

Abstract
We previously described isolation of a potentially new mammalian reovirus, designated BYD1, which can cause clinical symptoms similar to that of severe acute respiratory syndrome (SARS) in guinea pigs and macaques, from throat swabs of one SARS patient of Beijing, in 2003. For this study, we determined the genome sequences of BYD1 and the S1 gene sequences of other five mammalian reovirus isolates (BLD, JP, and BYL were isolated from different SARS patients during the outbreak, 302I and 302II were isolated from fecal specimens of two children of Beijing in 1982) to allow molecular comparison with other previously reported mammalian reoviruses (MRVs). Comparative analyses of the BYD1 genome with those of three prototype mammalian reovirus strains demonstrated that BYD1 is a novel reassortant virus, with its S1 gene segment coming from a previously unidentified serotype 2 isolate and other nine segments coming from ancestors of homologous T1L and T3D segments, which supports the hypothesis that mammalian reovirus gene segments reassort in nature. Further analyses of the S1 segments of the six isolates showed that all the isolates are novel serotype 2 MRVs based on their S1 gene sequences, which are markedly different from those of all previously reported, and the S1 genes of the four new isolates share more than 99% identity with each other, proving that they diverged from a common ancestor most recently, and the S1 genes of the four new isolates share about 65% identity with those of the two strains isolated in 1982.
Keywords: dsRNA Virus, Mammalian reovirus, Orthoreovirus, Virus genome
Introduction
Mammalian reoviruses (MRVs) are prototype members of the family Reoviridae, which contains segmented double-stranded RNA (dsRNA) viruses of both medical (rotavirus) and economic (bluetongue virus) importance (reviewed in [1–3]). They contain a genome of ten dsRNA segments encoding eight structural proteins and three nonstructural proteins [4]. Three large (L1–L3), three medium (M1–M3), and four small (S1–S4) segments can be distinguished in polyacrylamide gels [5]. MRV isolates (or “strains”) can be grouped into three serotypes, represented by three commonly studied prototype isolates: type 1 Lang (T1L), type 2 Jones (T2J), and type 3 Dearing (T3D). The genome sequences of three prototype MRV strains have been reported [6–8]. The lengths of six of the ten genome segments (L1, L3, M2, S2, S3, and S4) are invariant among the three prototype isolates. The lengths of three other segments (L2, M1, and M3) vary by only one to four bases, but the S1 genome segment is unique to each MRV strain and shows the greatest difference [9]. The S1 segment encodes the cell attachment protein σ1 which also specifies tissue tropism, serotype, and hemagglutination, and the nonstructural protein σ1s, while each of the other nine segments encodes one protein [10, 11].
MRV has a wide geographic distribution and can infect virtually all mammals including human beings [4]. Reoviruses were originally called respiratory enteric orphans on the basis of their repeated isolation from respiratory and enteric tracts of children with asymptomatic illness [12]. However, there have been several reports describing MRVs associated with meningitis in infants and children [13–15], and acute respiratory disease in adults [16]. We previously reported isolation and partial characterization of a new MRV strain, designated BYD1, isolated from throat swabs of one patient of Beijing with severe acute respiratory syndrome (SARS) in Hep-2 cell cultures, and SARS-Coronavirus (SARS-CoV) was also isolated from the same samples in Vero-E6 cell cultures [17]. In our previous study, three other MRV strains, designated BLD, JP, and BYL, were isolated from different SARS patients [18]. All the four MRV strains were purified by plaque assay and were identified as MRV serotype 2 members by virus neutralization. Preliminary experimental infections showed MRV BYD1 could cause SARS-like symptoms in macaques and guinea pigs [19, 20].
The aim of this study was to determine the genome sequences of BYD1 and the S1 gene sequences of BLD, JP, BYL, 302I, and 302II (the latter two were isolated in 1982) to allow molecular comparison with other previously reported MRV genes and proteins.
Materials and methods
Viruses and cell culture
MRV isolates BYD1, BLD, JP, and BYL were isolated, plaque purified, and characterized by Duan and co-authors [17, 21]. The BLD strain was isolated from throat swabs of one dead SARS patient in Beijing. The JP strain was isolated from throat swabs of one Guangzhou SARS patient. The BYL strain was isolated from a lung tissue sample of one dead SARS patient in Beijing Youan Hospital. MRV isolates 302I and 302II, which were isolated from fecal specimens of two children of Beijing in 1982 by Wu et al. [22], were kindly provided by Mao Panyong. All six MRV isolates were amplified in murine L929 cell monolayers in Joklik’s modified minimal essential medium (Gibco) supplemented to contain 2.5% fetal calf serum, 2.5% neonatal bovine serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 1 μg/ml amphotericin B. All cell culture manipulations were performed under biosafety level 3 conditions.
Viral dsRNA preparation
Infected cells were disrupted by three freeze-thaw cycles with 1% sodium dodecyl sulfate. Proteinase K was added to a final concentration of 1 mg/ml. After incubation for 1 h at 37°C, the total RNA was isolated by two consecutive water phenol–chloroform–isoamyl alcohol (25:24:1) extractions and recovered by precipitation with ethanol containing 0.3 M NaOAC (pH 5.2). The dsRNA was further purified by LiCl fractionation precipitation [23]. Viral genomic RNA was separated on a 1% TAE agarose gel and individual segments were excised and purified using Qiagen gel extraction kit (Qiagen).
cDNA preparation
Full-length cDNA copies of the individual dsRNA segments were prepared by using a modification of the Lambden method [24]. Briefly, a 3′-amino-blocked oligodeoxyribonucleotide (primer A: 5′ PO4-AggTCTCgTAgACCgTgCACC(A)17-NH2 3′) was ligated to both 3′-OH termini of the dsRNA with T4 RNA ligase at 37°C overnight according to the manufacturer’s directions (Promega). The tailed dsRNA was recovered by using Qiagen PCR purification kit and denatured by heating at 99°C for 1 min in the presence of 15% DMSO. cDNA copies of the tailed dsRNA were synthesized by using oligo d(T)18 and SuperScript reverse transcriptase (Invitrogen). Removal of excess RNA, cDNA annealing and filling in of partial duplexes was carried out as described by Lambden et al., except that cDNA hybridization was done for only 1 h at 65°C. Amplification of cDNA was performed by using Takara LA Taq (Takara) and a complementary primer (primer B: 5′ ggTgCACggTCTACgAgACCT 3′) according to procedures provided by Takara Corporation.
Cloning and sequencing of amplified cDNA products
Amplified cDNA products were separated on a 1% TAE agarose gel and purified using Qiagen gel extraction kit. Purified PCR products were cloned with a TOPO TA cloning kit (Invitrogen). Positive clones were identified on the basis of the size of the inserts. The cloned PCR products were sequenced with M13 forward and reverse primers using an ABI PRISM 377 DNA sequencer (Applied Biosystems).
Sequence analyses
Sequence alignments were generated by the ClustalX software [25]. Comparison of our sequences with those available from nucleic acid and protein databases was preformed by using the BLAST program (http://www.ncbi.nlm.nih.gov/blast). Synonymous and nonsynonymous substitution frequencies were calculated according to the methods of Nei and Gojobori [26] as applied by Dr. B. Korber at http://hcv.lanl.gov/content/hcv-db/SNAP/SNAP.html. Protein sequences were analyzed for conservative and nonconservative substitutions by pairwise ClustalW analyses, using BLOSUM matrix weighting [25]. Phylogenetic analyses were performed with the program MEGA [27] using the neighbor-joining methods. The branching orders of the phylograms were verified statistically by resampling the data 1,000 times in a bootstrap analysis as applied in MEGA 4.0.2. And the predicted secondary structures of σ1 proteins were analyzed using the MLRC secondary structure prediction program [28].
Results
Sequences of MRV BYD1 genome and five MRV S1 genes
The complete sequences of the BYD1 L1–L3, M1–M3, S1–S4 genes, and S1 genes of MRV 302I, 302II, BLD, BYL, JP have been deposited in the GenBank database under accession numbers DQ664184, DQ664185, DQ664186, DQ664187, DQ664188, DQ664189, DQ312301, DQ664190, DQ664191, DQ318037, EU049603, EU049604, EU049605, EU049606, and EU049607, respectively. The BYD1 genes were 3,854, 3,912, 3,901, 2,304, 2,203, 2,241, 1,437, 1,331, 1,198, and 1,196 nucleotides long, respectively, and all the S1 genes were 1,437 nucleotides long. All the genes contain the conserved nucleotides 5′ GATC-3′ at the 5′ terminus and 5′-TCATC 3′ at the 3′ terminus (identical to the terminal sequences of other MRV genes in GenBank). The BYD1 L1, L3, S2, S3, and S4 genes are the same length as the genes of the three prototype MRV strains T1L, T2J, and T3D, and corresponding BYD1 λ3, λ1, σ2, σNS and σ3 proteins that the genes encode are the same lengths as the cognate proteins of the prototype strains. The BYD1 L2 gene is the same length as that of T2J, the BYD1 M1 gene is the same length as those of T1L and T3D, the BYD1 M2 gene is the same length as those of T1L and T2J, and the BYD1 M3 gene is the same length as that of T1L. However, all the S1 genes in this work and encoded σ1 proteins differ in size from the S1 genes and σ1 proteins of the three prototype strains.
Comparative analysis of the BYD1 genome with those of three prototype strains
Pairwise comparisons of the nucleotide sequences of each of the ten genome segments between BYD1 and three prototype strains have shown that the L-class, M-class, S2–S4 segments of BYD1 share higher identity to those of T1L (82–92%) and T3D (77–92%) than to those of T2J (71–79%), while the S1 segment of BYD1 shares higher identity to that of T2J (61%) than to those of T1L and T3D (59 and 48%, respectively) (Table 1). Pairwise comparisons of the deduced amino acid sequences of cognitive proteins have shown that the sequence identities of λ3, λ2, λ1, μ2, μ1, μNS, σ2, σNS, and σ3 range from 75 to 98% between BYD1 and T1L, 81–97% between BYD1 and T2J, and 92–98% between BYD1 and T3D, while the σ1 of BYD1 share 52, 60, and 26% identities with three prototype strains, respectively.
Table 1.
Percent identity in pairwise comparisons of ten genes and ten deduced major proteins between MRV BYD1 and three prototype MRV strains
| BYD1 genes | Nucleotide identity (%) | BYD1 proteins | Amino acid identity (%) | |||||
|---|---|---|---|---|---|---|---|---|
| T1L | T2J | T3D | T1L | T2J | T3D | |||
| L1 | 92 | 75 | 92 | → | λ3 | 98 | 92 | 98 |
| L2 | 82 | 73 | 77 | → | λ2 | 96 | 87 | 92 |
| L3 | 92 | 77 | 92 | → | λ1 | 98 | 95 | 98 |
| M1 | 92 | 71 | 92 | → | μ2 | 96 | 81 | 95 |
| M2 | 85 | 77 | 92 | → | μ1 | 98 | 97 | 98 |
| M3 | 86 | 73 | 84 | → | μNS | 95 | 83 | 93 |
| S1 | 59 | 61 | 48 | → | σ1 | 52 | 60 | 26 |
| S2 | 86 | 77 | 86 | → | σ2 | 75 | 93 | 98 |
| S3 | 85 | 74 | 88 | → | σNS | 95 | 84 | 95 |
| S4 | 85 | 79 | 86 | → | σ3 | 95 | 89 | 95 |
The genetic diversity within MRVs is independent of viral serotype, and that MRV genes have distinct evolutionary histories has been demonstrated. For this study we examined the positions and types of nucleotide mismatches between BYD1 and the prototype strains to gain a comprehensive view of the evolutionary divergence of the protein-coding sequences. Most mismatches in any of the codon base positions between BYD1 and T2J segments are far greater than those between BYD1 and either T1L or T3D segments. Each of these mismatch percentages was converted to an evolutionary divergence value by multiplying mismatch percentage by 1.33 (Table 2). These values suggest that the L3, L1, M1, and M2 segments of BYD1 diverged from ancestors of homologous T1L and T3D segments most recently, while the S2, M3, L2, S3, and S4 segments of BYD1 diverged from longer ago. The consistently high values for divergence at third codon base positions among pairings with T2J genome segments (Table 2) indicate that the nine BYD1 segments diverged from ancestors substantially before their respective T2J homologs. Relative numbers of synonymous and nonsynonymous nucleotide changes identified in pairwise comparisons of the coding sequences of BYD1 and any of the prototype strains (Table 2) support the same conclusion.
Table 2.
Pairwise comparisons of variation at different codon positions in MRV genome segments between BYD1 and three prototype strains
| Codon position | Isolate | Variation (%) in the long open reading frame of genome segment | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| L1 | L2 | L3 | M1 | M2 | M3 | S2 | S3 | S4 | ||
| Firsta | T1L | 3.4 | 9.4 | 3.4 | 4.7 | 6.0 | 9.2 | 6.7 | 10.5 | 8.0 |
| T2J | 17.3 | 21.2 | 12.3 | 24.9 | 10.5 | 23.2 | 14.6 | 24.7 | 13.1 | |
| T3D | 3.9 | 14.7 | 3.1 | 5.8 | 3.4 | 8.1 | 6.4 | 7.3 | 5.8 | |
| Seconda | T1L | 1.0 | 2.3 | 1.5 | 2.0 | 1.1 | 1.7 | 1.6 | 3.3 | 2.9 |
| T2J | 5.1 | 7.8 | 3.9 | 11.2 | 1.9 | 9.8 | 4.5 | 9.8 | 6.6 | |
| T3D | 1.0 | 3.8 | 1.1 | 2.5 | 1.5 | 2.8 | 1.6 | 4.0 | 3.3 | |
| Thirda | T1L | 28.2 | 59.6 | 27.1 | 25.8 | 54.3 | 45.0 | 50.0 | 50.5 | 51.7 |
| T2J | 77.5 | 79.8 | 77.3 | 79.1 | 83.2 | 78.4 | 76.0 | 74.9 | 68.9 | |
| T3D | 28.7 | 72.6 | 28.4 | 26.0 | 26.5 | 52.2 | 50.9 | 41.1 | 51.4 | |
| Syn.b | T1L | 33.0 | 68.9 | 31.6 | 30.7 | 61.1 | 52.7 | 58.4 | 62.3 | 62.3 |
| T2J | 89.0 | 87.7 | 87.1 | 85.5 | 91.1 | 84.4 | 86.0 | 82.8 | 76.6 | |
| T3D | 33.7 | 81.6 | 32.3 | 31.3 | 30.0 | 59.6 | 60.9 | 48.7 | 60.2 | |
| Nonsyn.b | T1L | 0.9 | 2.5 | 0.8 | 1.6 | 1.0 | 2.6 | 1.4 | 2.6 | 2.4 |
| T2J | 6.1 | 9.1 | 4.2 | 12.4 | 3.0 | 11.2 | 4.9 | 11.1 | 6.6 | |
| T3D | 1.0 | 5.1 | 0.8 | 2.0 | 1.0 | 3.0 | 0.9 | 2.6 | 2.3 | |
| Cons.c | T1L | 64.0 | 68.4 | 42.9 | 73.1 | 38.5 | 74.4 | 75.0 | 41.1 | 52.6 |
| 1.3 | 3.0 | 0.7 | 2.6 | 0.7 | 4.0 | 2.2 | 1.9 | 2.7 | ||
| T2J | 66.7 | 61.9 | 49.2 | 63.8 | 60.0 | 60.5 | 59.3 | 56.1 | 63.1 | |
| 5.5 | 8.1 | 2.4 | 12.2 | 1.7 | 10.0 | 3.8 | 8.7 | 6.6 | ||
| T3D | 59.3 | 75.3 | 47.6 | 72.7 | 57.1 | 63.6 | 66.7 | 33.3 | 42.1 | |
| 1.3 | 5.7 | 0.8 | 3.3 | 1.1 | 3.9 | 1.4 | 1.6 | 2.2 | ||
| Noncon.c | T1L | 20.0 | 10.5 | 38.1 | 11.5 | 15.4 | 0.0 | 16.7 | 29.4 | 26.3 |
| 0.4 | 0.5 | 0.6 | 0.4 | 0.3 | 0.0 | 0.5 | 1.4 | 1.4 | ||
| T2J | 16.2 | 11.9 | 27.0 | 15.6 | 15.0 | 20.2 | 22.2 | 21.1 | 21.1 | |
| 1.3 | 1.6 | 1.3 | 3.0 | 0.4 | 3.3 | 1.4 | 3.3 | 2.2 | ||
| T3D | 18.5 | 8.2 | 33.3 | 9.1 | 21.4 | 11.4 | 22.2 | 27.8 | 42.1 | |
| 0.4 | 0.6 | 0.5 | 0.4 | 0.4 | 0.7 | 0.5 | 1.4 | 2.2 | ||
S1 not included because of uncertainty in where to place gaps
aValues determined for each pairwise comparison as: number of base changes/total such positions × 100 × 1.33
bValues determined as number of observed changes/number of positions at which changes could have occurred × 100
cUpper value indicates proportion of all amino acid substitutions that are conservative or nonconservative (using CLUSTAL W analysis with BLOSUM weighting); semi-conservative substitutions not included. Lower bold value indicates proportion of indicated types of alterations as a percentage of total number of amino acids within whole protein
The types of amino acid substitutions within each of the BYD1’ proteins were also examined. Pairwise analyses showed that most substitutions in most proteins were conservative (Table 2). Nonconservative substitutions were relatively rare in most proteins’ pairwise comparisons. For example, comparison of the BYD1 and T1L μNS proteins showed none (0.0%) of the 39 amino acid substitutions were nonconservative, and most BYD1:T1L comparisons gave low nonconservative substitution values ranging from 0.0 to 0.6% of total amino acid residues within the respective proteins. Only σNS and σ3 proteins demonstrated higher nonconservative variation, with values greater than 1.4% of total amino acid residues. Most of these higher nonconservative substitution values were observed when BYD1 proteins were compared to T2J proteins.
Sequence analysis of the MRV S1 genes and σ1 proteins
The MRV S1 segment, encoding viral attachment protein σ1 and nonstructural protein σ1s, is unique to each MRV prototype strain and determines the serotype and is also the major genetic determinant of neurovirulence in infected mice. When compared to other S1 genes in GenBank at the nucleotide level, the BYD1 S1 gene had the highest identity (about 63%) to the sequences of MRV strain T2/human/Netherlands/1/73 (GenBank Accession Number AY862137, serotype 2), about 59, 61, and 48% identity with three prototype strains, respectively. The predicted BYD1 S1 gene major product (σ1 protein) was 461 amino acids in length, and showed most identity (about 64%) to GenBank AAW58063 (serotype 2), about 52, 60, and 26% identity with three prototype MRV σ1 proteins, respectively. While the S1 gene of BYD1 showed surprisingly low homology with MRV strains reported before, we determined the full-length sequences of S1 genes of three other MRV strains isolated during the SARS period and two MRV strains isolated from fecal specimens of two children of Beijing in 1982 by Wu et al., described in the section “Materials and methods.” Sequence alignments showed that all the S1 genes of BLD, JP, BYL, and BYD1 shared more than 99% identity with each other, the S1 gene of 302I shared more than 99% identity with that of 302II and the S1 genes of the four strains isolated during SARS period shared about 65% identity with those of the two strains isolated in 1982. BLAST search analysis showed that the entire six S1 gene sequences of MRV strains used in this study were markedly different from those of all previously reported and all the six S1 genes were slightly closer to type 2 than to type 1 and type 3 strains. When compared to the BYD1 S1 gene, there were altogether six nucleotide base-pair differences (three amino acids) which were all located in the 5′ end 450 bp region of the S1 genes of BYL, BLD, and JP, and corresponding in the oligomer stability region of σ1 proteins, suggesting the N-terminal region of σ1 protein was perhaps more prone to variation than the C-terminal region. The high identities between the S1 genes of the four new MRV isolates indicated that the four strains diverged from a common ancestor most recently.
The predicted σ1 proteins of three serotype groups were 470–472, 460–462, and 455–456 amino acids long, respectively. The C-terminal “head” domains (about 156 amino acids long), which were the JAM-A (Junctional Adhesion Molecule A, a major receptor for all known MRVs) binding sites and determined the different serotypes [29], were highly conserved in each serotype, with extremely low identities between different serotypes (data not shown). The predicted σ1 proteins of the four new MRV isolates were 461 amino acids long, which were within the scope of serotype 2. And their “head” domains were also closer to those of serotype 2, sharing about 78% identity with GenBank AAW58063. Pairwise alignment with BYD1 σ1 and AAW58063 showed another highly homologous region (171–225 amino acids), which was in the “tail” domain and perhaps related with the capacity of MRV to bind carbohydrate receptors, sharing about 87% identity. These two regions of the four new isolates had low homology with those of type 1 or type 3 strains. The predicted σ1 proteins of the two 302 isolates were also 461 amino acids long, and shared about 76% identity with “head” domains of the four new isolates.
In order to define the evolutionary relationship of all the six new S1 genes with the S1 genes of other MRV strains whose sequences are deposited in GenBank, we constructed a phylogenetic tree by use of variation in the S1 gene nucleotide sequences and the neighbor-joining algorithm (Fig. 1). The most noteworthy feature of the S1 phylogenetic tree was that the S1 genes of the four new MRV isolates were substantially divergent from the type 1 and type 3 MRV strains. In 88% of the cases, the BYD1 S1 gene belonged to the type 2 clade, suggesting that the S1 gene segment diverged from a previously unidentified serotype 2 isolate. A phylogenetic tree generated by use of the σ1 protein amino acid sequences had a topology similar to the tree generated by using the S1 gene nucleotide sequences (data not shown). However, the preceding genome comparative analyses showed that other nine segments of BYD1 were closer to those of homologous T1L and T3D segments. Therefore, MRV BYD1 is a natural reassortant virus, thus providing evidence for the hypothesis that MRV gene segments reassort in nature.
Fig. 1.
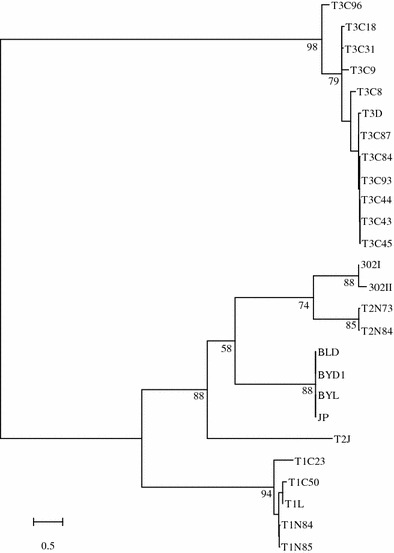
Phylogenetic tree analyses of six new MRV S1 genes, and homologous genes in other MRVs (based on available GenBank sequence data). Numbers shown at the branch nodes indicate percentage bootstrap support
Additional analysis of the functions and predicted secondary structures of the BYD1 proteins
Previous studies have comprehensively described the functions of proteins encoded by the ten genome segment of T3D [30, 31]. Pairwise comparisons have shown the high degree of conservation between λ3, λ2, λ1, μ2, μ1, μNS, σ2, σNS, and σ3 of BYD1 and cognate proteins of three prototype strains, exhibiting 95–98% deduced sequence identity (Table 1), so these nine BYD1 proteins most likely have the same structures of cognate T3D proteins. And the predicted secondary structures of these nine BYD1 proteins, made with the MLRC secondary structure prediction program, were also very similar to the cognate T3D proteins (data not shown). While σ1 is the most variable protein in the virus, PFAM analysis of the BYD1 σ1 reveals a match to PFAM domain Reo_sigma1 and alignments to 42 other sequences in the PFAM database bearing this domain. The predicted secondary structures of the BYD1 σ1 protein were analyzed and compared to the three prototype σ1 proteins (Fig. 2). The BYD1 σ1 N-terminal 150 amino acids, corresponding to the “stalk,” consisted primarily of α-helix and was very similar to this region in the σ1 proteins of the other MRVs. The region from amino acids 107 to 175 in BYD1 σ1 was predicted to be 1 consecutive α-helix. The region from amino acids 249 to 297 in BYD1 σ1 contained more predicted α-helix structure than this region in the other viruses, and the structure of C-terminal “head” domain was predicted to be very similar in the three prototype MRV σ1 proteins.
Fig. 2.
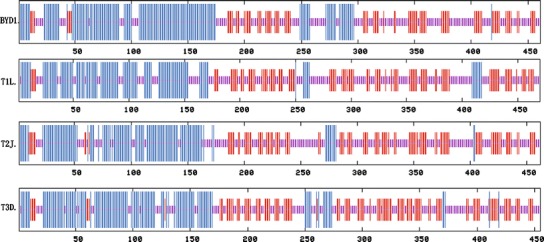
Secondary structure predictions of σ1 proteins of MRV BYD1 and the three prototype strains. Secondary structure predictions made with Network Protein Sequence Analysis [28], tall vertical lines (α-helix), medium lines (β-sheet), and short lines (random coil)
Discussion
MRVs share a number of common structural characteristics in the genome comprising ten segments of dsRNA and in the number and arrangement of structural proteins within particles [1, 4]. The MRV BYD1 strain analyzed here was isolated from the throat swabs of one SARS patient of Beijing. Comparative analyses of the BYD1 genome with those of three prototype strains demonstrated that this newly isolated virus is a novel human MRV isolate and also a reassortant virus, with the S1 segment coming from a previously unidentified serotype 2 isolate and other nine segments coming from ancestors of homologous T1L and T3D segments, which supports the hypothesis that MRV gene segments reassort in nature. Further analyses of the S1 segments of the four new MRV isolates and two earlier isolates confirmed that the four new isolates were new serotype 2 strains and showed greater sequence diversity of both the S1 genes and the deduced σ1 proteins. Compared to three prototype strains, the BYD1 strain had highly conserved proteins encoded by non-S1 segments, suggesting that the evolution of MRV non-S1 segments had reached equilibrium. Analyses of the S1 genes of the four newly isolated strains suggested that the N-terminal region of σ1 protein was perhaps more prone to variation than the C-terminal region. In these regards, this study enhanced our understanding of MRV evolution.
The sequences of the S1 segments of the other three new MRV isolates also proved that the four new isolates diverged from a common ancestor most recently. The BLD strain and JP strain were both isolated from throat swabs of SARS patients. Notably, the BYL strain was isolated from the lung tissue sample of one dead SARS patient. Although MRV infection has been shown quite common in the human population, no MRV isolates from human lung tissues had been reported before. Comparative analyses of the S1 genes of the six human MRV isolates used in this study support previous observations of the MRV protein σ1, including remarkable degree of variation in size, gene arrangement, and coding potential of polycistronic S-Class genome segments. The isolations of the four highly homologous MRVs from multiple relevant SARS patients suggest that the novel MRVs may simultaneously spread with SARS-CoV in certain cases and they might have been undiagnosed or misdiagnosed in more cases.
MRV infections have been associated with upper respiratory infections, fever, enteritis, febrile exanthema, and rare cases of encephalitis and meningitis in infants and children [13–15]. However, although MRV infections have been shown widely present in humans, these agents are isolated only rarely from clinical samples. In this regard, it is worth noting that the four novel MRV isolates used in this study were successively isolated from clinical samples of unambiguous SARS patients. It should be noted that all the four isolates were found to be mixed with poliovirus type 1 (vaccine strain) in our follow-up studies (data not shown). In a follow-up study, poliovirus type 1 RNAs were detected positive in four of eight fecal specimens from persons who had recovered from SARS for 2 years and two back mutations occurred in their primary attenuating mutation sites at nucleotide position 480 (G → A) in the 5′ UTR and nucleotide position 2795 (A → G) in the VP1 gene, while MRV and SARS-CoV RNAs were detected negative in all the specimens [32]. Although SARS-CoV has been accepted as the primary causative agent of SARS by the scientific community, this pathogen could not be identified from nearly 60% of the patients during the outbreak in Beijing [33]. In a previous review, Fan and Li [34] highlighted the possibility of MRV co-infections in SARS patients. However, at this stage we do not have serological evidence to prove poliovirus type 1 or MRV co-infections in SARS. Serological surveillance in SARS patients of Beijing will be necessary to resolve this issue.
In summary, in the present study the genome sequences of BYD1 and the S1 genes sequences of BLD, JP, BYL, 302I, and 302II were determined and analyzed. According the results the BYD1 is a novel human MRV isolate and also a reassortant virus, all the six strains are novel type 2 MRVs based on their S1 sequences, and the S1 genes of the four new isolates, which only share about 65% identity with those of 302I and 302II, diverged from a common ancestor most recently. Further serological surveillance in SARS patients will allow a better understanding of the epidemiology of the novel MRVs.
Acknowledgments
This study was supported by a National Natural Science Foundation of China (No. 30471555) and the Initiative Foundation for Scientific and Technological Innovation of Academic Military Medical Sciences (No. 200408183).
Contributor Information
Lihua Song, Email: songlihua@gmail.com.
Qing Duan, Phone: +86-10-63821339, FAX: +86-10-63821339, Email: duanq@nic.bmi.ac.cn.
References
- 1.Nibert M.L., Schiff L.A. Fields Virology. 4. Philadelphia: Lippincot Williams & Wilkins Inc; 2001. p. 1679. [Google Scholar]
- 2.Roy P. Fields Virology. 4. Philadelphia: Lippincot Williams & Wilkins Inc; 2001. p. 1835. [Google Scholar]
- 3.Estes M.K. Fields Virology. 4. Philadelphia: Lippincot Williams & Wilkins Inc; 2001. p. 1747. [Google Scholar]
- 4.Tyler K.L. Fields Virology. 4. Philadelphia: Lippincot Williams & Wilkins Inc; 2001. p. 1725. [Google Scholar]
- 5.Shatkin AJ, Sipe JD, Loh P. J. Virol. 1968;2:986–991. doi: 10.1128/jvi.2.10.986-991.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiener JR, Joklik WK. Virology. 1989;169:194–203. doi: 10.1016/0042-6822(89)90055-X. [DOI] [PubMed] [Google Scholar]
- 7.Breun LA, Broering TJ, McCutcheon AM, Harrison SJ, Luongo CL, Nibert ML. Virology. 2001;287:333–348. doi: 10.1006/viro.2001.1052. [DOI] [PubMed] [Google Scholar]
- 8.Yin P, Keirstead ND, Broering TJ, Arnold MM, Parker JS, Nibert ML, Coombs KM. Virol. J. 2004;1:6. doi: 10.1186/1743-422X-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang J, Hermann L, Coombs KM. Virus Genes. 2006;33:193–204. doi: 10.1007/s11262-005-0046-4. [DOI] [PubMed] [Google Scholar]
- 10.Fajardo E, Shatkin AJ. Virology. 1990;178:223–231. doi: 10.1016/0042-6822(90)90397-A. [DOI] [PubMed] [Google Scholar]
- 11.Belli BA, Samuel CE. Virology. 1993;193:16–27. doi: 10.1006/viro.1993.1099. [DOI] [PubMed] [Google Scholar]
- 12.Sabin AB. Science. 1959;130:1387–1389. doi: 10.1126/science.130.3386.1387. [DOI] [PubMed] [Google Scholar]
- 13.Tyler KL, Barton ES, Ibach ML, Robinson C, Campbell JA, O’Donnell SM, Valyi-Nagy T, Clarke P, Wetzel JD, Dermody TS. J. Infect. Dis. 2004;189:1664–1675. doi: 10.1086/383129. [DOI] [PubMed] [Google Scholar]
- 14.Johansson PJ, Sveger T, Ahlfors K, Ekstrand J, Svensson L. Scand. J. Infect. Dis. 1996;28:117–120. doi: 10.3109/00365549609049060. [DOI] [PubMed] [Google Scholar]
- 15.Hermann L, Embree J, Hazelton P, Wells B, Coombs RT. Pediatr. Infect. Dis. J. 2004;23:373–375. doi: 10.1097/00006454-200404000-00026. [DOI] [PubMed] [Google Scholar]
- 16.Chua KB, Crameri G, Hyatt A, Yu M, Tompang MR, Rosli J, McEachern J, Crameri S, Kumarasamy V, Eaton BT, Wang LF. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11424–11429. doi: 10.1073/pnas.0701372104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan Q, Zhu H, Yang Y, Li W, Zhou Y, He J, He K, Zhang H, Zhou T, Song L, Gan Y, Tan H, Jin B, Li H, Zuo T, Chen D, Zhang X. Chin. Sci. Bull. 2003;48:1369–1372. doi: 10.1007/BF03184165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuo T, Tan H, He J, Zhu H, Zhang H, Song L, Huang R, Duan Q. Bull. Acad. Mil. Med. Sci. 2003;27:241–243. [Google Scholar]
- 19.He C, Pang W, Yong X, Zhu H, Lei M, Duan Q. DNA Cell Biol. 2005;24:491–495. doi: 10.1089/dna.2005.24.491. [DOI] [PubMed] [Google Scholar]
- 20.Liang L, He C, Lei M, Li S, Hao Y, Zhu H, Duan Q. DNA Cell Biol. 2005;24:485–490. doi: 10.1089/dna.2005.24.485. [DOI] [PubMed] [Google Scholar]
- 21.Su Y, He J, Zhu H, Song L, Huang R, Duan Q. Bull. Acad. Mil. Med. Sci. 2005;29:418–420. [Google Scholar]
- 22.Wu S., Li Y., Fan W., Wu S., Wei X., Tong S., Wang Z., Zhang R., Li Y. Zhonghua Yi Xue Za Zhi. 1982;62:146–151. [PubMed] [Google Scholar]
- 23.Diaz-Ruiz JR, Kaper JM. Prep. Biochem. 1978;8:1–17. doi: 10.1080/00327487808068215. [DOI] [PubMed] [Google Scholar]
- 24.Lambden PR, Cooke SJ, Caul EO, Clarke IN. J. Virol. 1992;66:1817–1822. doi: 10.1128/jvi.66.3.1817-1822.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nei M, Gojobori T. Mol. Biol. Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 27.Kumar S, Tamura K, Nei M. Brief Bioinform. 2004;5:150–163. doi: 10.1093/bib/5.2.150. [DOI] [PubMed] [Google Scholar]
- 28.Combet C, Blanchet C, Geourjon C, Deleage G. Trends Biochem. Sci. 2000;25:147–150. doi: 10.1016/S0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- 29.Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. J. Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joklik WK, Roner MR. Prog. Nucleic Acid Res. Mol. Biol. 1996;53:249–281. doi: 10.1016/S0079-6603(08)60147-6. [DOI] [PubMed] [Google Scholar]
- 31.Becker MM, Peters TR, Dermody TS. J. Virol. 2003;77:5948–5963. doi: 10.1128/JVI.77.10.5948-5963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y., Xin S.J., Shen H.H., Hu Y., Xu D.P., Zhu H., Zhu L., Duan Q., Mao P.Y. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2006;20:66–68. [PubMed] [Google Scholar]
- 33.R. Walgate, Scientist (2003), http://www.biomedcentral.com/news/20030520/03/
- 34.Fen M., Li Z. Chin. J. Epidemiol. 2004;25:1078–1080. [Google Scholar]