reading frames [22, 23]. As a result, sites that are synonymous in one reading frame can be subject to purifying selection in the other reading frame [22, 23]. Because the unusual pattern of nucleotide substitution in such regions can obscure phylogenetic signals, overlapping portions of genes were excluded from the phylogenetic analyses of individual genes.
The number of synonymous substitutions per synonymous site (d S) and the number of nonsynonymous substitutions per nonsynonymous site (d N) were estimated by Li’s [24] method. This method was used because it accounts for transitional bias, and the transition:transversion ratio was estimated at 1.92 for the non-overlapping portions of protein-coding genes. Transitional bias can affect the estimation of d S and d N because transitions at two-fold degenerate sites are invariably synonymous [24, 25]. The variances of mean d S and d N were estimated by the bootstrap method [25].
Comparison of the topology of phylogenetic trees based on different genes was used to test the hypothesis that recombination among genes has occurred in the evolutionary history of these viral genomes. Bootstrapping of trees provided a test of the hypothesis that trees of different genes showed different topologies and thus of the hypothesis that recombination has occurred. The breakpoint for within-gene recombination events was tested by the maximum chi-square method [26] as implemented in the RDP3 program [27].
Results
Phylogenies of AIBV and TCoV genes
In the phylogenetic analysis of the non-overlapping portions of S gene sequences, the AIBV isolates clustered together and apart from TCoV isolates (Fig. 1A). The branch between the two clusters received 100% bootstrap support (Fig. 1A). The maximum chi-square method provided support (P < 0.0001) for a recombination event in the 3’ portion of the b portion of 1ab. Based on the breakpoint estimated by the latter method, a second phylogenetic tree was constructed based on the 3’ end of the b portion of 1ab (114 aligned nucleotides), excluding the portion that overlaps the S reading frame (Fig. 1B). In the latter tree also, the AIBV isolates clustered together and apart from TCoV isolates, although the bootstrap support for the branch separating the major clusters was only 86% (Fig. 1B).
Fig. 1.
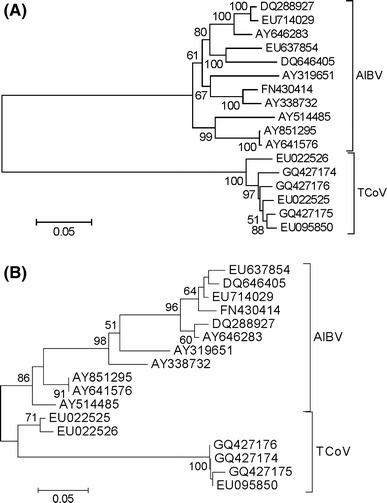
(A) Neighbor-joining tree of non-overlapping portions of the S gene based on MCL distance at 3310 aligned nucleotide sites. (B) Neighbor-joining tree of the 3′ end of the b portion of the 1 ab gene based on MCL distance at 123 aligned nucleotide sites. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch; only values ≥50% are shown
By contrast, in a phylogenetic tree based on the non-overlapping regions of 1a, the remainder of the b portion, 3a, 3b, and Env sequences, AIBV and TCoV did not form separate clusters (Fig. 2). Rather, the AIBV genome AY514485 (California 99) clustered within the cluster of TCoV genomes (Fig. 2). An internal branch receiving 100% bootstrap support clustered the latter genome with two TCov genomes, GQ427174 and GQ427176 (Fig. 2). The cluster including AY514485 and all TCoV isolates was separated from all other AIBV isolates by an internal branch that received 100% bootstrap support (Fig. 2). Thus, the AY514485 genome appeared to represent a recombinant between AIBV and TCoV. Phylogenetic trees based on the individual genes used in constructing the phylogeny shown in Fig. 2 showed similar topologies (not shown).
Fig. 2.
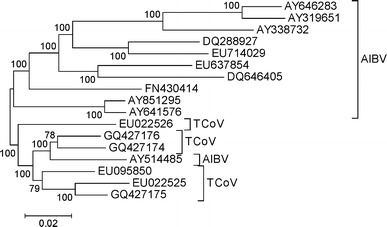
Neighbor-joining tree of non-overlapping portions of the 1 a, the b portion of 1ab (excluding the 3′ end; A), 3 a, 3 b, and Env sequences, based on MCL distance at 20,022 aligned sites. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch; only values ≥50% are shown
Other evidence of recombination was found in the phylogenies of the genes located downstream of Env. A phylogeny of the M gene showed several AIBV genes that clustered with TCoV (Fig. 3A). In the case of AY514485 and FN430414, support for clustering of the M genes with those of TCoV was weak (Fig. 3A). However, a cluster including the TCoV genome GQ427174 and the AIBV genomes AY641576 and AY851295 received 97% bootstrap support (Fig. 3A). In the case of the N gene, the AIBV genomes AY641576 and AY851295 clustered with TCoV genomes with weak support (Fig. 3B). However, the N gene of the AIBV genome EU637854 clustered with the TCoV genome GQ427174 with 100% bootstrap support (Fig. 3B). The number of nucleotide substitutions per site (estimated by the MCL method) between the N gene of EU637854 and that of GQ427174 was only 0.0017 ± 0.0011. On the other hand, the mean number of nucleotide substitutions per site between the N gene of EU637854 and the N genes of the main cluster of AIBV genomes (excluding EU637854, AY641576, and AY 851295; Fig. 3B) was 0.1164 ± 0.0114. The difference between these two values was highly significant (Z-test; P < 0.001).
Fig. 3.
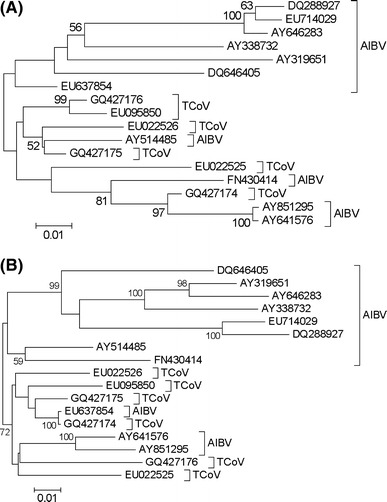
(A) Neighbor-joining tree of the non-overlapping portion of the M gene based on MCL distance at 668 aligned nucleotide sites. (B) Neighbor-joining tree of non-overlapping portions of N gene sequences based on MCL distance at 1170 aligned nucleotide sites. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch; only values ≥50% are shown
Phylogenetic trees of both 5a and 5b genes showed topologies supporting numerous recombinational events between AIBV and TCoV, although with relatively modest bootstrap support. In the 5a gene, there was 76% support for a cluster including all TCoV genomes along with four AIBV genomes (Fig. 4A). The 5a gene of the AIBV genomes AY514485 and EU637854 clustered with the TCoV genome GQ427175 with 82% bootstrap support (Fig. 4A). In the case of the 5b gene, the AIBV genomes AY514485 and EU637854 likewise clustered among the TCov genomes, though with weak (< 50%) bootstrap support (Fig. 4B). The poor resolution of the phylogenies of the 5a and 5b genes no doubt reflected the small numbers of nucleotide sites available in non-overlapping portions of these genes (198 and 180, respectively).
Fig. 4.
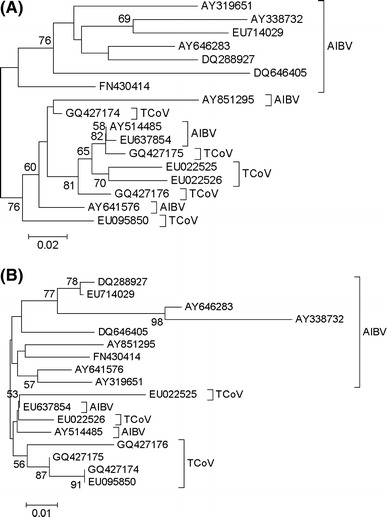
(A) Neighbor-joining tree of the non-overlapping portion of the 5a gene based on MCL distance at 198 aligned nucleotide sites. (B) Neighbor-joining tree of non-overlapping portion of the 5b gene based on MCL distance at 180 aligned nucleotide sites. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch; only values ≥50% are shown
Nucleotide substitution
Excluding the five AIBV genomes implicated in recombinational events with TCoV by the phylogenetic analyses (AY514485, AY641576, AY851295, EU637854, and FN430414), I estimated the number of synonymous substitutions per synonymous site (d S) and the number of nonsynonymous substitutions per nonsynonymous site (d N) for pairwise comparisons within and between AIBV and TCoV genomes (Table 3). Mean d S and mean d N were estimated separately for the non-overlapping portion of the S reading frame and for non-overlapping portions of other genes (excluding the 3’ end of the b portion of 1ab; Fig. 1B). In all comparisons, mean d S was significantly greater than mean d N (P < 0.001 in each case; Table 3), indicating that all of these coding regions are subject to purifying selection. In the comparison between AIBV and TCoV, both mean d S and mean d N were significantly greater in the S gene than in other coding regions (P < 0.001 in each case; Table 3). By contrast, in comparisons within AIBV, mean d S did not differ significantly between the S gene and other genes, and in comparisons within TCoV, mean d S in the S gene was significantly less than that in other genes (P < 0.001; Table 3). In AIBV, mean d N was significantly greater in the S gene than in other genes, but in TCoV, mean d N did not differ significantly between S and other genes (Table 3).
Table 3.
Mean numbers of synonymous (d S) nonsynonymous (d N) substitutions per site in non-overlapping portions of the S gene and other genes in pairwise comparisons within and between AIBV and TCov
| Comparison | S | Other genes | ||
|---|---|---|---|---|
| d S | d N | d S | d N | |
| AIBV | 0.419 ± 0.019 | 0.070 ± 0.005 | 0.401 ± 0.007 | 0.048 ± 0.001*** |
| TCoV | 0.094 ± 0.007 | 0.031 ± 0.003 | 0.164 ± 0.004*** | 0.029 ± 0.001 |
| AIBV vs. TCoV | 1.416 ± 0.121 | 0.775 ± 0.030 | 0.461 ± 0.009*** | 0.056 ± 0.001*** |
Analyses excluded five AIBV genomes (AY514485, AY641576, AY851295, EU637854, and FN430414) with histories of recombination with TCoV and the 3′end of the b portion of 1ab (Fig. 1B)
*** Z-tests of the hypothesis that d S or d N in the other genes is equal to the corresponding value in the S gene: P < 0.001
S protein phylogeny
In a phylogenetic tree of S proteins from coronaviruses of birds and mammals, coronaviruses from birds did not form a monophyletic group (Fig. 5). Two sequences from coronaviruses of passerine birds (munia and bulbul) clustered with those from carnivores (ferret badger and leopard cat), and this cluster received 100% bootstrap support (Fig. 5). A third sequence from a passerine bird (thrush) fell outside this cluster (Fig. 5). By contrast, AIBV and TCoV sequences, along with a sequence from a pigeon coronavirus, clustered together with 97% bootstrap support (Fig. 5). Thus, the phylogenetic tree supported the hypothesis that the S proteins of AIBV and TCoV were more closely related to each other than either was to any coronavirus derived from a mammalian host.
Fig. 5.
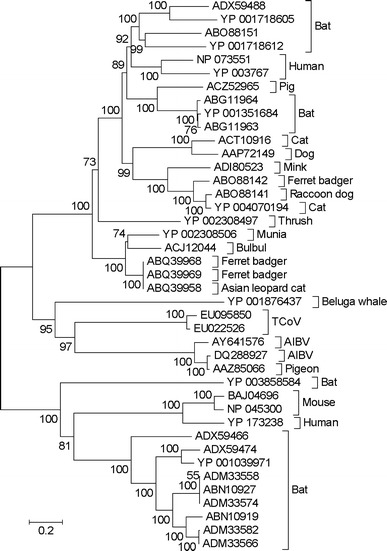
Neighbor-joining tree of S protein sequences of coronaviruses from avian and mammalian hosts based on the JJT distance at 796 aligned amino acid sites. Sequences are identified by accession number and host. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch; only values ≥50% are shown
Discussion
Sequence analyses (Fig. 1A; Table 3) supported the observation of Jackwood et al. (2010) that the S (spike) gene shows substantial sequence divergence between AIBV and TCov. One hypothesis to explain this observation is derivation of the S gene of AIBV or TCoV is that it has resulted from recombination [7]. Alternative hypotheses that do not invoke recombination include the following: (1) that amino acid sequence divergence in the spike protein is driven by positive selection or by relaxation of purifying selection; and (2) that the mutation rate in the S gene is unusually high.
The present analyses did not support the hypothesis that the S gene has been subject to positive Darwinian selection favoring amino acid replacements in the spike protein. Mean d S was significantly greater than mean d N in the S gene, even in the comparison between AIBV and TCoV, where a substantial amount of nonsynonymous substitution has occurred (Table 3). Moreover, in comparisons of TCoV isolates, mean d N in the S gene was very similar to that in the other genes (Table 3). In comparisons of AIBV isolates, mean d N was significantly higher in S than in other genes, but mean d N was still much lower than mean d S, even in the S gene (Table 3). These results imply that purifying selection (selection acting to eliminate deleterious mutations) has been the primary form of natural selection acting on the S gene. In general, purifying selection appears to have been less stringent on the S gene than on other genes, except within TCoV.
Because synonymous mutations are more likely to be neutral, or nearly so, than are nonsynonymous mutations, patterns of synonymous substitutions provide the most reliable index of the mutation rate in protein-coding genes [28]. The present results do not reveal a pattern of synonymous substitution in the S gene indicative of an unusually high mutation rate. In AIBV, mean d S was very similar in the S gene and other genes, while in TCoV, mean d S was actually significantly lower in the S gene than in other genes (Table 3).
The fact that neither selection on the spike protein nor a high mutation rate in the S gene can account for the observed results supports the hypothesis that the difference between the S genes and other genes with respect to the extent of divergence between AIBV and TCoV is the result of recombination. In the absence of a high mutation rate, the most reasonable explanation for an unusually high level of synonymous divergence in a given region of two related genomes is that the region in question is more anciently diverged than the remainder of the genome, and that recombination has brought together anciently diverged and more recently diverged genomic segments. However, in the case of AIBV and TCoV, the mechanism involved need not have involved recombination with a distantly related virus [7]. Rather, it is possible that the degree of sequence divergence seen in the S gene has been accumulated since the most recent common ancestor (MRCA) of AIBV and TCoV and that the other portions of the genome have been homogenized between these two lineages by repeated events of inter-lineage recombination since the MRCA.
Phylogenetic analyses of individual genes supported the hypothesis that recombination between the AIBV and TCoV lineages has been frequent. In the upstream portion of the genome, from 1a to the Env gene, excluding the S gene and the 3’ end of the b portion of the 1ab reading frame, the California 99 (AY514485) isolate clearly belongs to TCoV (Fig. 2). However, based on the S gene and the 3’ end of the b portion of the 1ab reading frame, the same isolate clearly belongs to AIBV (Fig. 1). The evolutionary relationships of AY514485 clearly indicate that the S gene and adjacent regions can be exchanged between AIBV and TCoV lineages.
In the downstream portion of the genome, there was evidence of multiple recombination events between the AIBV and TCoV lineages. Although AIBV-like in the upstream genes from 1a though Env, Peafowl/GD/KQ6/2006 (AY641576) and Mass 41 (AY851295) showed a close resemblance to TCoV in the M gene (Fig. 3A). Similarly, the N gene of the AIBV isolate CK/CH/LSD/05I (EU637854) was less than 1% different from that of the TCoV isolate TCoV/TX-GL/01 (GQ427174) but nearly two orders of magnitude more divergent from a typical AIBV N gene (Fig. 3B). These results imply that there have been recent recombination events, causing near-homogenization of certain genomic regions between genomes that continue to differ markedly in the S gene. Moreover, the occurrence of such events suggests an important role for recombination in homogenizing non-S portions of the genome between the AIBV and TCoV lineages.
In their present niches in the agricultural ecosystem, AIBV and TCoV infect different host species and different organ systems within those hosts. Under these circumstances, it seems unlikely that the two viruses have opportunities for recombination. However, the hypothesis proposed here does not require recombination at the present time, but only recombination in the evolutionary past between the lineage that gave rise to AIBV and the lineage that gave rise to TCoV. The history of these viruses prior to their appearance as agricultural pathogens is unknown, but recent analyses of viruses infecting wild birds suggest that AIBV and TCoV represent just the tip of the iceberg of a vast assemblage of avian coronaviruses awaiting discovery and characterization [29–31]. Moreover, individual bird species may be infected by a number of distinct coronaviruses [29–31], as would be required for recombination between different viral lineages.
A phylogenetic analysis of S proteins of coronaviruses of birds and mammals (Fig. 5) supported the hypothesis that the S proteins of AIBV and TCoV are closely related and that these two viruses belong to a clade of avian-specific coronaviruses. The fact that the S proteins of these two viruses clustered together in the phylogenetic tree provides evidence against the hypothesis that either of the two viruses obtained its S protein from a widely divergent coronavirus. The close phylogenetic relationship of AIBV and TCoV is consistent with the hypothesis that their ancestors shared hosts during their evolutionary history and thus with the hypothesis of recombination between these two coronavirus lineages.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This research was supported by grant GM43940 from the National Institutes of Health.
References
- 1.Cavanagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34(6):439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- 2.Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- 3.Ignjatović J, Sapats S. Avian infectious bronchitis virus. Rev Sci Tech. 2000;19(2):493–508. doi: 10.20506/rst.19.2.1228. [DOI] [PubMed] [Google Scholar]
- 4.Jones RC. Viral respiratory diseases (ILT, aMPV infections, IB): are they ever under control? Br Poultry Sci. 2010;51(1):1–11. doi: 10.1080/00071660903541378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villegas P. Viral diseases of the respiratory system. Poultry Sci. 1998;77:1143–1145. doi: 10.1093/ps/77.8.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guy JS. Turkey coronavirus is more closely related to avian infectious bronchitis virus than to mammalian coronaviruses: a review. Avian Pathol. 2000;29(3):207–212. doi: 10.1080/03079450050045459. [DOI] [PubMed] [Google Scholar]
- 7.Jackwood MW, Boynton TO, Hilt DA, McKinley ET, Kissinger JC, Paterson AH, Robertson J, Lemke C, McCall AW, Williams SM, Jackwood JW, Byrd LA. Emergence of a group 3 coronavirus through recombination. Virology. 2010;398:98–108. doi: 10.1016/j.virol.2009.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graham RL, Baric RS. Recombination, reservoirs, and the modular spike: mechanisms of coronavirus cross-species transmission. J Virol. 2010;84(7):3134–3146. doi: 10.1128/JVI.01394-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kusters JG, Jager EJ, Niesters HG, van der Zeijst BA. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine. 1990;8:605–608. doi: 10.1016/0264-410X(90)90018-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cavanagh D, Davis P. Evolution of avian coronavirus IBV: sequence of the matrix glycoprotein and intergenic region of several serotypes. J Gen Virol. 1988;69:621–629. doi: 10.1099/0022-1317-69-3-621. [DOI] [PubMed] [Google Scholar]
- 11.Wang L, Junker D, Collison EW. Evidence of natural recombination within the S1 gene of infectious bronchitis virus. Virology. 1993;192:710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- 12.Jia W, Karaca K, Parrish CR, Naqi SA. A novel variant of infectious bronchitis virus resulting from recombination among three different strains. Arch Virol. 1995;140:259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kottier SA, Cavanagh D, Britton P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virol. 1995;213:569–580. doi: 10.1006/viro.1995.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mardani K, Noormohammadi AH, Hooper P, Ignjatovic J, Browning GF. Infectious bronchitis viruses with a novel genome organization. J. Virol. 2008;82(4):2013–2024. doi: 10.1128/JVI.01694-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hewson KA, Ignjatovic J, Browning GF, Devlin JM, Noormohammadi AH. Infectious bronchitis viruses with naturally occurring genomic rearrangement and gene deletion. Arch Virol. 2011;156:245–252. doi: 10.1007/s00705-010-0850-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 18.Tamura K, Peterson D, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance (in press) [DOI] [PMC free article] [PubMed]
- 19.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 20.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10:512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 21.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–791. doi: 10.2307/2408678. [DOI] [PubMed] [Google Scholar]
- 22.Hughes AL, Westover K, da Silva J, O’Connor DH, Watkins DI. Simultaneous positive and purifying selection on overlapping reading frames of the tat and vpr genes of simian immunodeficiency virus. J Virol. 2001;75(17):7666–7672. doi: 10.1128/JVI.75.17.7966-7972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes AL, Hughes MA. Patterns of nucleotide difference in overlapping and non-overlapping reading frames of papillomavirus genomes. Virus Res. 2005;113(2):81–88. doi: 10.1016/j.virusres.2005.03.030. [DOI] [PubMed] [Google Scholar]
- 24.Li W-H. Unbiased estimates of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36(1):96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- 25.Nei M, Kumar S. Molecular evolution and phylogenetics. New York: Oxford University Press; 2000. [Google Scholar]
- 26.Maynard Smith J. Analyzing the mosaic structure of genes. J Mol Evol. 1992;34:126–129. doi: 10.1007/BF00182389. [DOI] [PubMed] [Google Scholar]
- 27.Heath L, van der Walt E, Vasani A, Martin DP. Recombination patterns in aphthoviruses mirror those found in other picornaviruses. J Virol. 2006;80:11827–11832. doi: 10.1128/JVI.01100-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes AL. Adaptive evolution of genes and genomes. New York: Oxford University Press; 1999. [Google Scholar]
- 29.Woo PC, Lau SK, Lan CS, Lai KK, Huang Y, Lee P, Luk GS, Dyrting KC, Chan K-H, Yuen K-Y. Comparative analysis of complete genome sequences of three avian coronaviruses reveals a novel group 3c coronavirus. J Virol. 2009;83:908–917. doi: 10.1128/JVI.01977-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hughes LA, Savage C, Naylor C, Bennett M, Chantrey J, Jones R. Genetically diverse coronaviruses in wild bird populations of northern England. Emerg Infect Dis. 2009;15:1091–1094. doi: 10.3201/eid1507.090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muradrasoli S, Bálint Á, Wahlgren J, Waldenström J, Belák S, Blomber J, Olsen B. Prevalence and phylogeny of coronaviruses in wild birds from the Bering Strait area (Beringia) PLoS One. 2010;5(10):e13640. doi: 10.1371/journal.pone.0013640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.