

Abstract
An avian infectious bronchitis virus (IBV) was isolated and identified from a commercial layer flock vaccinated with live attenuated H120 vaccine in China, designed as ck/CH/IBTZ/2012. To determine the origination and evolution of this isolated strain, we have carried out a complete genome sequencing of this strain. The genome of the ck/CH/IBTZ/2012 strain is 27,691 nucleotides in length and includes more than 10 open reading frames. Sequence comparison and phylogenetic analysis based on the full-length genomic sequences showed that ck/CH/IBTZ/2012 is mostly related to the LX4-like strains. However, sequence analysis based on the spike protein (S) gene sequences revealed that ck/CH/IBTZ/2012 possesses a distinct S gene setting it apart from the Massachusetts-type strains and LX4-type strains. The cleavage site within the spike protein (S) of ck/CH/IBTZ/2012 is HRRKR, which is different from the majority of the IBVs in China for their cleavage sits are HRRRR. Recombination analysis showed that ck/CH/IBTZ/2012 is a chimeric virus with a LX4-like backbone except S gene which might be from an unknown strain. Based on the data presented in this paper, it can be concluded that genetic changes due to adaptive evolution and recombination both contributed to the origin of strain ck/CH/IBTZ/2012, which belongs to a new genotype.
Electronic supplementary material
The online version of this article (doi:10.1007/s11262-014-1063-y) contains supplementary material, which is available to authorized users.
Keywords: Infectious bronchitis virus, Genome, Phylogenetic analysis, Recombination event
Avian infectious bronchitis virus (IBV), a typical species of the genus Coronavirus, can induce a highly contagious disease in chickens. The virus replicates not only in the epithelium of upper and lower respiratory tract tissues but also in many tissues along the alimentary tract, and elsewhere, e.g., kidney, oviduct, and testes [1]. IBV genome consists of a single-stranded RNA with a high mutation frequency. Molecular studies have shown that new IBV strains can emerge due to the changes in the genome, including insertions, deletions, and point mutations, in some cases, recombination [2–4]. A number of IBV serotypes have been identified worldwide, and some of these serotypes cannot be controlled by heterologous serotype vaccines, making this virus difficult to identify and extremely difficult to control [5]. Infectious bronchitis (IB) was first described in the early 1980s in China. Vaccination programs based on Massachusetts-type live attenuated vaccine (H120, H52, Ma5, and W93) and the inactivated oil-emulsion vaccine containing M41 strain have been used for many years to prevent and control of IB. In spite of extensive vaccination, IB is still epidemic due to the serotypes of the vaccines used to differ from those of the predominant IBV isolates in China [6, 7]. Therefore, it is very important to determine the genetic characteristics of IBV isolates for the control of IB. Studies on the molecular epidemiology of IBVs have been reported that LX4-type strains are predominant in China in recent years [8]. For the lack of vaccines against the endemic strains, IB remains a problem in the Chinese poultry industry and has caused severe economic losses in recent years [9, 10].
The ck/CH/IBTZ/2012 virus was detected in commercial layer flocks vaccinated with live attenuated H120 vaccine in Jiangsu province, China, and clinical signs in the birds were mild egg drop and abnormal egg shells. To further determine the origination and evolution of this isolated strain, we have carried out a complete genome sequencing of this strain. 18 pairs of overlapping primers encompassing the entire genome were designed in regions that are conserved among most of the IBV strains available through GenBank database. Three additional primers were specifically designed to amplify viral genomic RNA 5′- and 3′-end sequences (Table S1). Viral RNA was extracted from ck/CH/IBTZ/2012 virus-infected allantoic fluid with Trizol Reagent (TaKaRa, Japan) following the manufacturer’s instruction. The first-strand cDNA was synthesized using PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa, Japan) according to the manufacturer’s instruction. The PCR reaction was carried out at 95 °C for 1 min followed by 25 cycles of 95 °C for 30 s, 50 °C for 30 s, 72 °C for 3 min, and finally, 72 °C for 10 min. The 5′- and 3′-end of the genome were obtained by using a rapid amplification of cDNA ends (RACE) kit (TaKaRa, Japan) following the manufacturer’s instruction. Twenty overlapped PCR fragments spanning the entire viral genome were amplified using specific primer sets. The PCR products were extracted from agarose gels by using a Gel Extraction Kit (TaKaRa, Japan) and cloned into pMD18-T vector (TaKaRa, Japan) following the manufacturer’s instruction. Positive clones were screened by PCR followed by sequencing. At least three clones of each fragment were sequenced. The sequences were analyzed using the Sequencher 5 sequence analyses program, and a single contiguous sequence comprises the entire genome of ck/CH/IBTZ/2012 virus which was constructed. The full-length genome of isolate ck/CH/IBTZ/2012 is 27,691 nucleotides (nt) in length and includes 10 open reading frames (ORFs) (Fig. S1). The complete sequence has been deposited in the GenBank database under the accession number of KF663559. The spike protein (S) gene of ck/CH/IBTZ/2012 is 3,516 nts long. The cleavage site within the spike protein of ck/CH/IBTZ/2012 is HRRKR, which is different from the majority of the IBVs in China for their cleavage sits are HRRRR [11, 12]. The genomic sequence of the IBV strain ck/CH/IBTZ/2012 was analyzed with Lasergene software V7.1 (DNASTAR, Madison, WI, USA). A total of 53 IBV reference strains for which the entire genome sequences were available through GenBank database were selected for analysis (Table S2). In the comparison of full-length genomes, the highest similarity (92.9 %) was found in the sequence of isolate ck/CH/LJL/110302 (KC136209), and the lowest (80.9 %) was found to that of isolate MG10 (EU095850). Unlike the full genome sequence, the S1 and S2 sequences of ck/CH/IBTZ/2012 were markedly different from those of 53 IBV reference strains (Table 1). Phylogenetic trees were constructed with the neighbor-joining method by using MEGA5.2 software (www.megasoftware.net) with the Kimura 2-parameter nt substitution model [13]. Phylogenetic analysis based on the genomic sequences divided 54 IBV strains into two main groups, group I and group II (Fig. 1). The group I was composed of diverse genotypes, including Massachusetts, Iowa, Gray, Arkansas, California, Delaware, MG10, Georgia. Most of them were standard strains or vaccine strains. The group II was composed of four subclusters, including 22 novel strains isolated in China in recent years. The ck/CH/IBTZ/2012 has been shown to be closely related to this group based on the full-length genomic sequence. The phylogenetic trees constructed based on the 1a region, 1b region, 3b gene, E gene, M gene, 5a gene, 5b gene, and N gene were consistent with that obtained from full-length genomic sequence. The exceptions were the phylogenetic trees constructed using the S1 gene, S2 gene, and 3a gene, in which ck/CH/IBTZ/2012 was branched as a separate genotype (Fig. S2). The aligned nt sequences of genome were analyzed with the Recombination Detection Program (RDP4, Version 4.26) to detect potential recombination events [14]. Two major recombination events were detected in the 1a gene and S gene of the isolate ck/CH/IBTZ/2012. The first major recombination region was located in the 1a gene, with beginning breakpoint position at nt position 959 and ending breakpoint at nt position 5269. It indicated that ck/CH/IBTZ/2012 was a potential recombinant strain between the A2 (EU526388) and SAIBk (DQ288927) strains (Fig. S3). The second major recombination region was located at nt position 20147–23793 in the S gene. Its major parent was the GX-YL9 (HQ850618) strain, but its minor parent was unknown. Subsequently, BLASTN (http://blast.ncbi.nlm.nih.gov/Blast.cgi) analyses using the S1 nt sequence were conducted. The S1 sequence of ck/CH/IBTZ/2012 (located at nt 20371–22011) showed the highest identities (98–99 %) to strains TC07-2 (GQ265948.1), CK/CH/GD/KP10 (HQ018919.1), GX-NN09032 (JX292013.1), CK/CH/SD09/005 (HM230749.1), CK/CH/GD/NC10 (HQ018903.1), ck/CH/LHB/110615 (JQ739243.1), and ck/CH/LHB/110617 (JQ739244.1), which were isolated in China in recent years [10, 15]. The S1 sequence of ck/CH/IBTZ/2012 also showed 98 % identities to strains K23/10 (JF804677.1), K46/10 (JF804679.1), K308/09 (JF804689.1), K26/10 (JF804678.1), and K273/09 (JF804687.1), which were isolated in Korea from 2009 to 2010 [16]. This strikingly high identity of S1 gene implied a close genetic relationship and possibly indicated a common origin.
Table 1.
The nt sequence pairwise comparison of percentage identity based on different regions between ck/CH/IBTZ/2012 and other IBV strains
| Strain | Genome | 1a | 1b | S1 | S2 | 3a | 3b | E | M | 5a | 5b | N |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| California99 | 84.6 | 84.8 | 88.7 | 65.0 | 75.7 | 82.1 | 79.4 | 85.5 | 87.5 | 83.1 | 94.3 | 88.4 |
| Cal56b | 84.7 | 84.9 | 89.1 | 65.3 | 76.5 | 81.6 | 78.9 | 79.5 | 87.2 | 85.4 | 94.3 | 89.1 |
| FL18288 | 84.6 | 85.3 | 88.8 | 61.7 | 76.0 | 81.6 | 80.5 | 81.3 | 87.5 | 82.7 | 95.5 | 89.2 |
| ck/CH/LJL/111054 | 84.6 | 85.0 | 88.9 | 62.2 | 76.3 | 82.1 | 80.5 | 81.6 | 87.5 | 83.1 | 95.5 | 89.1 |
| ArkDPI101 | 84.7 | 84.9 | 88.9 | 65.2 | 76.5 | 84.4 | 80.0 | 81.9 | 87.3 | 83.1 | 94.7 | 88.9 |
| ArkDPI11 | 84.7 | 84.9 | 88.9 | 65.1 | 76.5 | 84.4 | 80.0 | 81.9 | 87.3 | 83.1 | 95.1 | 89.0 |
| Arkansas DPI | 84.8 | 85.1 | 89.0 | 65.2 | 76.5 | 85.0 | 80.0 | 81.9 | 87.3 | 83.1 | 95.1 | 88.9 |
| Arkansas Vaccine | 84.7 | 85.2 | 89.0 | 64.8 | 77.2 | 82.7 | 80.0 | 82.2 | 87.3 | 81.8 | 95.1 | 88.8 |
| H120 | 84.3 | 84.7 | 89.0 | 63.4 | 75.8 | 76.4 | 82.0 | 83.7 | 87.3 | 84.0 | 93.5 | 87.4 |
| ck/CH/LNM/091017 | 84.3 | 84.7 | 89.0 | 63.3 | 75.9 | 76.4 | 82.0 | 83.7 | 87.3 | 84.0 | 93.5 | 87.4 |
| ck/CH/LDL/101212 | 84.2 | 84.6 | 89.0 | 63.2 | 75.9 | 76.4 | 82.0 | 83.7 | 87.3 | 83.6 | 93.5 | 87.3 |
| ZJ971 | 84.2 | 84.6 | 89.0 | 63.4 | 75.8 | 76.4 | 82.0 | 83.7 | 87.3 | 84.0 | 93.5 | 87.5 |
| H52 | 84.1 | 84.6 | 88.8 | 63.3 | 75.7 | 75.8 | 81.5 | 84.3 | 87.3 | 84.0 | 93.1 | 87.1 |
| Mass41 Vaccine | 83.5 | 84.3 | 88.2 | 63.0 | 75.7 | 76.4 | 81.5 | 83.7 | 87.3 | 81.3 | 92.7 | 87.5 |
| Georgia 1998 pass8 | 83.9 | 85.1 | 88.8 | 54.8 | 75.5 | 82.1 | 81.5 | 85.8 | 88.3 | 82.2 | 95.5 | 87.7 |
| Georgia 1998 Vaccine | 83.9 | 85.0 | 88.7 | 54.9 | 75.5 | 82.1 | 80.5 | 84.9 | 87.3 | 80.4 | 95.5 | 89.1 |
| Delaware072 | 83.7 | 84.4 | 88.6 | 54.7 | 75.1 | 83.3 | 79.4 | 84.3 | 87.0 | 82.2 | 95.5 | 88.5 |
| ck/CH/LDL/97I P5 | 84.8 | 85.3 | 89.0 | 63.0 | 75.0 | 82.7 | 78.4 | 81.3 | 88.1 | 82.7 | 91.5 | 89.3 |
| ck/CH/LDL/97I P115 | 84.8 | 85.3 | 89.0 | 62.9 | 75.0 | 82.7 | 78.4 | 81.0 | 88.2 | 82.7 | 91.5 | 89.2 |
| M41 | 84.2 | 85.4 | 88.8 | 62.6 | 75.6 | 79.3 | 81.5 | 84.6 | 87.5 | 80.0 | 93.1 | 88.3 |
| ck/CH/LHLJ/100902 | 84.1 | 85.3 | 88.8 | 62.5 | 75.7 | 79.3 | 81.5 | 84.9 | 87.3 | 80.9 | 93.5 | 88.2 |
| ck/CH/LHLJ/07VII | 84.0 | 85.2 | 88.9 | 62.2 | 75.7 | 74.7 | 81.5 | 84.6 | 87.2 | 80.4 | 93.5 | 87.5 |
| Beaudette | 84.4 | 85.3 | 88.6 | 62.5 | 75.4 | 80.4 | 81.0 | 84.9 | 87.3 | 84.5 | 96.3 | 88.0 |
| Iowa97 | 84.8 | 85.7 | 89.2 | 63.7 | 75.3 | 79.3 | 79.4 | 83.7 | 85.0 | 82.7 | 92.7 | 87.7 |
| Gray | 84.6 | 85.6 | 88.7 | 62.9 | 76.3 | 84.4 | 79.4 | 84.3 | 85.7 | 83.1 | 93.5 | 87.8 |
| JMK | 84.3 | 85.6 | 88.6 | 62.9 | 76.5 | 84.4 | 80.0 | 84.3 | 87.2 | 83.1 | 93.5 | 88.2 |
| MG10 | 80.9 | 85.2 | 88.6 | 36.9 | 54.6 | 83.3 | 78.9 | 81.3 | 86.9 | 84.5 | 92.7 | 87.5 |
| NGA/A116E7/2006 | 84.5 | 85.0 | 89.2 | 64.0 | 75.3 | 78.7 | 79.4 | 77.4 | 85.6 | 78.6 | 91.9 | 89.5 |
| SNU8067 | 84.8 | 85.2 | 89.5 | 64.3 | 76.5 | 80.4 | 82.0 | 87.6 | 88.2 | 81.8 | 93.5 | 88.7 |
| KM91 | 85.0 | 85.3 | 89.2 | 64.4 | 76.7 | 67.2 | 85.7 | 89.1 | 89.8 | 81.8 | 93.9 | 88.7 |
| ITA/90254/2005 | 86.0 | 85.4 | 92.7 | 66.9 | 75.5 | 79.8 | 76.0 | 84.0 | 87.9 | 80.4 | 93.9 | 89.0 |
| ck/SWE/0658946/10 | 85.1 | 84.8 | 91.7 | 66.3 | 75.1 | 78.7 | 76.5 | 81.9 | 83.1 | 78.1 | 91.1 | 88.5 |
| TW2575-98 | 84.1 | 83.7 | 89.4 | 64.8 | 75.6 | 82.1 | 80.5 | 83.7 | 87.3 | 84.5 | 93.5 | 87.5 |
| ck/CH/LHB/100801 | 84.3 | 83.9 | 89.7 | 64.1 | 75.5 | 84.4 | 84.1 | 83.7 | 87.6 | 85.9 | 93.9 | 87.5 |
| Ck/CH/LSD/05I | 84.7 | 84.4 | 89.6 | 64.6 | 75.8 | 75.2 | 77.9 | 83.7 | 87.9 | 83.1 | 95.5 | 89.2 |
| BJ | 87.5 | 87.7 | 93.2 | 63.5 | 75.2 | 81.0 | 77.6 | 81.9 | 89.4 | 85.9 | 89.5 | 91.9 |
| A2 | 87.4 | 87.7 | 93.4 | 62.9 | 75.7 | 80.4 | 77.6 | 82.2 | 88.2 | 92.7 | 91.1 | 91.5 |
| ck/CH/IBWF/2007 | 87.3 | 89.1 | 91.4 | 62.9 | 76.5 | 83.9 | 87.3 | 84.9 | 86.9 | 91.8 | 98.3 | 88.3 |
| YN | 87.2 | 88.9 | 91.4 | 63.0 | 76.4 | 83.9 | 87.3 | 84.9 | 87.0 | 84.0 | 98.3 | 88.6 |
| SC021202 | 87.2 | 89.0 | 91.3 | 63.3 | 76.4 | 82.1 | 87.3 | 84.9 | 87.0 | 91.3 | 98.3 | 88.6 |
| SAIBK | 86.2 | 88.9 | 89.5 | 64.1 | 76.4 | 83.9 | 84.6 | 84.0 | 86.9 | 86.3 | 97.9 | 89.7 |
| DY07 | 92.2 | 96.0 | 96.4 | 66.3 | 76.1 | 81.0 | 78.1 | 81.6 | 87.3 | 91.8 | 91.9 | 93.9 |
| Sczy3 | 92.1 | 95.8 | 96.1 | 66.3 | 76.1 | 81.0 | 78.1 | 81.6 | 87.2 | 88.1 | 92.7 | 93.5 |
| ck/CH/LDL/091022 | 92.8 | 96.8 | 97.2 | 66.4 | 76.2 | 81.6 | 78.1 | 81.6 | 87.3 | 85.0 | 90.3 | 92.7 |
| ck/CH/LJL/110302 | 92.9 | 97.2 | 96.3 | 66.5 | 76.0 | 81.6 | 78.1 | 81.3 | 87.3 | 85.0 | 90.7 | 96.0 |
| ck/CH/IBYZ/2011 | 92.5 | 96.2 | 97.0 | 66.2 | 76.3 | 81.6 | 77.6 | 81.9 | 86.9 | 92.2 | 90.7 | 95.5 |
| YX10 | 92.4 | 96.2 | 97.1 | 66.4 | 76.1 | 81.0 | 78.1 | 81.6 | 87.2 | 85.9 | 95.5 | 90.5 |
| ck/CH/LZJ/111113 | 92.6 | 97.5 | 95.9 | 65.3 | 76.3 | 82.7 | 77.6 | 81.6 | 87.2 | 85.0 | 91.5 | 94.7 |
| ck/CH/LGD/120723 | 92.3 | 96.3 | 96.8 | 63.1 | 76.4 | 85.0 | 73.1 | 85.8 | 87.2 | 88.6 | 90.7 | 92.5 |
| ck/CH/LGD/120724 | 92.3 | 96.4 | 96.7 | 63.0 | 76.5 | 85.0 | 73.1 | 85.8 | 87.2 | 88.6 | 90.7 | 92.5 |
| GX-YL5 | 92.4 | 96.8 | 97.3 | 63.1 | 76.0 | 85.6 | 71.3 | 86.4 | 89.4 | 93.1 | 95.9 | 88.3 |
| GX-YL9 | 91.9 | 96.7 | 96.8 | 62.7 | 76.4 | 81.0 | 86.8 | 84.9 | 86.4 | 93.6 | 92.7 | 87.6 |
| CQ04-1 | 90.6 | 92.6 | 97.5 | 62.9 | 76.2 | 83.9 | 85.2 | 79.0 | 85.9 | 90.9 | 97.1 | 88.3 |
Boldface indicates the highest, and italic, the lowest, nt sequence identity in different genes
Fig. 1.
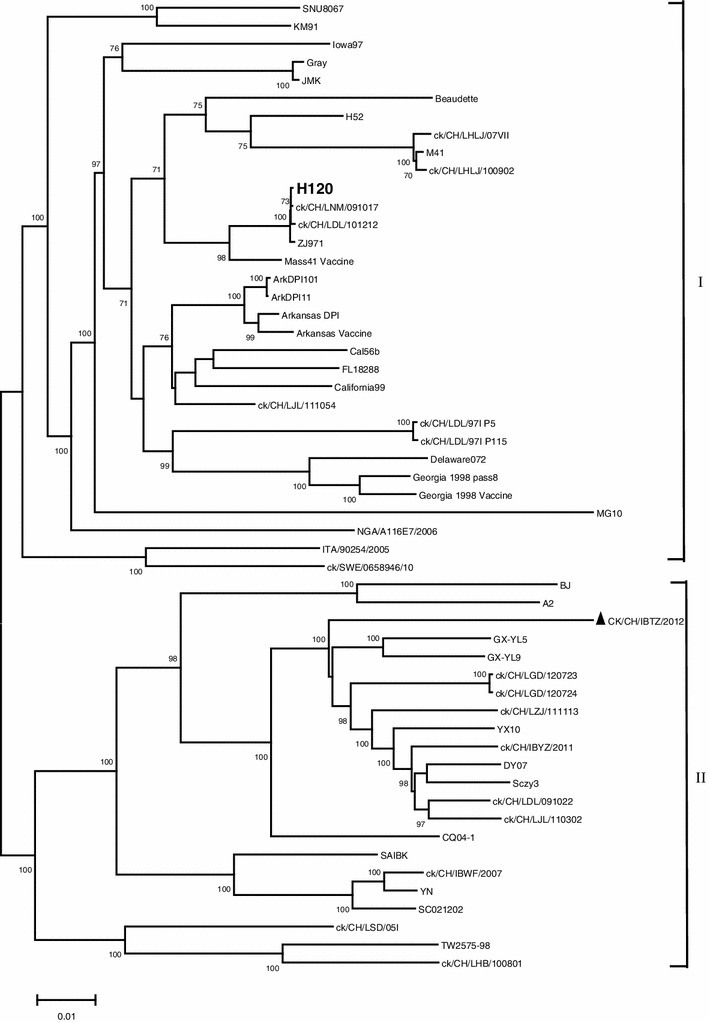
Phylogenetic tree constructed based on the full-length genomes of isolate ck/CH/IBTZ/2012 and 53 published IBV reference strains using the neighbor-joining method (Mega5.2). Numbers on the branches represent the percentage of 1,000 bootstrap samples supporting the branch, only values >70 % are shown. The isolate ck/CH/IBTZ/2012 is marked with a filled triangle, and the H120 vaccine strain used in this flock is marked with boldface
In conclusion, sequence comparison and phylogenetic analysis based on the full-length genomic sequences showed that ck/CH/IBTZ/2012 was mostly related to the LX4-like strains. However, phylogenetic trees are constructed using the S1 and S2 gene sequences, in which ck/CH/IBTZ/2012 was branched as a separate genotype. The spike protein is a glycoprotein on the virus envelope surface and a determinant of the serotype, most molecular epidemiology studies on IBV were focused on S gene [9, 10]. Sequence analysis based on the S gene sequences revealed that ck/CH/IBTZ/2012 possesses a distinct S gene setting it apart from the Massachusetts-type strains and LX4-type strains. Recombination analysis showed that ck/CH/IBTZ/2012 is a chimeric strain derived from LX4-like strains which are predominant in China in recent years [8]. Based on the data presented in this paper, it can be concluded that genetic changes due to adaptive evolution and recombination both contributed to the origin of strain ck/CH/IBTZ/2012, which belongs to a new genotype. Vaccination programs based on Massachusetts-type live attenuated vaccine H120 cannot control this new epidemic strain for which they have different genotypes. The sequence information provided in this article will contribute to future studies on the molecular epidemiology and be able to develop better measures to control IB.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
This work was supported by Grants from the National Natural Science Foundation of China (31101832) and Jiangsu Province Agricultural Science & Technology Support Program (No. BE2012369).
References
- 1.Cook JK, Jackwood M, Jones RC. Avian Pathol. 2012;41:239–250. doi: 10.1080/03079457.2012.680432. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Wang HN, Wang T, Fan WQ, Zhang AY, Wei K, Tian GB, Yang X. Virus Genes. 2010;41:377–388. doi: 10.1007/s11262-010-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu X, Shao Y, Ma H, Sun C, Zhang X, Li C, Han Z, Yan B, Kong X, Liu S. Infect. Genet. Evol. 2013;14:29–38. doi: 10.1016/j.meegid.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song JE, Jeong WG, Sung HW, Kwon HM. Virus Genes. 2013;46:371–374. doi: 10.1007/s11262-012-0856-0. [DOI] [PubMed] [Google Scholar]
- 5.Jackwood MW. Avian Dis. 2012;56:634–641. doi: 10.1637/10227-043012-Review.1. [DOI] [PubMed] [Google Scholar]
- 6.Liu S, Kong X. Avian Pathol. 2004;33:321–327. doi: 10.1080/0307945042000220697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu S, Zhang X, Wang Y, Li C, Liu Q, Han Z, Zhang Q, Kong X, Tong G. Vet. J. 2009;179:130–136. doi: 10.1016/j.tvjl.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou NL, Zhao FF, Wang YP, Liu P, Cao SJ, Wen XT, Huang Y. Virus Genes. 2010;41:202–209. doi: 10.1007/s11262-010-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Z, Sun C, Yan B, Zhang X, Wang Y, Li C, Zhang Q, Ma Y, Shao Y, Liu Q, Kong X, Liu S. Infect. Genet. Evol. 2011;11:190–200. doi: 10.1016/j.meegid.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo H, Qin J, Chen F, Xie Q, Bi Y, Cao Y, Xue C. Virus Genes. 2012;44:19–23. doi: 10.1007/s11262-011-0657-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan F, Zhao Y, Yue W, Yao J, Lihua L, Ji W, Li X, Liu F, Wu Q. Avian Dis. 2011;55:451–458. doi: 10.1637/9446-070510-ResNote.1. [DOI] [PubMed] [Google Scholar]
- 12.Ji J, Xie J, Chen F, Shu D, Zuo K, Xue C, Qin J, Li H, Bi Y, Ma J, Xie Q. Virol. J. 2011;8:184. doi: 10.1186/1743-422X-8-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S, Stecher G, Peterson D, Tamura K. Bioinformatics. 2012;28:2685–2686. doi: 10.1093/bioinformatics/bts507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin D, Rybicki E. Bioinformatics. 2000;16:562–563. doi: 10.1093/bioinformatics/16.6.562. [DOI] [PubMed] [Google Scholar]
- 15.He K, Li M, Wei P, Mo ML, Wei TC, Li KR. J. Virol. 2012;86:13887–13888. doi: 10.1128/JVI.02722-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim TH, Kim MS, Jang JH, Lee DH, Park JK, Youn HN, Lee JB, Park SY, Choi IS, Song CS. Poult. Sci. 2012;91:89–94. doi: 10.3382/ps.2011-01739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.