

Abstract
Inhibition of the androgen receptor (AR) is the mainstay treatment for advanced prostate cancer. Ralaniten (formally EPI-002) prevents AR transcriptional activity by binding to its N-terminal domain (NTD) which is essential for transcriptional activity. Ralaniten acetate (EPI-506) the triacetate pro-drug of ralaniten, remains the only AR-NTD inhibitor to have entered clinical trials (NCT02606123). While well tolerated, the trial was ultimately terminated due to poor pharmacokinetic properties and resulting pill burden. Here we discovered that ralaniten was glucuronidated which resulted in decreased potency. Long-term treatment of prostate cancer cells with ralaniten results in upregulation of UGT2B enzymes with concomitant loss of potency. This has proven to be a useful model with which to facilitate the development of more potent second-generation AR-NTD inhibitors. Glucuronidated metabolites of ralaniten were also detected in the serum of patients in Phase 1 clinical trials. Therefore, we tested an analogue of ralaniten (EPI-045) which was resistant to glucuronidation and demonstrated superiority to ralaniten in our resistant model. These data support that analogues of ralaniten designed to mitigate glucuronidation may optimize clinical responses to AR-NTD inhibitors.
Keywords: androgen receptor, drug resistance, ralaniten, UDP-glucuronosyltransferase (UGT2B)
The androgen receptor (AR) signaling axis has long been recognized as a key driver of prostate cancer growth and survival. Therefore, androgen deprivation therapy (ADT) remains the primary treatment option for patients who demonstrate biochemical relapse, and those presenting with locally advanced or metastatic disease.1−3 While ADT has been proven to provide an initial immediate benefit with respect to tumor burden, the disease ultimately progresses to a lethal form termed metastatic castration-resistant prostate cancer (mCRPC), with an average life expectancy of 2–3 years.2−5
mCRPC is defined as disease progression despite castrate levels of testosterone. Most mCRPC is characterized by the restoration of AR transcriptional activity evidenced by rising serum levels of prostate-specific antigen (PSA).2−7 In accordance with these observations, the standard of care for men with mCRPC following ADT revolves around sustained AR inhibition using antiandrogens which competitively bind the ligand-binding domain (LBD) of the AR8 or CYP17A1 inhibitors4,9 that inhibit steroidogenesis. However, both of these approaches either directly or indirectly target the AR LBD. Mutations in this domain10−12 as well as constitutively active AR-splice variants lacking the LBD13,14 have been directly implicated in the clinic as being associated with both primary and acquired resistance. Indeed, nearly all patients who initially respond to AR targeted therapies will ultimately relapse and develop acquired resistance within 12–24 months.2,5,15
An alternative approach is to target the N-terminal domain (NTD) of the AR. The NTD contains key motifs responsible for interacting with coregulatory molecules, and its function is essential for the transcriptional activity of the AR.16,17 This rationale was validated by demonstrating reduced tumor burden and decreased serum PSA in mice bearing human prostate cancer xenografts which expressed, or were treated with, a decoy molecule corresponding to the NTD of the AR (AR1–558).18,19 This has led to the discovery of a new class of small molecules (anitens) which have recently received “first-in-class” status as novel inhibitors of the AR-NTD.20−22 Importantly, the NTD shares very little sequence homology with other related nuclear receptors,19,20,23 and the specificity of the ralaniten derivatives for the AR has been previously described.20 Reports have demonstrated the antitumor efficacy of the lead compound, ralaniten/RAL (previously EPI-002), in a PDX, CRPC xenograft models,20,21 and more recently using an enzalutamide-resistant LNCaP95 xenograft model which expresses the AR-V7 variant.24 As a result, a Phase I clinical trial that investigated the safety profile of ralaniten acetate (the prodrug of ralaniten) in the context of abiraterone or enzalutamide resistance was initiated in November, 2015 (ClinicalTrials.gov identifier: NCT02606123, ESSA Pharma, Inc.).
While ralaniten was well tolerated and imparted PSA responses and stable disease in some patients, its poor pharmacokinetic profile prevented further advancement into Phase II. Here we show that ralaniten is subject to glucuronidation by UDP-glucuronosyltransferase (UGT2B) enzymes resulting in loss of potency. These enzymes target nucleophilic hydroxyl groups and mediate the conjugation of glucuronic acid to suitable substrates. Glucuronidation is typically a detoxifying pathway and usually renders the target physiologically nonfunctional and highly hydrophilic.25,26 Clinical samples from CRPC patients who were resistant to enzalutamide and/or abiraterone confirm that ralaniten was glucuronidated in humans. Despite this setback small molecule inhibitors of the AR-NTD have considerable therapeutic potential—especially in the context of resistance to existing AR-targeted therapies. Analogues designed to mitigate glucuronidation showed improved potency in the context of elevated UGT activity. This may have clinical relevance as overexpression of UGT genes has been reported in CRPC tissues.27−29
Results
Loss of Potency with Long-Term Exposure to Ralaniten
To understand how mechanisms of resistance may arise in response to chronic treatment with an AR-NTD inhibitor, we generated a model using LNCaP cells. Much of the preclinical data for ralaniten was completed in this cell line with validation using VCaP, LNCaP95, PDXs, and the Herschberger assay.20,21,24 LNCaP cells provide a clear and well-defined baseline with which to compare any future characterization in the resistant line, at the transcriptional level as well as for measuring cellular proliferation in vitro and in vivo. In addition, LNCaP cells have been previously used to identify resistance mechanisms including increased levels of AR and expression of consistutively active AR splice variants.30−32
Parental LNCaP cells were maintained in growth media supplemented with increasing concentrations of ralaniten until they demonstrated stable growth at 50 μM at approximately 18 months (Supplementary Figure S1A–D). The resulting resistant line was termed LNCaP-RALR, and was initially characterized in terms of androgen responsiveness and sensitivity to ralaniten or antiandrogens. Dose escalation experiments were employed to measure sensitivity to antagonists targeting the NTD or LBD using ralaniten, or antiandrogens bicalutamide (BIC) and enzalutamide (ENZ). LNCaP-RALR cells demonstrated significantly reduced sensitivity to ralaniten than parental LNCaP cells with an IC50 of 50.65 μM versus 9.98 μM, respectively (Figure 1A). Importantly, both bicalutamide and enzalutamide had similar IC50 values in each cell line (BIC: LNCaP, 2.11 μM; vs LNCaP-RALR, 1.42 μM. ENZ: LNCaP, 0.92 μM; vs LNCaP-RALR, 1.01 μM, Figure 1A), indicating that cross-resistance between these antagonists targeting alternative regions of the AR did not occur.
Figure 1.

Ralaniten no longer inhibits the growth of LNCaP-RALR cells. (A) Dose response curves of LNCaP and LNCaP-RALR cells treated with ralaniten, bicalutamide, or enzalutamide and stimulated with 0.1 nM R1881. Data presented as mean ± SEM and analyzed by two-way ANOVA with Sidak’s test applied post hoc, n = 4 independent experiments. (B) Transcript levels of AR, PSA, FKBP5, and RHOU normalized to SDHA from LNCaP and LNCaP-RALR cells treated with ralaniten (35 μM), enzalutamide (5 μM), or DMSO control and stimulated with 1 nM R1881 (black bars) or EtOH vehicle (white bars). LNCaP-RALR cells show reduced sensitivity to ralaniten compared to LNCaP cells. Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied post hoc, n = 3 independent experiments. (C) Protein levels of AR and AR regulated genes from whole cell lysates treated as in panel B. (D) Western blots of AR levels in LNCaP and LNCaP-RALR cells transfected with control or AR siRNA (10 nM) for 24–96 h. (E) Crystal violet assay measuring proliferation of cells 96 h post-transfection with either scrambled (SCRM) or AR siRNA (10 nM) and stimulated with 0.1 nM R1881 or EtOH vehicle. Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied post hoc, n = 3 independent experiments. (F) Protein levels of AR and AR-variant from whole cell lysates treated as in panel B. Lysates harvested from LNCaP95 and VCaP cells serve as positive controls for AR-variant (AR-V). *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; n.s., not significant: RAL, ralaniten; ENZ, enzalutamide.
Both LNCaP and LNCaP-RALR cells showed induction of expression of well-characterized full-length AR (ARFL) regulated genes following stimulation with synthetic androgen R1881 confirming the presence of functional ARFL (Figure 1B,C). As expected, parental LNCaP cells were sensitive to both ralaniten and enzalutamide which blocked AR transcriptional activity. Conversely, LNCaP-RALR cells failed to respond to ralaniten while retaining sensitivity to enzalutamide (Figure 1B,C) consistent with dose response data. We next employed targeted siRNA to the AR. Levels of AR protein were undetectable after 24 h and remained so through to 96 h in both cell lines treated with AR siRNA (Figure 1D). Both cell lines treated with AR siRNA followed by stimulation with 0.1 nM R1881 demonstrated significantly reduced levels of proliferation, and were comparable to cells treated in the absence of androgen (Figure 1E). Thus, androgen-induced proliferation of LNCaP-RALR cells remain dependent upon ARFL as expected, and agrees well with previous observations of sensitivity to antiandrogens. Surprisingly, we noted a specific silencing of the classical androgen induced gene, PSA, in the resistant line. While protein and mRNA expression could be detected, it was substantially reduced (Figure 1B,C). As yet the mechanism by which this occurs has not been deduced. It may involve epigenetic silencing of the PSA gene as the AR is clearly driving the growth of these cells, and is functioning similarly to the parental line with respect to other AR regulated genes.
Amplification of ARFL and expression of constitutively active AR splice variants have both been associated with poor clinical outcomes and resistance to hormonal therapies.13,14,33 We therefore probed whole cell lysates harvested from both LNCaP and LNCaP-RALR cells using an antibody to the AR NTD to detect ARFL and truncated AR-splice variants. LNCaP95 and VCaP cells were used as positive controls for AR-splice variants and VCaP cells also have amplification of AR.(34,35) We found levels of AR protein to be stable across treatments for both the parental and resistant LNCaP cell lines; suggesting a unique mechanism as compared to previous reports of increased AR as a mechanism of resistance to first generation antiandrogens in LNCaP cells.30,31 Importantly there was no evidence that either cell line expressed any AR-variant at the protein level (Figure 1F). Nor could any mRNA transcript be detected for the clinically relevant AR-V7 splice variant under conditions used, whereas mRNA transcript levels for ARFL were similar in both cell lines regardless of the addition of androgen, enzalutamide, or ralaniten (Figure 1B).
Gain-of-function mutations represent a clinically relevant mechanism of resistance to both inhibitors of steroidogenesis as well as antiandrogens.3,7,10−12 Ralaniten specifically binds the TAU-5 domain within the AF-1 region of the AR-NTD.20−22 Therefore, any gain-of-function mutation(s) selected as a resistance mechanism would likely reside in or near this region. cDNA isolated from both LNCaP and LNCaP-RALR cells was cloned and sequenced. No mutations were identified in the NTD. However, the well-known T877A lesion in the C-terminal LBD was present in all clones as expected, considering the parental LNCaP cell line harbors this LBD mutation.5,6,24,36 LNCaP cells are sensitive to ralaniten; therefore this lesion was not considered to be associated with resistance to ralaniten (Supplementary Figure S2A,B). Collectively these data show that LNCaP-RALR cells retain functional ARFL, and imply that alternative AR targeting strategies would be effective in the context of ralaniten resistance.
Loss of Efficacy of Ralaniten in LNCaP-RALR Xenografts
To test if enzalutamide would retain activity in vivo in the context of ralaniten resistance, castrated mice bearing LNCaP-RALR xenografts were randomized into three treatment groups: ralaniten (200 mg/kg), enzalutamide (10 mg/kg), or vehicle. Xenografts of the parental LNCaP cells were also employed as a control for ralaniten sensitivity. LNCaP xenografts are highly sensitive to enzalutamide at a dose of 10 mg/kg.37
Mice bearing LNCaP xenografts showed little to no tumor growth with ralaniten treatment, however LNCaP-RALR xenografts were unresponsive to ralaniten (Figure 2A,B). Mouse body weight was regularly recorded over the course of the experiment (Figure 2C). Enzalutamide treatment blocked LNCaP-RALR tumor growth (Figure 2A,B). LNCaP-RALR xenografts maintained expression of ARFL at the protein (Figure 2D) and mRNA levels (Figure 2E). As seen in earlier experiments, classical ARFL regulated genes were reduced by enzalutamide but not by ralaniten (Figure 2D,E). Interestingly, PSA expression was readily detected in LNCaP-RALR xenografts both by immunohistochemistry and qRT-PCR, indicating that expression was context dependent. Importantly only enzalutamide treatment was sufficient to reduce both protein and mRNA expression of PSA. These data confirm that LNCaP-RALR xenografts retain biological resistance to ralaniten, yet remain dependent upon ARFL and are sensitive to enzalutamide.
Figure 2.

LNCaP-RALR xenografts retain biological resistance to ralaniten and sensitivity to enzalutamide in vivo. (A) LNCaP and LNCaP-RALR tumor growth in castrated mice treated daily with ralaniten (200 mg/kg), enzalutamide (10 mg/kg), or vehicle by oral gavage. Tumors were harvested 2 days after last treatment. Data presented as mean ± SEM and analyzed by two-way ANOVA with Sidak’s test applied post hoc. (B) Representative photographs of LNCaP-RALR tumors. Scale bars: 10 mm. (C) Body weight change over the course of the experiment. Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied post hoc (n = 5 mice per group). (D) Representative examples of immunohistochemistry (IHC) from LNCaP-RALR tumors showing AR and PSA expression levels (n = 2 tumors per group). (E) Real-time PCR of AR, FKBP5, RHOU, and PSA transcript normalized to SDHA harvested from LNCaP-RALR tumors (n = 8 tumors/sample for all genes except FKBP5, n = 6 for RAL). *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; n.s., not significant: RAL, ralaniten; ENZ, enzalutamide; VEH, vehicle.
Several UGT2B Isoforms Are Enriched in LNCaP-RALR Cells and May Provide a Mechanism for Loss of Potency to Ralaniten
To reveal a potential mechanism for loss of potency, we employed a whole transcriptome microarray on cDNA isolated from LNCaP and LNCaP-RALR cells treated with enzalutamide (5 μM), ralaniten (35 μM), or DMSO vehicle in the presence or absence of R1881 (1 nM). Initially, the ability of ralaniten or enzalutamide to inhibit AR mediated gene transcription on a global level was assessed. Protein coding genes which displayed ≥2 fold increase in the presence of DMSO/R1881 compared to DMSO/EtOH were defined as androgen induced. In total, 243 genes were identified in LNCaP cells, and 260 in LNCaP-RALR with 156 genes common to both cell lines. Of these subsets, genes which were inhibited ≥2 fold by either ralaniten or enzalutamide in the presence of R1881 were identified. This analysis revealed that on a global scale, the ability of ralaniten to block AR mediated transcription was largely blunted in the resistant line compared to LNCaP cells (LNCaP-RALR, 13.1% genes inhibited vs LNCaP, 44.8% genes inhibited). Conversely, enzalutamide retained its ability to block transcription of AR regulated genes in both cell lines (LNCaP-RALR, 64.6% genes inhibited vs LNCaP, 68.3% genes inhibited, Figure 3A).
Figure 3.

Enrichment of UGT2B isoforms may provide a mechanism for loss of potency to ralaniten in LNCaP-RALR cells. (A) Analysis of global gene expression identified genes which were specifically induced by R1881 (LNCaP = 243; LNCaP-RALR = 260). The Venn diagrams show the percentage of androgen induced genes in this subset, the expression of which decreased by ≥2 fold in the presence of enzalutamide or ralaniten. (B) Heatmap representing differentially regulated genes between LNCaP-RALR (group 1/blue) and LNCaP (group 2/black) cells treated with ralaniten (35 μM), enzalutamide (5 μM), or DMSO vehicle and stimulated with 1 nM R1881 (+) or EtOH vehicle (−). Four UGT2B isoforms (UGT2B11, UGT2B15, UGT2B17, and UGT2B28 were identified which were highly enriched in the resistant line (between 10- and 18-fold increase) compared to matched LNCaP samples (n = 2 per treatment group). (C) Transcript levels of UGT2B isoforms normalized to SDHA in additional samples validated the microarray data. Data are presented as mean ± SEM, n = 3 independent experiments. (D) Microsomes harvested from untreated parental LNCaP and LNCaP-RALR cells revealed increased basal expression of UGT2B isoforms in the resistant line. (E) The UGT-Glo assay quantified the amount of UGT enzymatic activity, by measuring the amount of a luciferin substrate which could be consumed through glucuronidation (unpaired Student’s t-test; n = 4; p = 0.0003). Data are presented as mean ± SEM: RAL, ralaniten; ENZ, enzalutamide.
Hierarchical clustering revealed 208 differentially regulated genes between LNCaP and LNCaP-RALR which showed an increase in expression >2 fold in the resistant line compared to matched controls. Of note, four of the five top hits were all members of the UDP-glucuronoslytransferase (UGT2B) family (UGT2B15, 18.38 fold; UGT2B17, 16.99 fold; UGT2B11, 13.99 fold; UGT2B28, 9.35 fold; Figure 3B). These genes are responsible for phase II metabolism of a wide variety of lipophilic compounds including drugs, environmental pollutants, and steroid hormones.36−40
UGT2B enzymes target nucleophilic hydroxyl groups,25,26 a behavior that agrees well with the fact that ralaniten has three hydroxyl groups which could be potentially glucuronidated. UGT enzymes are found mainly in the liver and gastrointestinal tract; however, UGT2B15, UGT2B17, and UGT2B28 are also expressed in the prostate.36−39,41−44 Studies using cell lines have shown disparate trends for expression of UGT2B15 and UGT2B17 when comparing androgen-sensitive LNCaP and C4–2 cells to androgen-independent CRPC models such as LNCaP-abl, DU145, and PC3 cells.39,45 Additional studies using clinical samples have also reported increased levels of UGT isoforms including UGT2B15, UGT2B17, and UGT2B28 studied here with clinical progression, biochemical relapse, CRPC, and metastases.27−29 Thus, deregulation of UGT2B enzymes has clinical relevance.
Microarray data was validated using qRT-PCR and Western blot in additional samples (Figure 3C,D). Each of the UGT2B isoforms were highly enriched in the resistant line compared to parental LNCaP cells. UGT2B11 and UGT2B28 were strongly induced by androgen in LNCaP-RALR cells but poorly expressed in parental cells regardless of androgen. Consistent with previous reports that UGT2B15 and UGT2B17 are androgen-repressed genes, we too observed that expression of these genes was inhibited by synthetic androgen R1881, and that enzalutamide partially restored levels of expression.39,42,43 Enzymatic activity was similarly increased in LNCaP-RALR microsomes displaying a 6-fold increase (p = 0.0003) in the amount of substrate consumed compared to those isolated from LNCaP cells (Figure 3E).
We next asked if UGT2B isoforms were also enriched in additional clones, and if expression was related to the degree of ralaniten resistance (Supplementary Figure S3A). While not significant, there was a trend in which clones with the lowest UGT2B expression (clones E9 and F9) showed the greatest sensitivity to ralaniten (Supplementary Figure S3B,C). Collectively these results point to a mechanism of resistance whereby ralaniten is preferentially metabolized by one or more UGT2B isoforms, allowing cells to maintain AR transcriptional activity.
Loss of Potency to Ralaniten Is Dependent upon Expression of UGT2B15 or UGT2B17
To test the hypothesis that overexpression of UGT2B genes would result in loss of potency to ralaniten, we endeavored to restore sensitivity to ralaniten by knocking down expression of specific isoforms of UGT2B. To this end, pooled siRNA against UGT2B15 or UGT2B17 was used, as these isoforms had the greatest expression in the resistant line. Cross-reactivity was noted for both siRNAs as UGT2B15 and UGT2B17 were each inhibited to a similar extent, preventing the ability to distinguish the contributions of each gene. Conversely expression of UGT2B11 and UGT2B28 were not affected by siRNA treatment (Figure 4A). Western blot confirmed that protein levels could be silenced for the entirety of the treatment regimen, prior to beginning the proliferation experiment (Figure 4B). Attenuation of expression of UGT2B15 or UGT2B17 was sufficient to restore sensitivity of LNCaP-RALR cells to ralaniten (25 μM) in blocking cellular growth to levels that were comparable to that in the absence of androgen (Figure 4C). When cells were treated with siRNAs to both UGT2B15 and UGT2B17, no additive or synergistic effect was measured which may be due to the cross-reactivity between the two siRNA pools.
Figure 4.

Sensitivity to ralaniten is dependent upon expression of UGT2B15orUGT2B17. (A) Cells treated with 15 nM siRNA targeting UGT2B15 and UGT2B17 efficiently silenced mRNA expression. Significant cross-reactivity was observed with respect to UGT2B15 and UGT2B17; however, no other isoforms were silenced by siRNA treatment. Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied post hoc, n = 4 independent experiments. (B) Western blots of UGT2B15/17 protein levels in LNCaP-RALR cells transfected with 15 nM control, UGT2B15 (top), or UGT2B17 (bottom) siRNA for 24–96 h. (C) Crystal violet assay measuring proliferation of cells 96 h post-transfection with either scrambled (SCRM), UGT2B15, and/or UGT2B17 siRNA (15 nM). Twenty-four hours after transfection, cells were treated with ralaniten (25 μM) or DMSO vehicle and stimulated with 1 nM R1881 (black bars) or EtOH (white bars). Silencing of either UGT2B15 or UGT2B17 was sufficient to restore sensitivity to ralaniten (n = 3 independent experiments). Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied posthoc,n = 3 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; n.s., not significant; RAL, ralaniten.
We attempted to discern whether ectopic UGT2B15 or UGT2B17 expression could reduce sensitivity to ralaniten. Parental LNCaP cells were transduced with a recombinant lentivirus containing an expression vector coding for HA-tagged human UGT2B15, UGT2B17, or GFP under the control of a CMV promoter (Figure 5A,B) and enzymatic activity was confirmed (Figure 5C). Both LNCaP-2B15 (clone 2/C2) and LNCaP-2B17 (clone 1/C1) demonstrated increased ability to glucuronidate a fluorescent substrate compared to control LNCaP-GFP (clone 1/C1) cells. However, enzymatic activity was also significantly lower when compared to LNCaP-RALR cells (2B15_C2, p = 0.0220; 2B17_C1, p = 0.0005) possibly due to the influence of additional UGT2B isoforms (2B11 and 2B28) specifically expressed in the LNCaP-RALR cell line (Figure 5C).
Figure 5.

Ectopic expression of UGT2B15 reduces sensitivity to ralaniten. (A) Real-time PCR of UGT2B11, UGT2B15, UGT2B17, and UGT2B28 mRNA transcript normalized to levels of SDHA transcript harvested from untreated clones isolated following lentiviral transduction, and LNCaP-RALR cells. Data presented as mean ± SEM and analyzed by one-way ANOVA with Dunnett’s test applied post hoc, n = 3 independent experiments. (B) Western blots of ectopic and endogenous UGT2B15/UGT2B17 in LNCaP-RALR and lentiviral transduced clones. (C) UGT2B enzymatic activity quantified using microsomes harvested from LNCaP-RALR, 2B15-HA (C2), 2B17-HA (C1), and GFP-HA (C1) cells. Data presented as mean ± SEM and analyzed by two-way ANOVA with Tukey’s test applied post hoc, n = 3 independent experiments. (D) Dose response curves showing androgen dependent growth of LNCaP-RALR and lentiviral transduced clones treated with ralaniten and stimulated with 0.1 nM R1881. IC50 values were calculated using a linear regression and interpolating/extrapolating where the line crossed 50% growth. GFP-HA (C1), 18.15 μM; LNCaP-RALR, 39.68 μM; 2B15-HA (C2), 32.48 μM; 2B17-HA (C1), 28.14 μM. Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied post hoc, n = 5 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; n.s., not significant.
Lastly we challenged LNCaP-RALR, 2B15_C2, 2B17_C1, and GFP_C1 clones with increasing concentrations of ralaniten. LNCaP-GFP_C1 cells displayed an IC50 of 18.15 μM, comparable to previous experiments using parental LNCaP cells (16.91 μM). Ralaniten treatment was significantly associated with lower levels of androgen-dependent growth in LNCaP-GFP cells compared to all three cell lines at 25 μM and 35 μM concentrations, and as low as 10 μM for both LNCaP-RALR and 2B15_C2 (Figure 5D). This discrepancy could occur because UGT2B17 expression was less robust than UGT2B15 in transduced clones (Figure 5A,B). Taken together, these data further implicate UGT2B15, and to a lesser extent, UGT2B17 in mediating loss of potency to ralaniten.
Detection of Ralaniten or Its Analogue EPI-045, and Its Glucuronides
LNCaP-RALR cells could provide a preclinical model for structure activity relationship (SAR) studies to test if potential chemical modifications to drugs that are predicted to reduce glucuronidation, have improved potency compared to ralaniten. For example we pursued EPI-045, an analogue of ralaniten which lacks the primary alcohol on C1 that we suspected to be the major site of glucuronidation (Figure 6A). The ability of mouse liver microsomes to effectively glucuronidate ralaniten or EPI-045 in the presence and absence of uridine 5′-diphosphoglucuronic acid (UDPGA) cofactor was analyzed by high-performance liquid chromatography (HPLC). A single peak corresponding to ralaniten–glucuronide was measured when microsomes were incubated with UDPGA, and this peak disappeared following cotreatment with β-glucuronidase (Figure 6B, left column, bottom). The small peaks detected from EPI-045 treated microsomes that were incubated with UDPGA also disappeared with β-glucuronidase treatment. These data suggest that EPI-045 is a poor substrate for glucuronidation compared to ralaniten (Figure 6B, right column, bottom).
Figure 6.

Ralaniten is a substrate for O-glucuronidation by UGT2B enzymes. (A) Chemical structures of ralaniten, EPI-045, and major glucuronide of ralaniten M15 (EPI-002053). Potential sites of glucuronidation are shown in red. (B) HPLC chromatograms show glucuronidated products following incubation in the presence of UDPGA cofactor. While some minor peaks are observed in samples incubated with EPI-045 (labeled 045-G), the intensity is substantially lower than that seen in those incubated with ralaniten. Cotreatment with β-glucuronidase is sufficient to remove glucuronides from substrate, and thus only peaks indicating unbound drugs are seen. (C) Detection of ralaniten metabolites using LC/MS following incubation with human hepatocytes. Direct glucuronidation of ralaniten represents the most prevalent metabolite (M15). Another notable metabolite, M4, represents dechlorination followed by cysteine conjugation. (D) LC–MS chromatographs showing ralaniten metabolites isolated from pooled human plasma samples (ESSA Pharma, Inc.). Patients were treated with 3600 mg of ralaniten acetate daily. M15 represents direct glucuronidation, M19 is an oxidized metabolite. (E) Dose response curves measuring androgen-dependent growth of LNCaP cells treated with escalating concentrations of ralaniten or EPI-002053 (synthetic ralaniten glucuronide structurally equivalent to M15) and stimulated with 0.1 nM R1881. IC50 values were calculated using a linear regression and interpolation/extrapolation where the line crossed 50% growth. RAL, 14.78 μM; EPI-002053, >35 μM; p = 0.007. Data presented as mean ± SEM and analyzed by two-way ANOVA with Sidak’s test applied post hoc, n = 4 independent experiments. (F) Transcript levels of AR, PSA, FKBP5, and RHOU normalized to SDHA harvested from LNCaP cells treated with RAL (35 μM), EPI-002053 (35 μM) or v/v DMSO vehicle and stimulated with 1 nM R1881. Data presented as mean ± SEM and analyzed by one-way ANOVA with Dunnett’s test applied post hoc, n = 4 independent experiments. (G) Protein levels of AR and AR regulated genes from whole cell lysates (LNCaP) treated as in part F. *p < 0.05; **p < 0.01; ***p < 0.001; #p < 0.0001; n.s., not significant. RAL, ralaniten.
We next attempted to delineate which hydroxyl specie(s) contained on ralaniten was the primary target for glucuronidation. The HPLC experiment was repeated so that the glucuronide fraction could be isolated and analyzed by 1H NMR and then compared to individual synthetic monoglucuronides of ralaniten (Supplementary Figure S4). Given that preliminary in vitro work suggested EPI-045 retained sensitivity in the LNCaP-RALR cell line, we expected that the primary alcohol on C1 would be the main target for glucuronidation of ralaniten. Instead we found that the primary glucuronide product corresponded to M15, a glucuronide metabolite formed from the secondary alcohol on C20 (Supplementary Figure S4). This finding helps explain why EPI-045 may be partially glucuronidated. However, considering that EPI-045 also contains this functional group, this does raise the question as to why it is glucuronidated to a lesser extent than ralaniten. A possible explanation is that altering the primary alcohol restricts recognition or binding of EPI-045 by UGT2B enzymes due to changes in the hydrophobicity compared to ralaniten (ralaniten, C log P = 2.7958; EPI-045: C log P = 4.7136). Further work will be required to definitively understand this mechanism. Consistent with these data, the profiling of ralaniten metabolites using UPLC-HRMS/MS following incubation with human hepatocytes revealed that one of the major pathways of ralaniten clearance in humans was by direct glucuronidation. Furthermore, the most abundant metabolite was a glucuronide formed from the secondary alcohol on C20 and is structurally identical to M15 (Figure 6A,C).
A Phase I clinical trial investigating ralaniten acetate (EPI-506) in CRPC patients who were resistant to enzalutamide and/or abiraterone was completed in November 2017 (NCT02606123). Ralaniten acetate was given orally at escalating doses. The maximum dose tested of 3600 mg/day was well tolerated and imparted PSA responses and stable disease in some patients, but unfortunately it had poor pharmacokinetic properties. Plasma samples collected from patients treated at 3600 mg/day were analyzed by LC–MS and identified a number of metabolites of ralaniten. Ion spectra of the metabolite products were obtained using UPLC–HRMS/MS to profile individual metabolites. While the most prevalent metabolite was formed following oxidation of ralaniten (M19), direct glucuronidation (M15) was also identified as a major metabolic pathway in humans (Figure 6D). This implies that possibly hepatic and gastrointestinal mediated glucuronidation of ralaniten may be a significant mechanism for its clearance. Collectively these data demonstrate that ralaniten is a good substrate for glucuronidation, that this seems to preferentially involve the alcohol group on C20, and occurs readily in humans. Thus, glucuronidation of ralaniten has clinical relevance.
Glucuronidation is typically thought of as a detoxifying pathway, with the biological activity of the substrate reduced due to both the steric hindrance of the glucuronide adduct, as well as increased clearance from the cell. An important exception is the glucuronide metabolite of the opioid morphine (morphine-6β-glucuronide) which has nearly a 100-fold increase in pharmacological activity than the parental compound.46 Therefore, we wanted to confirm that glucuronidation of ralaniten did in fact reduce or impair its ability to function as an AR-NTD antagonist.
Synthetic EPI-002053 is structurally identical to M15—the glucuronide metabolite formed on C20 following incubation of ralaniten with liver microsomes, and also found in patient plasma. LNCaP cells were used for these experiments, as they have been repeatedly shown to be sensitive to ralaniten. Unlike ralaniten, EPI-002053 had no effect on AR-mediated transcriptional activity nor androgen-dependent growth of LNCaP cells (Figure 6E–G). Unsurprisingly, these data confirm that glucuronidation of ralaniten abrogates its activity, and supports our hypothesis that loss of potency to ralaniten was mediated through metabolism by UGT2B enzymes.
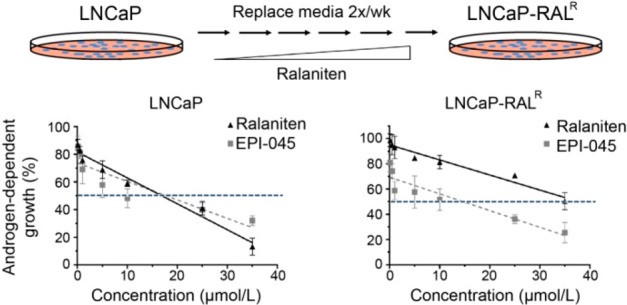
EPI-045 Effectively Inhibits AR Signaling in LNCaP-RALR Cells in Vitro and in Vivo
Like ralaniten, EPI-045 specifically targets the AR and does not prevent growth of the AR-negative PC3 cell line (Supplementary Figure S5A). Similarly, EPI-045 specifically targets the AR-NTD and does not bind the LBD (Figure 7A and Supplementary Figure S5B). Accordingly, EPI-045 is capable of targeting AR-V7 transcriptional activity and inhibits growth of variant expressing LNCaP 95 cells (Figure 7B,C). Sensitivity to EPI-045 was also readily measured in both LNCaP and LNCaP-RALR cells (LNCaP IC50, 17.79 μM vs LNCaP-RALR IC50, 14.70 μM; p = 0.96, Figure 7D). Unlike ralaniten, EPI-045 inhibited AR transcriptional activity in the LNCaP-RALR line (Figure 7E,F).
Figure 7.

EPI-045 effectively inhibits AR signaling in LNCaP-RALR cells in vitro. (A) Competition binding curve showing inhibition of ligand (2 nM) binding to the recombinant AR-LBD (20 nM) by antiandrogen bicalutamide, agonist R1881, and ralaniten analogue EPI-045. EPI-045 does not bind the AR-LBD and therefore does not inhibit ligand binding. Data presented as mean ± SD, n = 2 independent experiments. (B) LNCaP cells cotransfected with the AR-V7 driven synthetic UBE2C-luciferase reporter (consisting of 3 tandem repeats of an AR-V7-specific promoter element of the UBE2C gene) reporter and AR-V7 expression vectors. Cells were subsequently treated with DMSO, enzalutamide (5 μM), ralaniten (35 μM), or EPI-045 (25 μM) for 24 h. Western blot shows AR-V7 protein expression in matched samples cotransfected with UBE2C-luciferase reporter and AR-V7 expression vectors and treated as above. Data presented as mean ± SD and analyzed by one-way ANOVA with Dunnett’s test applied post hoc, n = 3 independent experiments. (C) Crystal violet assay measuring proliferation of LNCaP 95 cells. Cells were pretreated with DMSO, enzalutamide (5 μM), ralaniten (35 μM), or EPI-045 (35 μM) for 1 h prior to stimulation with 0.1 nM R1881 or EtOH. Absorbance was measured 96 h following drug treatment. Data presented as mean ± SD and analyzed by one-way ANOVA with Dunnett’s test applied post hoc, n = 4 independent experiments. (D) Dose response curves of the growth of LNCaP and LNCaP-RALR cells treated with EPI-045 and stimulated with 0.1 nM R1881. Data presented as mean ± SEM and analyzed by two-way ANOVA with Sidak’s test applied post hoc, n = 4 independent experiments. (E) Transcript levels of AR, NKX3.1, FKBP5, and RHOU normalized to SDHA from LNCaP and LNCaP-RALR cells treated with EPI-045 (35 μM), RAL (35 μM), or v/v DMSO and stimulated with 1 nM R1881 (black bars) or EtOH (white bars). AR transcriptional activity is inhibited by EPI-045 in both cell lines Data presented as mean ± SEM and analyzed by two-way ANOVA with Sidak’s test applied post hoc, n = 3 independent experiments. (F) Protein levels of AR and AR regulated genes from whole cell lysates treated as in panel E. Data are presented as mean ± SEM ***p < 0.001; #p < 0.0001; n.s., not significant. BIC, bicalutamide; ENZ, enzalutamide; RAL, ralaniten.
The in vivo efficacy of EPI-045 to inhibit the growth of LNCaP-RALR xenografts was next assessed. For this study castrated mice bearing LNCaP-RALR xenografts were randomized to receive ralaniten (50 mg/kg), EPI-045 (50 mg/kg), or vehicle every other day by tail vein injection. Drugs were administered in this way to limit the amount of first-pass metabolism, and avoid potential differences in oral bioavailability between the two compounds. EPI-045 had significant antitumor activity compared to both vehicle (p < 0.0001) and ralaniten (p = 0.0016, Figure 8A,B). Some sensitivity of LNCaP-RALR xenografts to ralaniten was observed compared to vehicle treatment when ralaniten was administered by i.v. (p = 0.0263) unlike when ralaniten was dosed orally (Figure 2A). This observation also agrees with the clinical data and suggests that ralaniten is subjected to hepatic and possibly gastrointestinal glucuronidation. Both ralaniten and EPI-045 were well-tolerated at 50 mg/kg (Figure 8C).
Figure 8.

EPI-045 has antitumor activity in LNCaP-RALR xenografts. (A) LNCaP and LNCaP-RALR tumor growth in castrated mice treated every other day with ralaniten (50 mg/kg), EPI-045 (50 mg/kg), or vehicle. To reduce first pass metabolism and hepatic mediated glucuronidation, drugs were administered by tail vein injection. Tumors were harvested 2 days after last treatment. Data presented as mean ± SEM and analyzed by two-way ANOVA with Tukey’s test applied post hoc. (B) Representative photographs of LNCaP-RALR tumors. Scale bars: 10 mm. (C) Body weight change over the course of the experiment. n = 10 (VEH), n = 11 (EPI-045), n = 4 (RAL). (D) IHC from LNCaP-RALR tumors showing AR, UGT2B15, Ki67, and TUNEL counterstained with DAPI. (E) Quantification of proliferative and apoptotic indices from tumors. Data presented as mean ± SEM and represents three sections from three different tumors per treatment group. Ki67:2732 (vehicle); 2514 (ralaniten); and 2619 (EPI-045) cells in total were counted. For TUNEL staining, 3123 (vehicle), 3538 (ralaniten), and 3400 (EPI-045) cells were counted. (F) Real-time PCR of AR, PSA, FKBP5, and RHOU transcript normalized to SDHA harvested from LNCaP-RALR tumors. Data presented as mean ± SEM and analyzed by two-way ANOVA with Dunnett’s test applied post hoc (n = 8 tumors/sample except RAL, n = 7). *p < 0.05; **p < 0.01; #p < 0.0001; n.s., not significant. RAL, ralaniten; VEH, vehicle.
Neither ralaniten nor EPI-045 had any significant effect on the protein levels of AR or UGT2B15 when measured by immunohistochemistry (IHC) in harvested tumors from castrated hosts (Figure 8D). The antitumor effect of EPI-045 was further demonstrated, as markers associated with proliferation (Ki67) and apoptosis (TUNEL) were decreased and increased, respectively, compared to levels in tumors harvested from mice treated with vehicle (Figure 8D,E). Additionally EPI-045 significantly reduced levels of mRNA of AR-target genes such as FKBP5 and RHOU in harvested tumors, whereas ralaniten had no significant effect (Figure 8F). These data support that resistant LNCaP-RALR cells and tumors have sensitivity to EPI-045 which is an AR-NTD antagonist that is poorly glucuronidated.
Discussion
The discovery of constitutively active AR-splice variants (e.g., AR-V7, and AR-V567es) and their clinical relevance in driving resistance to current AR-targeted therapies has been reported by numerous institutions.13,14 A therapeutic niche clearly exists for small molecules targeting the AR-NTD in the post-enzalutamide or abiraterone setting.20−22,24,36,47
Phase I data from the clinical trial indicated ralaniten was well tolerated in patients. Additionally, some evidence of therapeutic effect was indicated despite suboptimal steady state blood levels, as PSA declines and stable disease were observed in some patients within the higher dose cohorts.48 However, this therapeutic response was limited by a poor pharmacokinetic profile, possibly due to the first-pass metabolism of ralaniten by the liver or gastrointestinal tract.
Here we have identified a subset of genes (UGT2B) whose expression was causatively associated with loss of potency to ralaniten. We have demonstrated that ralaniten is a substrate for UGT2B mediated metabolism, revealed the preferred target moiety for glucuronide conjugation, and show that the resulting adduct had no activity with respect to antagonizing the AR, or preventing proliferation following androgen stimulation. Importantly, the glucuronide metabolite identified in our in vitro work was structurally identical to M15 identified in human plasma samples. Collectively these data demonstrate that ralaniten is a good substrate for glucuronidation, that this seems to preferentially involve the alcohol group on C20, and occurs readily in humans. Thus, glucuronidation of ralaniten has clinical relevance.
Interestingly, antiandrogens enzalutamide and bicalutamide retained their antagonistic effects in the LNCaP-RALR cell line. The mechanism of clearance of both (R)-bicalutamide49 and enzalutamide50 occurs primarily by CYP mediated Phase-I metabolism, rather than glucuronidation which agrees well with our model. This, taken together with the fact that LNCaP-RALR cells remain dependent upon AR transcriptional activity to promote their proliferation, provides a strong rationale to pursue combinatorial or sequential therapies targeting both the LBD and NTD.
We have characterized EPI-045, a derivative of ralaniten which is resistant to UGT2B mediated glucuronidation. EPI-045 showed increased potency as compared to ralaniten against a background of elevated UGT2B activity both in vitro and in vivo. This work represents a proof-of-concept study demonstrating that modification of functional groups on ralaniten can reduce metabolic liabilities and increase potency. Preclinical studies investigating the safety and efficacy of additional ralaniten analogues are ongoing. Here we have laid the foundation for characterizing next generation inhibitors and describe the first model of acquired resistance to any small molecule targeting the AR-NTD. Accordingly, the LNCaP-RALR cell line represents a unique asset with which to screen second-generation compounds against a background of increased glucuronidation.
Interestingly, the expression of PSA was significantly reduced in LNCaP-RALR cells when cultured in vitro, although expression was restored in vivo. We speculate that the reason is possibly due to epigenetic silencing of the PSA gene, and that this is context dependent. While the mechanism is outside the scope of this study, it will be interesting to discover if this is specific to ralaniten, a common response following chronic treatment with AR-NTD inhibitors, or simply long-term culture as has been previously reported.30 PSA expression is used clinically as a surrogate for AR activity and disease progression, therefore this observation highlights the need to identify additional biomarkers.
In summary, glucuronidation of ralaniten is a metabolic liability resulting in its reduced potency as we have shown in vitro and in vivo. Glucuronidation of ralaniten was identified both in our model of acquired resistance to ralaniten, as well as in clinical samples from prostate cancer patients. Small molecules targeting of the AR-NTD have considerable clinical potential as they have the ability to overcome many mechanisms of resistance to existing AR-targeted therapies.20,24,47 However, this study underscores the need to develop second generation AR-NTD inhibitors which avoid the glucuronidation pathway. Doing so will hopefully improve the clinical utility of these compounds both in the context of first pass metabolism, and possibly mediated by the tumor itself.
Methods
Cell Culture and Generation of a Ralaniten-Resistant Cell Line
LNCaP cells were from Dr. Leland Chung (Cedars-Sinai Medical Center, Los Angeles, CA) and maintained in phenol red-free RPMI 1640 supplemented with 5% FBS (VWR). LNCaP-RALR cells were generated by exposing parental LNCaP cells with increasing concentrations of ralaniten beginning September, 2012. Once stable growth of these cells was seen at 25 μM ralaniten (approximately 18 months), highly resistant clones were selected by serial dilution. Cells which formed colonies were then challenged with increasing concentrations of ralaniten up to 50 μM. Of the six clones which were isolated, clone D7 had the fastest doubling time in the presence of 50 μM ralaniten and was chosen for further characterization (hereafter referred to as LNCaP-RALR). LNCaP-RALR cells were maintained in RPMI 1640 supplemented with 5% FBS and 50 μM ralaniten. Media was replaced once weekly with fresh compound. The remaining clones were cryopreserved in liquid nitrogen.
LNCaP-GFP, LNCaP-2B15, and LNCaP-2B17 clones were maintained in phenol red-free RPMI 1640 supplemented with 5% heat-inactivated FBS and 1.5 μg/mL puromycin (Sigma). Cells were resuscitated immediately prior to experimentation, maintained for no more than 10 to 15 passages, and periodically tested to ensure they were mycoplasma-free (VenorGem Mycoplasma Detection Kit, Sigma-Aldrich). Authentication was done by short tandem repeat analysis in June, 2017.
Gene and Protein Expression in Parental and LNCaP-RALR Cell Lines
AR, FKBP5, and PSA were probed using AR-N20, FKBP51 (H-100), and PSA (C-19) antibodies (Santa Cruz Biotechnology). Antibodies against UGT2B11 (ab116019), UGT2B15 (ab89274), UGT2B17 (ab92610), and UGT2B28 (ab129729) were obtained from Abcam. HA was probed using HA-Tag (C29F4) from Cell Signaling. β-Actin was used as a loading control, and membranes were probed using the mouse monoclonal anti-β-actin antibody (a5316 from Sigma). Transcripts were measured by qRT-PCR QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems by Life Technology) and gene expression was normalized to the house keeping gene SDHA. Further details and primer sequences are given in the Supporting Information.
siRNA Transfection and Proliferation Assay
Pooled siRNA against AR (L-003400-00-0005), UGT2B15 (L-020194-02-0005), UGT2B17 (L-020195-00-0005) and nontargeting control (D-001810-10-05) were purchased from Dharmacon Research (Layfayette, USA). Lipofectamine RNAiMAX (Invitrogen) was used to transfect 10 nM (AR) or 15 nM (UGT2B15/17) siRNA into cells. For the proliferation assay, cells were treated with 25 μM ralaniten and 0.1 nM R1881 (UGT2B15/17 knock-down experiment), or 0.1 nM R1881 only (AR knock down experiment) 24 h post-transfection. After 72 h post-treatment, cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) and incubated with 0.1% crystal violet solution (Sigma). Dye was solubilized using a 1% SDS solution and absorbance was read using a VersaMax Microplate Reader (Molecular Devices) at 595 nm. Further details are described in the Supporting Information.
Global Gene Expression Analysis
Total RNA was extracted from both LNCaP and LNCaP-RALR cells treated with ralaniten (35 μM), enzalutamide (5 μM) or DMSO vehicle and stimulated with either 1 nM R1881 or EtOH vehicle. RNA was reverse transcribed and cDNA was hybridized to the GeneChip Human Transcriptome Array 2.0 from Affymetrix. RT-PCR, cDNA hybridization, and chip reading was carried out at CDRD’s Target Validation Division at the University of British Columbia (Vancouver, BC, Canada; www.cdrd.ca). Analysis of raw signal output was done using GeneSpring software (version 13.1). Clustered data was generated by conducting a 2-Way ANOVA on data with significance threshold set at 0.05. The Benjamini–Hochberg correction was applied to reduce the false discovery rate.
HPLC of Drugs and Their Glucuronide Metabolites
Mouse liver microsomes were incubated with ralaniten or EPI-045 in reaction buffer for 16 h at 37 °C. The reaction mixture contained 50 μg of mouse liver, 15.8 μg of respective compounds, 4 mM UDPGA, 50 mM TES (pH 7.5), 8 mM MgCl2, 25 μg/mL alamathecin, and 30U β-glucuronidase or v/v phosphate buffer (3.24 mM, pH 6.8) to a total volume of 200 μL. Following incubation, the reaction mixture was centrifuged for 15 min at 12 000 rcf, and supernatant was stored on ice until samples were analyzed via C18 reversed-phased HPLC using an InertSustain 5 μM column (25 × 0.4 cm). An isocratic gradient system of 67:33 was employed containing (0.05% TFA/H2O)/MeCN for 28 min followed by a linear gradient to 11:9 (0.05% TFA/H2O)/MeCN over an additional 20 min (flow rate 1 mL/min), with UV detection at 201 nM. Synthetic ralaniten G-1, G-2, and G-3, and EPI-045 G-1 and G-2 glucuronides were used as standards for retention time comparison and coinjection (Supplementary Figures S6 and S7).
Identification of Ralaniten Metabolites in Human Hepatocytes and Patient Serum
All samples were prepared and analyzed at RMI Laboratories. Briefly, ralaniten (10 μM) was incubated with hepatocytes in incubation media (inVitroGRO KHB buffer, IVT) for 4 h at 37 °C. The sample was quenched and centrifuged at 13 000 rpm and supernatant was analyzed by UPLC–HRMS using a C18 reverse-phased Phenomenex Kinetex BP 1.7 μM column (2.1 × 50 mm) and run on a Waters ACUITY I-Class system. An isocratic gradient system of 90:10 for 0.5 min; 58:42 for 3 min; 45:55 for 13 min; 25:75 for 1.5 min; 5:95 for 1 min; and 90:10 for 1 min was employed containing (0.1% formic acid/H2O)/(0.1% formic acid/MeCN) at a flow rate of 0.5 mL/min. Product ion spectra were acquired by a separate UPLC–HRMS/MS run to assign structures of metabolites by MS/MS conducted on a Waters Xevo G2 XS system. Plasma samples were pooled using the Hamilton pooling method. A 50 μL aliquot was quenched with two volumes of ACN and centrifuged for 5 min at 13 000 rpm, and supernatant from each sample was analyzed by LC–MS as above. Product ion spectra were acquired by a separate LC–MS/MS run to assign structures of metabolites by MS/MS conducted on a Waters Xevo G2 XS system.
Fluorescence Polarization and Gene Reporter Assays
The Polar Screen Androgen Receptor Competitor Assay kit (Invitrogen) was employed according to the manufacturer’s protocol with 20 nM AR-LBD and 2 nM Fluoromone. The reactions were done in 40 mL aliquots in triplicate in Greiner 384 black clear bottom plates, and fluorescence polarization was read using the Infinite M1000 (Tecan) with excitation at 530 nM and emission at 590 nM.
LNCaP cells (35 000 cells/well) in 24-well plates were cotransfected with synthetic UBE2C-luciferase reporter (consisting of 3 tandem repeats of an AR-V7-specific promoter element of the UBE2C gene) reporter, AR-V7, or empty vector and treated the next day with DMSO, enzalutamide (5 μM), ralaniten (35 μM) or EPI-045 (25 μM) under serum-free and phenol red-free conditions. After 24 h of incubation, cells were harvested and luciferase activities were measured and normalized to protein concentration.
Xenografts and Animal Experiments
Male NOD/SCID mice at 6 to 8 weeks of age were subcutaneously injected with LNCaP or LNCaP-RALR cells (1 × 107 cells/site) using Matrigel (Becton Dickinson). Mice were castrated once tumors reached ∼100 mm3 and randomized into treatment groups to receive ralaniten (200 mg/kg), enzalutamide (10 mg/kg), or vehicle control (5% DMSO/1% CMC/0.1% Tween-80) once daily by oral gavage. Treatment began 1 week following castration. For the second experiment, treatments were administered once every other day by tail vein injection; ralaniten (50 mg/kg), EPI-045 (50 mg/kg), or vehicle (15% DMSO/30% PEG-400). Mouse body weight and tumor volume (defined as volume = length × width × height × 0.5236) were regularly recorded and tumors were excised 2 days after the last treatment in both experiments.
Gene and Protein Expression in Xenografts
To analyze tumor gene expression, tumors were flash frozen and ∼100 mg was pulverized under liquid nitrogen. Samples were added to 1 mL of TRIzol reagent (Invitrogen) and homogenized using a prechilled Dounce tissue grinder. RNA was extracted and reverse transcribed as detailed above. Further details are given in the Supporting Information and Methods describing IHC.
Statistical Analysis
A one- or two-way ANOVA statistical test was used to determine significance for all comparisons unless specifically stated otherwise (Graphpad Prism, version 7.0). p-Value corrections were applied for all multiple comparisons (Tukey, Sidak, or Dunnett as appropriate), and a p-value < 0.05 was considered statistically significant.
Study Approval
Blood plasma specimens used in this work were deidentified samples obtained from patients enrolled in Phase I clinical trial (NCT02606123), which received Research Ethics Board approval at all participating sites. All patients provided written informed consent prior to inclusion in the study. All experiments involving animals were conducted in compliance with, and the approval of, the Animal Care Committee of the University of British Columbia (A18-0077).
Acknowledgments
Clinical samples were obtained from the clinical trial of EPI-506 (NCT02606123) conducted by ESSA Pharma, Inc. and supported in part by a grant from the Cancer Prevention Institute of Texas (CPI30020). This work was supported by the US National Cancer Institute (#R01 CA105304) awarded to M.D.S.
Glossary
Abbreviations
- ADT
androgen deprivation therapy
- AR
androgen receptor
- BIC
bicalutamide
- ENZ
enzalutamide
- LBD
ligand-binding domain
- mCRPC
metastatic castration-resistant prostate cancer
- NTD
N-terminal domain
- PSA
prostate specific antigen
- RAL
ralaniten
- UDPGA
uridine diphosphate glucuronic acid
- UGT2B
UDP glycosyltransferase 2 family, polypeptide B
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsptsci.9b00065.
Supporting materials and methods relating to AR cloning and sequencing, lentiviral transduction, siRNA knockdown, microsomal isolation and enzymatic activity, synthesis of ralaniten derivatives, and tumor immunohistochemistry; supporting tables (Table S1); supporting figures and associated legends (Figures S1–S7) (PDF)
Author Contributions
J.K.O. was responsible for concept formation, data generation and collection, data analysis, and manuscript composition. M.D.S. was the supervisory author on this manuscript and was responsible for the concept data analysis and manuscript composition. J.W. and A.H.T. performed the animal experiments. T.T. generated samples for AR sequencing and completed the V7 reporter experiment. N.M. and Y.C.Y. were responsible for early maintenance of the resistant cell line, and N.M. performed the lentiviral transduction of LNCaP cells, the initial EPI-045 work with PC3 cells, and resuscitated frozen cell lines for culture. R.J.A., D.W., and K.J. provided experimental compounds including glucuronidated standards, and performed the HPLC and 1H NMR experiments. K.N.C. and B.M. oversaw the clinical trial and provided samples for metabolic analysis.
The authors declare the following competing financial interest(s): JKO, JW, KJ, DW, AHT, NM, YCY, RJA, MDS are inventors of licensed technology to ESSA Pharma, Inc. JKO, JW, DW, NRM, RJA, MDS have stock equity. MDS and RJA are consultants of ESSA Pharma Inc. TT, KNC and BM have nothing to declare.
Supplementary Material
References
- Huggins C.; Hodges C. V. (1941) Studies of prostatic cancer, I. The effect of castration, of estrogen and of androgen injection on serum phosphatase in metastatic carcinoma of the prostate. Cancer Res. 1, 293–297. [DOI] [PubMed] [Google Scholar]
- Harris W. P.; Mostaghel E. A.; Nelson P. S.; Montgomery B. (2009) Androgen deprivation therapy, progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 6, 76–85. 10.1038/ncpuro1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karantanos T.; Corn P. G.; Thompson T. C. (2013) Prostate cancer progression after androgen deprivation therapy, mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 32, 5501–5511. 10.1038/onc.2013.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fizazi K.; Scher H. I.; Molina A.; Logothetis C. J.; Chi K. N.; Jones R. J.; Staffurth J. N.; North S.; Vogelzang N. J.; Saad F.; Mainwaring P.; Harland S.; Goodman O. B. Jr; Sternberg C. N.; Li J. H.; Kheoh C. M.; Haqq T.; de Bono J. S. (2012) Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer, Final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 13, 983–992. 10.1016/S1470-2045(12)70379-0. [DOI] [PubMed] [Google Scholar]
- Cai C.; Chen S.; Ng P.; Bubley G. J.; Nelson P. S.; Mostaghel E. A.; Marck B.; Matsumoto A. M.; Simon N. I.; Wang H.; Chen S.; Balk S. P. (2011) Intratumoral De Novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 71, 6503–6513. 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman B. J.; Feldman D. (2001) The Development of Androgen-Independent Prostate Cancer. Nat. Rev. Cancer 1, 34–45. 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Scher H. I.; Sawyers C. L. (2005) Biology of progressive, castration-resistant prostate cancer, Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 23, 8253–8261. 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Scher H. I.; Fizazi K.; Saad F.; Taplin M.-E.; Sternberg C. N.; Miller K.; de Wit R.; Mulders P.; Chi K. N.; Shore N. D.; Armstrong A. J.; Flaig T. W.; Flechon A.; Mainwaring P.; Fleming M.; Hainsworth J. D.; Hirmand M.; Selby B.; Seely L.; de Bono J. S. (2012) Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. N. Engl. J. Med. 367, 1187–1197. 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- Ryan C. J.; Smith M. R.; Fizazi K.; Saad F.; Mulders P. F.; Sternberg C. N.; Miller K.; Logothetis C. J.; Shore N. D.; Small E. J.; Carles J.; Flaig T. W.; Taplin M. E.; Higano C. S.; de Souza P.; de Bono J. S.; Griffin T. W.; de Porre P.; Yu M. K.; Park Y. C.; et al. (2015) Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302), Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 16, 152–160. 10.1016/S1470-2045(14)71205-7. [DOI] [PubMed] [Google Scholar]
- Chen E. J.; Sowalsky A. G.; Goa S.; Cai C.; Voznesensky O.; Schaefer R.; Loda M.; True L. D.; Ye H.; Troncoso P.; Lis R. L.; Kantoff P. W.; Montgomery R. B.; Nelson P. S.; Bubley G. J.; Balk S. P.; Taplin M. E. (2015) Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 21, 1273–1280. 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph J. D.; Lu N.; Qian J.; Sensintaffar J.; Shao G.; Brigham D.; Moon M.; Maneval E. C.; Chen I.; Darimont B.; Hager J. H. (2013) A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discovery 3, 1020–1029. 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Taplin M. E.; Bubley G. J.; Ko Y. J.; Small E. J.; Upton M.; Rajeshkumar B.; Balk S. P. (1999) Selection for Androgen Receptor Mutations in Prostate Cancers Treated with Androgen Antagonist. Cancer Res. 59, 2511–2515. [PubMed] [Google Scholar]
- Antonarakis E. S.; Lu C.; Luber B.; Wang H.; Chen Y.; Zhu Y.; Silberstein J. L.; Taylor M. N.; Maughan B. L.; Denmeade S. R.; Pienta K. J.; Paller C. J.; Carducci M. A.; Eisenberger M. A.; Luo J. (2017) Clinical Significance of Androgen Receptor Splice Variant-7 mRNA Detection in Circulating Tumor Cells of Men With Metastatic Castration-Resistant Prostate Cancer Treated With First- and Second-Line Abiraterone and Enzalutamide. J. Clin. Oncol. 35, 2149–2156. 10.1200/JCO.2016.70.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonarakis E. S.; Changxue L.; Wang H.; Luber B.; Nakazawa M.; Roeser J. C.; Chen Y.; Mohammad T. A.; Chen Y.; Fedor H. L.; Lotan T. L.; Zheng Q.; de Marzo A. M.; Isaacs J. T.; Isaacs W. B.; Nadal R.; Paller C. J.; Denmeade S. R.; Carducci M. A.; Eisenberger M. A.; et al. (2014) AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 371, 1028–1038. 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.; Ryan C. J. (2012) Androgen receptor directed therapies in castration-resistant metastatic prostate cancer. Curr. Treat Options Oncol. 13, 189–200. 10.1007/s11864-012-0188-2. [DOI] [PubMed] [Google Scholar]
- Jenster G.; van der Korput H. A. G. M.; van Vroonhoven C.; van der Kwast T. H.; Trapman J.; Brinkmann A. O. (1991) Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization. Mol. Endocrinol. 5, 1396–1404. 10.1210/mend-5-10-1396. [DOI] [PubMed] [Google Scholar]
- Jenster G.; van der Korput H. A. G. M.; Trapman J.; Brinkmann A. O. (1995) Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J. Biol. Chem. 270, 7341–7346. 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- Myung J. K.; Wang G.; Chiu H. H. L.; Wang J.; Mawji N. R.; Sadar M. D. (2017) Inhibition of androgen receptor by decoy molecules delays progression to castration recurrent prostate cancer. PLoS One 12, 1–17. 10.1371/journal.pone.0174134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quayle S. N.; Mawji N. R.; Wang J.; Sadar M. D. (2007) Androgen receptor decoy molecules block the growth of prostate cancer. Proc. Natl. Acad. Sci. U. S. A. 104, 1331–1336. 10.1073/pnas.0606718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen R. J.; Mawji N. R.; Wang J.; Wang G.; Haile S.; Myung J.-K.; Watt K.; Tam T.; Yang Y. C.; Banuelos C. A.; Williams D. E.; McEwan I. J.; Wang Y.; Sadar M. D. (2010) Regression of Castrate-Recurrent Prostate Cancer by a Small-Molecule Inhibitor of the Amino-Terminus Domain of the Androgen Receptor. Cancer Cell 17, 535–546. 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- Myung J. K.; Bañuelos C. A.; Fernandez J. G.; Mawji N. R.; Wang J.; Tien A. H.; Yang Y. C.; Tavakoli I.; Haile S.; Watt K.; McEwan I. J.; Plymate S.; Andersen R. J.; Sadar M. D. (2013) An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Invest. 123, 2948–2960. 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mol E.; Fenwick R. B.; Phang C. T.; Buzón V.; Szulc E.; de la Fuente A.; Escobedo A.; García J.; Bertoncini C. W.; Estébanez-Perpiñá E.; McEwan I. J.; Riera A.; Salvatella X. (2016) EPI-001, A Compound Active against Castration-Resistant Prostate Cancer, Targets Transactivation Unit 5 of the Androgen Receptor. ACS Chem. Biol. 11, 2499–2505. 10.1021/acschembio.6b00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan I. J. (2012) Intrinsic disorder in the androgen receptor, identification, characterisation and drugability. Mol. BioSyst. 8, 82–90. 10.1039/C1MB05249G. [DOI] [PubMed] [Google Scholar]
- Yang Y. C.; Banuelos C. A.; Mawji N. R.; Wang J.; Kato M.; Haile S.; McEwan I. J.; Plymate S.; Sadar M. D. (2016) Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin. Cancer Res. 22, 4466–4477. 10.1158/1078-0432.CCR-15-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiang T. K. L.; Ensom M. H. H.; Chang T. K. H. (2005) UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol. Ther. 106, 97–132. 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Tukey R. H.; Strassburg C. P. (2000) Human UDP-glucuronosyltransferases, metabolism, expression, and disease. Annu. Rev. Pharmacol. Toxicol. 40, 581–616. 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- Montgomery R. B.; Mostaghel E. A.; Vessella R. L.; Hess D. L.; Kalhorn T. F.; Higano C. S.; True L. D.; Nelson P. S. (2008) Maintenance of intratumoral androgens in metastatic prostate cancer, A mechanism for castration-resistant tumor growth. Cancer Res. 68, 4447–4454. 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pâquet S.; Fazli L.; Grosse L.; Verreault M.; Têtu B.; Rennie P. S.; Bélanger A.; Barbier O. (2012) Differential expression of the androgen-conjugating UGT2B15 and UGT2B17 enzymes in prostate tumor cells during cancer progression. J. Clin. Endocrinol. Metab. 97, 428–432. 10.1210/jc.2011-2064. [DOI] [PubMed] [Google Scholar]
- Kaushik A. K.; Vareed S. K.; Basu S.; Putluri V.; Putluri N.; Panzitt K.; Brennan C. A.; Chinnaiyan A. M.; Vergara I. A.; Erho N.; Weigel N. L.; Mitsaides N.; Shojaie A.; Palapattu G.; Michailidis G.; Sreekumar A. (2014) Metabolomic profiling identifies biochemical pathways associated with castration-resistant prostate cancer. J. Proteome Res. 13, 1088–1100. 10.1021/pr401106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culig Z; Hoffmann J; Erdel M; Eder I E; Hobisch A; Hittmair A; Bartsch G; Utermann G; Schneider M R; Parczyk K; Klocker H (1999) Switch from antagonist to agonist of the androgen receptor blocker bicalutamide is associated with prostate tumour progression in a new model system. Br. J. Cancer 81, 245–251. 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. D.; Welsbie D. S.; Tran C.; Baek S. H.; Chen R.; Vessella R.; Rosenfeld M. G.; Sawyers C. L. (2004) Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 10, 33–39. 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Hu R.; Dunn T. A.; Wei S.; Isharwal S.; Veltri R. W.; Humphreys E.; Han M.; Partin A. W.; Vessella R. L.; Isaacs W. B.; Bova G. S.; Luo J. (2009) Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 69, 16–22. 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostaghel E. A.; Marck B. T.; Plymate S. R.; Vessella R. L.; Balk S.; Matsumoto A. M.; Nelson P. S.; Montgomery R. B. (2011) Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer, Induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res. 17, 5913–5925. 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R.; Lu C.; Mostaghel E. A.; Yegnasubramanian S.; Gurel M.; Tannahill C.; Edwards J.; Isaacs W. B.; Nelson P. S.; Bluemn E.; Plymate S. R.; Luo J. (2012) Distinct transcriptional programs mediated by the ligand dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 72, 3457–3462. 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C.; He H. H.; Chen S.; Coleman I.; Wang H.; Fang Z.; Chen S.; Nelson P. S.; Liu X. S.; Brown M.; Balk S. P. (2011) Androgen Receptor Gene Expression in Prostate Cancer is Directly Suppressed by the Androgen Receptor Through Recruitment of Lysine Specific Demethylase 1. Cancer Cell 20, 457–471. 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan A.; McEwan I. (2016) A sting in the tail, the N-terminal domain of the androgen receptor as a drug target. Asian J. Androl. 18, 687–694. 10.4103/1008-682X.181081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg N. J.; Wongvipat J.; Joseph J. D.; Tran C.; Ouk S.; Dilhas A.; Chen Y.; Grillot K.; Bischoff E. D.; Cai L.; Aparicio A.; Dorow S.; Arora V.; Shao G.; Qian J.; Zhao H.; Yang G.; Cao C.; Sensintaffar J.; Wasielewska T.; et al. (2012) ARN-509, A novel antiandrogen for prostate cancer treatment. Cancer Res. 72, 1494–1503. 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger A.; Pelletier G.; Labrie F.; Barbier O.; Chouinard S. (2003) Inactivation of androgens by UDP-glucuronosyltransferase enzymes in humans. Trends Endocrinol. Metab. 14, 473–479. 10.1016/j.tem.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Bao B. Y.; Chuang B. F.; Wang Q.; Sartor O.; Balk S. P.; Brown M.; Kantoff P. W.; Lee G. S. (2008) Androgen Receptor Mediates the Expression of UDP Glucuronosyltransferase 2B15 and B17 Genes. Prostate 68, 839–848. 10.1002/pros.20749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanioka N.; Naito T.; Narimatsu S. (2008) Human UDP-glucuronosyltransferase isoforms involved in bisphenol A glucuronidation. Chemosphere 74, 33–36. 10.1016/j.chemosphere.2008.09.053. [DOI] [PubMed] [Google Scholar]
- Belledant A.; Hovington H.; Garcia L.; Caron P.; Brisson H.; Villeneuve L.; Simonyan D.; Têtu B.; Fradet Y.; Lacombe L.; Guillemette C.; Lévesque E. (2016) The UGT2B28 sex-steroid inactivation pathway is a regulator of steroidogenesis and modifies the risk of prostate cancer progression. Eur. Urol. 69, 601–609. 10.1016/j.eururo.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Goodwin J. F.; Kothari V.; Drake J. M.; Zhao S.; Dylgjeri E.; Dean J. L.; Schiewer M. J.; McNair C.; Jones J. K.; Aytes A.; Magee M. S.; Snook A. E.; Zhu Z.; Den R. B.; Birbe R. C.; Gomella L. G.; Graham N. A.; Vashisht A. A.; Wohlschlegel J. A.; Graeber T. G.; et al. (2015) DNA-PKcs mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell 28, 97–113. 10.1016/j.ccell.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosse L.; Pâquet S.; Caron P.; Fazli L.; Rennie P. S.; Bélanger A.; Barbier O. (2013) Androgen glucuronidation, An unexpected target for androgen deprivation therapy, with prognosis and diagnostic implications. Cancer Res. 73, 6963–6971. 10.1158/0008-5472.CAN-13-1462. [DOI] [PubMed] [Google Scholar]
- Beaulieu M.; Levesque E.; Hum D. W.; Belanger A. (1996) Isolation and characterization of a novel cDNA encoding a human UDP-glucuronosyltransferase active on C19 steroids. J. Biol. Chem. 271, 22855–22862. 10.1074/jbc.271.37.22855. [DOI] [PubMed] [Google Scholar]
- Zhu Z.; Chung Y. M.; Sergeeva S.; Kepe V.; Berk M.; Li J.; Ko H. K.; Li Z.; Petro M.; DiFilippo F. P.; Lee Z.; Sharifi N. (2018) Loss of dihydrotestosterone-inactivation activity promotes prostate cancer castration resistance detectable by functional imaging. J. Biol. Chem. 293, 17829–17837. 10.1074/jbc.RA118.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D.; Standifer K. M.; Inturrisi C. E.; Pasternak G. W. (1989) Pharmacological Characterization of Morphine-63-GIucuronide, a Very Potent Morphine Metabolite. J. Pharmacol Exp Ther. 251, 477–483. [PubMed] [Google Scholar]
- Antonarakis E. S.; Chandhasin C.; Osbourne E.; Luo J.; Sadar M. D.; Perabo F. (2016) Targeting the N-Terminal Domain of the Androgen Receptor, A New Approach for the Treatment of Advanced Prostate Cancer. Oncologist 21, 1427–1435. 10.1634/theoncologist.2016-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaishampayan U.; Montgomery R. B.; Gordon M. S.; Smith D. C.; Barber K.; de Haas-Amatsaleh A.; Thapar N.; Chandhasin C.; Perabo F.; Chi K. N. (2017) 794EPI-506 (ralaniten acetate), a novel androgen receptor (AR) N-terminal domain (NTD) inhibitor, in men with metastatic castration-resistant prostate cancer (mCRPC), phase 1 update on safety, tolerability, pharmacokinetics and efficacy. Ann. Oncol. 28, mdx370.011. 10.1093/annonc/mdx370.011. [DOI] [Google Scholar]
- Cockshott I. D. (2004) Bicalutamide Clinical Pharmacokinetics and Metabolism. Clin. Pharmacokinet. 43, 855–878. 10.2165/00003088-200443130-00003. [DOI] [PubMed] [Google Scholar]
- Gibbons J. A.; de Vries M.; Krauwinkel W.; Ohtsu Y.; Noukens J.; van der Walt J. S.; Mol R.; Mordenti J.; Ouatas T. (2015) Pharmacokinetic Drug Interactions Studies with Enzalutamide. Clin. Pharmacokinet. 54, 1057–1069. 10.1007/s40262-015-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.