

Summary
Fibroblast growth factor receptor 1 (FGFR1) is frequently amplified in human small-cell lung cancer (SCLC), but its contribution to SCLC and other lung tumors has remained elusive. Here, we assess the tumorigenic capacity of constitutive-active FGFR1 (FGFR1K656E) with concomitant RB and P53 depletion in mouse lung. Our results reveal a context-dependent effect of FGFR1K656E: it impairs SCLC development from CGRPPOS neuroendocrine (NE) cells, which are considered the major cell of origin of SCLC, whereas it promotes SCLC and low-grade NE bronchial lesions from tracheobronchial-basal cells. Moreover, FGFR1K656E induces lung adenocarcinoma (LADC) from most lung cell compartments. However, its expression is not sustained in LADC originating from CGRPPOS cells. Therefore, cell context and tumor stage should be taken into account when considering FGFR1 inhibition as a therapeutic option.
Graphical Abstract
Highlights
-
•
FGRF signaling impairs SCLC development initiated from CGRPPOS NE cells
-
•
FGRF signaling promotes development of NE tumors initiated from K14POS cells
-
•
Rb;p53;Fgfr1 mice develop ADCs that retain signatures of their cell of origin
-
•
Low expression of Fgfr1 in progressed tumors suggests a role in tumor initiation
Ferone et al. show that inducing FGFR signaling in lung neuroendocrine (NE) cells of Rb;p53 mice results in impairment of SCLC and enhancement of peripheral NE lesions. The location and tumor characteristics depend on the targeted cell type. Rb;p53;Fgfr1 mice also develop adenocarcinomas with the incidence depending on the targeted cell type.
Introduction
Small-cell lung cancer (SCLC) constitutes 15% of all lung cancer cases and is the most aggressive subtype with a 5- year survival rate of 2%–8% for stage III/IV disease. Patients, frequently diagnosed with extensive disease, receive chemotherapy, often leading to a remarkable initial response. Unfortunately, patients almost invariably relapse within months with resistant disease. The standard of care first-line treatment has not changed in over 30 years, and despite ongoing efforts, no molecularly targeted drugs have been approved to date for the treatment of SCLC. However, immunotherapy with anti-PD1 antibody pembrolizumab for treating metastatic SCLC has been recently approved by the Food and Drug Administration (FDA) for patients with disease progression or after platinum-based chemotherapy and at least one other first-line treatment.
Mechanisms underlying the initial sensitivity to chemotherapy and the invariably subsequent resistance are not well understood. This highlights the importance of deeper understanding of the basic biology of SCLC, studying its initiation and progression, defining functional contribution of key drivers, and identification of the cells of origin for the tumor.
SCLC belongs to the broader class of tumors with neuroendocrine (NE) differentiation. Lung tumors with a NE phenotype can be divided in human into two major categories: (1) high-grade NE carcinomas consisting of SCLC and large cell neuroendocrine carcinoma (LCNEC) and (2) low-grade NE tumors consisting of typical and atypical carcinoids (Arrigoni et al., 1972, Mills et al., 1982).
Transformation and growth of NE tumors may be promoted by autocrine and paracrine signaling of secreted neuropeptides (Kazanjian et al., 2004, Koutsami et al., 2002). However, it is still questionable whether all NE tumors arise from the same bronchial NE cells or if cells committed to other lineages are involved (Park et al., 2011, Sutherland and Berns, 2010). It is also controversial whether the diverse NE tumors require the same molecular aberrations. So far, precursor lesions, such as tumorlets or diffuse idiopathic pulmonary neuroendocrine cell hyperplasia (DIPNECH), have been observed in man in association with carcinoids but never with other NE tumors including SCLC (Gazdar and Brambilla, 2010, Rizvi et al., 2009, Travis, 2010).
Transformation of lung cells into SCLC is promoted by the biallelic inactivation of the tumor suppressors TP53 and RB1 (George et al., 2015). In RB1-proficient tumors, overexpression of cyclin D1 may constitute an alternative mechanism, but this is relatively rare (George et al., 2015). Mice in which Rb1 and Trp53 are biallelically inactivated in lung cells (Meuwissen et al., 2003) recapitulate the development of human SCLC, but acquire additional lesions that are also recurrently found in human SCLC. These latter lesions were shown to accelerate tumor development and/or metastatic growth in these models. This is the case for mice that overexpress MycL on top of the biallelic inactivation of Rb1 and Trp53, which show an earlier onset of SCLC and shortened latency of tumor development (Huijbers et al., 2014). MYCL is a transcription factor member of a family of oncogenes found amplified in ~9% of human SCLC (Calbo et al., 2011, George et al., 2015, Iwakawa et al., 2013).
The concomitant overexpression of NFIB, a transcription factor whose focal amplification was found at high frequency in Rb1flox/flox;Trp53flox/flox mice (hereafter referred to as RP mice), not only promoted the earlier onset of SCLC but it also enhanced the metastatic dissemination of SCLC in mouse (Denny et al., 2016, Semenova et al., 2016).
Other mouse models based on the conditional inactivation of Rb1 and Trp53 in combination with either p130 (Schaffer et al. (2010) or Pten (Cui et al., 2014, McFadden et al., 2014) showed the whole spectrum of NE tumors, including LCNEC and adenocarcinoma (ADC) with NE elements pointing to the substantial plasticity of lung cells.
In contrast with the genetic lesions discussed above, much less is understood about the role of fibroblast growth factor receptor 1 (FGFR1) amplification and therefore the activation of the mitogen-activated protein kinase (MAPK) downstream signaling pathway in SCLC. Intriguingly, in spite of its occurrence in human SCLC (Peifer et al., 2012, Voortman et al., 2010), we never found spontaneous copy number variation of Fgfr1 in RP mice. Hence, it remained unclear whether its activation is beneficial to the development of SCLC, because there is evidence that the MAPK signaling pathway may play both a promoting as well as an antagonistic role.
For instance, other genetic alterations in molecules signaling through the MAPK pathway, besides FGFR1 amplification, are rarely seen in human SCLC (George et al., 2015, Peifer et al., 2012, Rudin et al., 2012). This is the case for epidermal growth factor receptor (EGFR) amplification that has been found only in the combined subtype of SCLC with lung adenocarcinoma (LADC) (Tatematsu et al., 2008). Consistently, treatment of LADC carrying EGFR mutations with EGFR inhibitors showed a low but significant transition to SCLC (Niederst et al., 2015), suggesting that the suppression of MAPK signaling pathway might facilitate the development of SCLC. Furthermore, in vitro activation of this pathway resulted in impaired proliferation, growth arrest, and in some cases in loss of NE features (Calbo et al., 2011, Ravi et al., 1998, Ravi et al., 1999, Sriuranpong et al., 2001).
However, other data suggest that the MAPK pathway can promote proliferation and survival in SCLC cell populations. As mentioned before, FGFR1 has been found focally amplified in 6% to 30% of SCLC human samples (Peifer et al., 2012, Voortman et al., 2010). Moreover, SCLC xenograft growth was impaired by inhibition of both FGFR1 and FGFR2 (Pardo et al., 2009). In vitro data showed that FGF2 promotes the survival of SCLC cells (Pardo et al., 2001, Pardo et al., 2003, Pardo et al., 2006). Furthermore, FGF2 can also contribute to the metastatic dissemination of NE tumor sub-clones (Kwon et al., 2015).
In order to unravel the contribution of FGFR1 amplification and its downstream pathway to the development of SCLC, we generated mice that conditionally overexpressed a dominant active mutant form of Fgfr1 on top of the biallelic inactivation of Trp53 and Rb1. We targeted different subpopulations of lung cells to address the potential importance of the cellular context in conjunction with these specific genetic alterations to assess how different lung cell populations respond to Fgfr1 overexpression.
Results
FGFR1K656E Expression Selectively Promotes NE Bronchial Lesions, whereas It Impairs SCLC
In this study, we compared the phenotype of lung tumors in the established RP mouse model, which constitutes our control (Meuwissen et al., 2003), with the newly generated Rb1flox/flox;Trp53flox/flox;LSL-Fgfr1K656E mouse model (hereafter RP-Fgfr1) (see Figure S1 and STAR Methods for description). First, we targeted all lung cell types of RP-Fgfr1 and RP mice by intratracheal delivery of Ad5-CMV-Cre virus. Adenoviral delivery of Cre recombinase in pulmonary cells caused biallelic Rb1 and Trp53 inactivation, with concomitant induction of Fgfr1K656E expression along with yellow fluorescent protein (YFP) from the Col1A1 locus.
12 out of 14 RP-Fgfr1 mice injected with Ad5-CMV-Cre showed breathing difficulties within 135 days due to the development of tumors in the nasal cavity (Table S1). Although none of the RP-Fgfr1 mice developed SCLC within this early time-window, 58% (7 out of 12) showed neoplastic NE lesions lining the bronchi/bronchioles (BLs) (Figure 1C; Table S1), and 42% developed peripherally located NE alveolar lesions (ALs). Moreover, 33% of RP-Fgfr1 (4 out of 12) also developed LADC.
Figure 1.

FGFR1 Overexpression Selectively Promotes NE Bronchial Lesions whereas It Impairs SCLC
(A–H) Scan image of CGRP staining of coronal sections of lungs from RP (A, B, E, and F) and RP-Fgfr1 (C, D, G, and H) mice injected with either Ad5-CMV-Cre (A–D) or Ad5-CGRP-Cre (E–H) and collected at humane endpoint. Scale bar indicates 2 mm.
(I and J) Quantification of NE tumor burden in Ad5-CMV-Cre (CMV)-injected (I) or Ad5-CGRP-Cre (CGRP)-injected RP and RP-Fgfr1 mice (J). Insets represent BL and ADC (C) and AL (D). Scale bar insets indicate 50 μm (C and D).
(K and L) Quantification of ADC tumor burden (K) and tumor-free survival curve (L) of Ad5-CMV-Cre- and Ad5-CGRP-Cre-injected RP-Fgfr1 mice (Fgfr1).
By comparing age-matched RP and RP-Fgfr1 mice, we noticed a shift in the type of CGRP-positive NE lung lesions from the typical central SCLC in 90% of RP mice to BLs and ALs in 80% and 70% of the RP-Fgfr1 mice, respectively (Figures 1A–1D; Tables S1 and S2). Only 20% (2 out of 10) RP-Fgfr1 mice developed central SCLC (Table S1). In those two mice, only a small fraction of SCLC lesions was positive to FGFR1 expression (data not shown). Furthermore, the overall tumor burden of NE lesions in RP-Fgfr1 mice was considerably lower as compared to RP mice (Figure 1I). RP-Fgfr1 mice that survived longer than 135 days also showed an increased penetrance of LADC whereas none of RP mice showed LADC (Tables S1 and S2).
Taken together, these data suggest that FGFR1 is disadvantageous for the development of typical SCLC, whereas it promotes BLs, ALs, and LADC when RB1 and TP53 are inactivated.
In order to study the contribution of FGFR1 to NE lesions without the confounding development of LADC, we specifically targeted NE cells of RP-Fgfr1 and RP mice by intratracheal injection of Ad5-CGRP-Cre. We compared the presence of lung NE lesions in age-matched RP-Fgfr1 and RP mice (170 days upon Ad5-CGRP-Cre injection). Before this time point, mice did not develop readily detectable lesions except for nasal NE tumors (Tables S3 and S4). We found that only 18% (3 out of 17) of RP-Fgfr1 mice developed SCLC compared to 52% of RP mice (15 out of 29). As seen before for Ad5-CMV-Cre-injected RP-Fgfr1 mice, in this case, only a small fraction of SCLC lesions was positive to FGFR1 expression (data not shown). The penetrance of ALs was slightly higher in RP-Fgfr1 as compared to RP mice (35% versus 28%, respectively). Surprisingly, BLs were rarely found in Ad5-CGRP-Cre-injected mice as compared to Ad5-CMV-Cre-injected mice, suggesting the existence of a different cell of origin, which was not targeted by Ad5-CGRP-Cre virus. Yet, FGFR1K656E expression promoted to some extent also BLs originating from CGRPPOS cells: 29% of RP-Fgfr1 mice developed BLs compared to only 7% of RP mice. Overall, the burden of NE lesions was reduced in RP-Fgfr1 compared to RP mice (Figures 1E–1H and 1J), and the type of NE lesions shifted from central to peripheral (Figures 1E–1H; Tables S3 and S4).
Unexpectedly, 41% of Ad5-CGRP-Cre treated RP-Fgfr1 mice (7 out of 17) also developed LADC in spite of Rb1 deletion and the NE origin of the targeted cell (Table S3). Nevertheless, the tumor burden and penetrance of LADC originating from CGRPPOS cells were much lower compared to the Ad5-CMV-Cre-induced LADC (Figure 1K; Tables S1 and S3). The onset of LADC originating from CGRPPOS cells occurred considerably later, allowing mice to survive longer (Figure 1L).
Taken together, our data indicate that FGFR1K656E expression in RB1, TP53-deficient NE cells, imposes a shift from high grade SCLC to low grade NE tumors located mostly in the peripheral lung. Furthermore, FGFR1K656E expression drives the transformation of a subset of CGRPPOS cells into LADC.
Our data also show that CGRPPOS cells are not the predominant cell of origin of BLs; nevertheless, FGFR1K656E expression enables a sub-fraction of these cells to transform into BLs.
Bronchial Lesions Are Prominently Found in Ad5-CMV-Cre-Injected RP-Fgfr1 Mice and Share the Molecular Circuitry with Cisplatin-Resistant Alveolar Lesions
We compared the number of mice with BLs (penetrance) as well as the number of BLs per mouse (Figures 2A and 2B; Tables S1, S2, S3, and S4) in the different cohorts of mice and found that both were higher in RP-Fgfr1 as compared to RP mice. This difference was particularly pronounced when mice were injected with Ad5-CMV-Cre and to a lesser extent with Ad5-CGRP-Cre (Figures 2A and 2B). In contrast, FGFR1K656E expression had little or no impact on the number of ALs per mouse (Figures 2C and 2D), although the penetrance was slightly increased in both Ad5-CGRP-Cre- and Ad5-CMV-Cre-injected RP-Fgfr1 mice (Tables S1, S2, S3, and S4).
Figure 2.

Bronchial Lesions Are Prominently Found in Ad5-CMV-Cre-Injected RP-Fgfr1 Mice
(A–D) Quantification of BLs (A and B) and ALs (C and D) in either Ad5-CMV-Cre (CMV; A and C) or Ad5-CGRP-Cre (CGRP; B and D)-injected RP and RP-Fgfr1 mice.
(E–H) A representative BL of RP-Fgfr1 mice stained for CGRP (E), CDH1 (F), FGFR1 (G), GFP (H). Scale bar, 100 μm.
(I) CGRP and FGFR1 staining of NE (left) and ADC lesions (right) of RP-Fgfr1 mice injected with Ad5-CMV-Cre and collected at Humane Endpoint.
(J) PCR analysis on genomic DNA isolated from lung lesions (NE and ADC) of RP and RP-Fgfr1 mice. Two Primer sets were designed to distinguish floxed from recombinant (Rec) Fgfr1 alleles in the Col1A1 locus. As expected, both primer sets did not give any PCR product when DNA from RP mice was used as negative control, whereas in both NE (RP-Fgfr1-NE 4, RP-Fgfr1-NE 5) and ADC (RP-Fgfr1-ADC 3) lesions of RP-Fgfr1 mice, the primer set for the Rec Fgfr1 allele gave a PCR product of the proper length. The primer set for the floxed allele gave only a faint band in the line of ADC sample. The image is representative of n = 2 ADC samples and n = 4 SCLC samples.
Independently of the genotype, BLs expressed a high level of CGRP and CDH1 (Figures 2E and 2F and data not shown). BLs in RP-Fgfr1 mice expressed FGFR1K656E in patches along with YFP, as visualized by antibodies against GFP (Figures 2G and 2H).
To exclude that the sporadic expression of FGFR1K656E in some BLs was due to partial recombination of Fgfr1 allele, we isolated genomic DNA from FGFR1POS lesions such as LADC derived from Ad5-CMV-Cre-injected mice and from FGFR1NEG NE lesions (Figure 2I). We used SCLC isolated from RP mice as negative control of Fgfr1K656E ectopic allele recombination. PCR analysis (Figure 2J) showed that both NE and LADC lesions with very different levels of FGFR1 protein expression had recombined the ectopic Fgfr1K656E allele. This result indicates that the transgenic Fgfr1K656E allele recombination is very efficient. Therefore, the absence of FGFR1K656E protein expression is likely the result of silencing of the allele by epigenetic mechanisms. Due to insufficient material, we could not successfully isolate genomic DNA directly from BLs. However, the finding that a wide collection of samples either negative or positive to FGFR1 staining all showed a near-complete activating recombination of Fgfr1 allele suggests that the same has likely happened for BLs.
The discovery that BLs were mostly found in mice injected with Ad5-CMV-Cre and only very rarely in mice injected with Ad5-CGRP-Cre (Figures 1E–1H, 2A, and 2B), points to the existence of an unknown cell of origin that does not express CGRP, but, upon deletion of Rb1 and Trp53, activates CGRP expression.
To gain insight into the cell of origin of BLs, and in order to define these lesions molecularly, we performed RNA sequencing (RNA-seq) of BLs and compared their expression profile to SCLC and ALs. The expression profile of ALs has been described recently for cisplatin-treated Ad5-CMV-Cre-injected RPM (Rb1/Trp53/Mycl) mice (Böttger et al., 2019). For consistency, we isolated BLs from RPM mice and compared their expression profile with that of either ALs or SCLC of RPM mice. Our analysis identified 471 genes differentially expressed (DE) between SCLC and BLs, with 79 upregulated and 392 downregulated genes. We also compared SCLC to ALs and found a total of 834 DE genes, with 232 upregulated and 602 downregulated genes (Figures S2A, S2B, S2D, and S2E). The expression profile of BLs and ALs showed considerable overlap: only 81 genes were differentially expressed, with 52 downregulated and 29 upregulated genes in ALs lesions as compared to BLs (Figures S2C and S2F).
A principal component analysis (PCA) to determine the variance among all samples and also to define relationships between the three groups showed that BL and AL samples clustered in close proximity and distant from SCLC samples (Figure 3A). The list of genes included in the PC1 analysis revealed 70 commonly upregulated genes in ALs and BLs compared to SCLC and 15 genes commonly downregulated (Figure 3B). Among commonly upregulated genes, we found genes encoding transmembrane receptors involved in lung development, such as Egfr, Fgfr2, Robo1, and secreted factors, such as Igf2 and Wnt4 (Figure 3B).
Figure 3.

Bronchial Lesions Share the Molecular Circuitry with Cisplatin-Resistant Alveolar Lesions
(A) Principal component analysis on triplicates of SCLC, BLs, and ALs samples. Plotted are the principle components 1 and 2 (PC1, PC2).
(B) Heatmap of genes with the highest rotation value for PC1.
(C) Gene set enrichment analysis (GSEA) using the HALLMARKS gene sets of ALs versus SCLC (left graph) and BLs versus SCLC (right graph); FDR q value <0.25, normalized enrichment score (NES) ≥1.
(D–K) ALDH1A1 (D–G) and EGFR (H–K) staining on lung sections indicating whole lung of RP-Fgfr1 mice (E and I) and RPM mice as positive control (D and H) collected at humane endpoint. High magnification images of RP-Fgfr1 lungs show ALDH1A1 staining of ADC and BL (F) or AL (G) and EGFR staining of ADC and BL (J) or AL (K). Scale bars indicate 2 mm (D, E, H, and I) and 50 μm (F, G, J, and K). Arrows in (D) and (H) indicate central and peripheral lesions. Arrows in (E) and (I) indicate peripheral BL and AL lesions. Rectangles in (E) and (I) indicate regions that are shown with higher magnification in (F), (G), (J), and (K).
See also Figure S2.
Gene set enrichment analysis identified gene sets involved in xenobiotic metabolism and the oxidative stress response, such as aldehyde dehydrogenase family members (Aldh1a1, Aldh1a7), glutathione S-transferase family members (Gstm1, Gstm2), and cytochrome P450 2f2 (Cyp2f2) (Figures 3B and 3C).
Among other commonly upregulated gene sets in BLs and ALs versus SCLC, we found INNATE IMMUNE RESPONSE and KRAS SIGNALING UP, which suggest an involvement of MAPK pathway in both low-grade NE lesions compared to SCLC.
Gene set enrichment analysis of downregulated genes in ALs versus SCLC identified, among the most negatively enriched gene sets, E2F TARGETS, G2M CHECKPOINT, MYC TARGETS V1, mTORC1 SIGNALING, and MITOTIC SPINDLE (Figure 3C). In BLs versus SCLC, we found the same downregulated gene sets, but only E2F TARGETS and MYC TARGETS V1 were prominently downregulated (false discovery rate [FDR] <0.25).
Taken together, our sequencing data show that the expression profile of BLs greatly overlaps with that of ALs. In both cases, we found gene profiles suggesting reduced proliferation and altered metabolism. The remarkable difference of these low-grade NE lesions with SCLC suggests that they represent a distinct class of neoplastic lesions and, most importantly, that they have a different cell of origin.
In order to assess whether BLs and ALs of RP-Fgfr1 mice share the expression profile of low-grade NE lesions found in RPM mice, we performed immunostaining for ALDH1A1 and EGFR, which were highly expressed in BLs and ALs compared to SCLC in RPM mice (Figures 3D and 3H). Although the overall lung phenotype of RP-Fgfr1 mice appeared very different from that of RPM mice, mainly because of the reduced SCLC and the presence of LADC (Figures 3D and 3E), we detected enhanced expression of ALDH1A1 in BLs and ALs (Figures 3E–3G); BLs and ALs of RP-Fgfr1 mice also showed high levels of EGFR, this in striking difference with LADC (Figures 3I–3K).
Our data indicate that low-grade NE lesions found in RP-Fgfr1 mice likely represent the same type of lesions found in RPM mice and are histologically indistinguishable from lesions found in the BLs and ALs in the various genotypes. Only the frequency in which these lesions occur appears to be influenced by the additional driver genes and the cell-type specificity of the promoter driving Cre in the adenoviral vectors (Table S11).
The notion that our expression data did not reveal any gene signature specific for known lung cell subtypes suggests that the target cells do not constitute an identified lung progenitor cell. More detailed future investigations are required to address this issue.
FGFR1K656E Expression Drives the Transformation of K14-Expressing Cells into SCLC and, to a Lesser Extent, ADC
Because our aim was to gain insight into the role of FGFR1 in human SCLC, it appeared critical to identify the lung cell compartment sensitive to transformation into SCLC upon FGFR1-mediated signaling. Hence, we targeted a larger array of distinct lung cell lineages using an additional set of adenoviral vectors permitting Cre recombinase-mediated switching, specifically in K14-, SPC-, and CC10-expressing cells.
Injection of RP and RP-Fgfr1 mice with Ad5-K14-Cre specifically causes recombination in basal cells (Ferone et al., 2016). Remarkably, both RP and RP-Fgfr1 mice developed SCLC with a penetrance of 60% and 50%, respectively (Tables S5 and S6). Tumors were invasive in both RP and RP-Fgfr1 mice (Figures 4A, 4B, 4D, and 4E). Invariably, we found intrapulmonary dissemination (IPD) of SCLC (Tables S5 and S6). RP-Fgfr1 mice also developed LADCs that were completely absent in RP mice (Figures 4A and 4D; Tables S5 and S6).
Figure 4.

FGFR1 Overexpression Drives the Transformation of K14-Expressing Cells into SCLC and, to a Lesser Extent, ADC
(A–F) Scan image of HE (A and D), CGRP (B and E), and FGFR1 (C and F) staining of coronal sections of lungs from RP (A–C) and RP-Fgfr1 mice (D–F) injected with Ad5-K14-Cre and collected at humane endpoint. Scale bar indicates 2 mm. Inset in (D) represents SCLC and the marginal ADC compartment. Scale bar inset indicates 50 μm.
(G) Lung cancer-free survival curve of mice with indicated genotypes.
(H–J) Quantification of BLs (H), ALs (I), and NE tumor burden (J) of mice with indicated genotypes.
Tumor-free survival of RP-Fgfr1 mice was shorter compared to RP mice (Figure 4G). We found peripheral low-grade NE lesions to be the major difference between Ad5-K14-Cre-injected RP and RP-Fgfr1 mice. BLs and ALs were found in 33% and 58% of RP-Fgfr1 mice, respectively, compared to 7% and 20% in RP mice (Tables S5 and S6). The number of BLs and ALs per mouse was also increased by FGFR1K656E expression (Figures 4H and 4I). NE tumor burden was comparable between RP and RP-Fgfr1 mice (Figure 4J). Contrary to CGRPPOS cells, K14POS cells were tolerating high levels of FGFR1K656E expression as assessed by immunostaining of RP and RP-Fgfr1 lung (Figures 4C and 4F).
Taken together, our data show that in case of RP-Fgfr1 mice, the primary cell of origin of SCLC is a K14POS lung cell rather than the typical CGRPPOS NE cell, which has been considered the predominant SCLC initiating cell in all mouse models described to date. Our data also show that activation of FGFR1 signaling is able to promote BLs and ALs in K14-expressing cells although K14POS cells might not be the exclusive cell of origin of these low-grade NE lesions.
FGFR1K656E Expression Promotes Early Transformation of CC10POS and SPCPOS Cells in LADC and Not SCLC
Because K14POS, and not CGRPPOS, cells are the cell of origin of SCLC in RP-Fgfr1 mice, we wanted to know whether other predominant lung cell lineages could also give rise to NE lesions and to what extent. Therefore, we specifically switched conditional alleles of RP-Fgfr1 and RP control mice in club secretory cells by using Ad5-CC10-Cre and in alveolar type 2 (AT2) cells by using Ad5-SPC-Cre (Sutherland et al., 2011).
RP mice developed SCLC exclusively when either CC10POS or SPCPOS cells were targeted (Figures 5A–5D). However, tumor onset was much slower as compared to RP mice injected with Ad5-CGRP-Cre, resulting, on average, in a longer survival (Tables S4, S8, and S10). RP mice injected with Ad5-CC10-Cre showed a tendency of an even lower penetrance of SCLC compared to RP mice injected with Ad-SPC-Cre (Tables S8 and S10) in line with previous observations (Table S12). Low-grade NE lesions were spuriously present in RP mice injected with Ad5-CC10-Cre and virtually absent in RP mice injected with Ad5-SPC-Cre (Tables S8 and S10).
Figure 5.

FGFR1K656E Expression Promotes Early Transformation of CC10POS and SPCPOS Cells in LADC and Not SCLC
(A–H) HE staining of RP (A–D) and RP-Fgfr1 mice (E–H) injected with either Ad5-CC10-Cre (A, B, E, and F) or Ad5-SPC-Cre (C, D, G, and H) collected at humane endpoint. Images show the whole lung (A, E, C, and G) or ADC lesions at higher magnification (B, F, D, and H).
(I and J) NE tumor burden (I) and ADC tumor burden (J) of mice with the indicated genotypes.
(K) SCLC-free survival curve of RP mice injected with either Ad5-CC10-Cre (CC10) or AD5-SOC-Cre (SPC).
(L) ADC-free survival curve of RP-Fgfr1 mice injected with either Ad5-CC10-Cre (CC10) or AD5-SOC-Cre (SPC). Scale bars indicate 2 mm (A, C, E, and G) and 50 μm (B, D, F, and H).
The concomitant expression of FGFR1K656E in either CC10POS and SPCPOS cells gave rise to LADC (Figures 5E–5H). Due to the quick appearance of massive lesions of LADC in RP-Fgfr1 mice (Figures 5K and 5L; Tables S7, S8, S9, and S10), we cannot exclude the potential for late onset of SCLC following Ad5-CC10-Cre and Ad5-SPC-Cre injection of RP-Fgfr1 mice.
The penetrance of LADC was slightly lower upon Ad5-CC10-Cre injection (89%) as compared to 100% upon Ad5-SPC-Cre injection (Tables S7 and S9). Switched SPCPOS cells resulted in a higher tumor burden as compared to CC10POS cells. This was the case both for SCLC in RP mice and for LADC in RP-Fgfr1 mice (Figures 5I and 5J), although the difference was less pronounced for SCLC. The higher tumor burden of both RP and RP-Fgfr1 mice injected with Ad5-SPC-Cre was associated with a shorter survival (Figures 5K and 5L).
Taken together, our data show that activation of FGFR1 in RB1 and TP53 deficient secretory club and AT2 cells is imposing LADC. AT2 cells seem more prone to transformation compared to club secretory cells, either into SCLC or LADC.
Gene Expression Profile of LADC Arising from SPCPOS and CGRPPOS Reveals Differences Associated with the Cell of Origin
We showed that Ad5-CGRP-Cre-injected RP-Fgfr1 mice developed LADC (Figures 1K and 1L). In order to identify potential differences due to the distinct cells of origin giving rise to LADC, we compared the expression profile of LADC originating from SPCPOS cells and CGRPPOS cells with SCLC originating from CGRPPOS cells of RP-Fgfr1 mice. Our aim was to identify commonly regulated genes associated with either tumor histology (LADC from SPCPOS and CGRPPOS cells) or the cell of origin (LADC and SCLC originating from CGRPPOS cells).
Gene expression data showed that LADC samples, although originating from two different cells of origin, clustered together, while SCLC samples formed a distinct cluster (Figure S3A). We then performed a PCA on the data. The first principle component separated LADC and SCLC samples and explained 68% of the variation in the dataset (Figure 6A), not surprisingly the largest variation in the dataset.
Figure 6.

Gene Expression Profile of ADC Arising from SPCPOS and CGRPPOS Reveals Differences Associated with the Cell of Origin
(A) PCA analysis on triplicates of SCLC, ADC developed by Ad5-CGRP-Cre-injected RP-Fgfr1 mice (SCLC-CGRP; ADC-CGRP), and ADC developed by Ad5-SPC-Cre-injected RP-Fgfr1 mice (ADC-SPC), defined by variables PC1 and PC2.
(B) Heatmap of significantly (FDR <0.05) upregulated and downregulated genes between SCLC and ADC originating from CGRPPOS cells (C-ADC) versus ADC initiated from SPCPOS cells (S-ADC). Gene ontology and functional analysis performed with DAVID program identified the indicated terms (legend with different colors).
(C–N) FGFR1 (C–E), SOX2 (F–H), and SOX9 (I–K) staining on SCLC (CGRP-SCLC; C, F, I, and L) and ADC (CGRP-ADC; D, G, J, and M) developed by Ad5-CGRP-Cre-injected RP-Fgfr1 mice, and ADC developed by Ad5-SPC-Cre-injected RP-Fgfr1 mice (SPC-ADC; E, H, K, and N). TTF1 staining is used as positive control of ADC (L–N). Scale bar indicates 50 μm.
See also Figure S6.
We subsequently performed gene enrichment and functional analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) program on the list of genes with major variation among samples extracted from PC1 (Figure S3B).
Among commonly upregulated genes in LADC versus SCLC, we mainly found genes involved in gas exchange such as Sftpa1, Sftpb, Sftpc, and Sftpd, and in the regulation of autophagy like Dram1 and Lamp3 (Figure S3B).
Downregulated genes were mostly represented by cellular functions associated to neuronal migration (Lmx1b, Sox1, Celsr3, and Cdk5r2), nerve-growth processing (Pcsk1 and Pcsk2), secretion of neuropeptides (Scg1 and Scg2), and secretory granule biogenesis (Chga). We also found downregulation of neuronal lineage genes like Ascl1 and genes encoding for neuropeptides, such as Calca.
Taken together, our data show that LADC derived from either SPCPOS or CGRPPOS cells of RP-Fgfr1 mice are very similar in terms of expression profile.
However, we also identified a number of DE genes common to LADC and SCLC originating from CGRPPOS cells versus LADC originating from SPCPOS cells (Figure 6B). Among the commonly upregulated genes in LADC and SCLC originating from CGRPPOS cells, we found genes involved in the oxidoreduction process (Ndufs5, Ndufa12, and Ndufa1), in DNA damage repair, metal ion binding, and most importantly, in neurogenesis, such as Dlg3, Dab1, and Lhx2 (Figure 6B). Among the upregulated transcription factors, we found that Sox2 was consistently upregulated in both LADC and SCLC originating from CGRPPOS cells as compared to LADC from SPCPOS cells (Figure 6B) and also confirmed by IHC staining (Figures 6F–6H); TTF1 staining is used as biomarker of LADC (Figures 6L–6N). Among the commonly downregulated genes in LADC and SCLC originating from CGRPPOS cells, we found genes involved in the activation of MAPK pathway (Alk), lung alveolus development (Pdpn), cell adhesion (Col6a6 and Itga6), stem cell maintenance (Nanog and Lif), and cell fate specification, such as Sox9 and Fgfr1. The latter two were also confirmed by IHC (Figures 6C–6E and 6I–6K).
Our data indicate that FGFR1 signaling can drive initiation of LADC in CGRPPOS cells of RP-Fgfr1 mice, whereas they also show that both SCLC and LADC originating from CGRPPOS cells do not tolerate persistent FGFR1 signaling.
FGFR1K656E Promotes the Development of Tumors in the Nasal Cavity of RP-Fgfr1 Mice
Besides lung tumors, RP-Fgfr1 mice injected with Ad5-CMV-cre, Ad5-CGRP-Cre, and Ad5-K14-Cre viruses also developed nasal tumors.
Strikingly, 64% of RP-Fgfr1 mice (14 out of 22) injected with Ad5-CMV-Cre developed nasal tumors compared to only 15% (3 out of 19) of RP mice (Figure S4; Tables S1 and S2). Nasal tumors in both RP and RP-Fgfr1 mice expressed synaptophysin (SYP), a recognized biomarker of NE tumors (Figures S4C and S4H). However, the nasal tumors from RP-Fgfr1 mice showed a great degree of pleomorphism containing numerous single and multinucleated giant bizarre cells (Figures S4A, S4B, S4F, and S4G). In general, the nasal tumors of RP-Fgfr1 mice were invasive and often grew into surrounding tissues and organs such as orbit, sinus, and even brain, whereas nasal tumors of RP mice were smaller and less invasive. As expected, nasal tumors of RP-Fgfr1 mice expressed high levels of FGFR1K656E (Figures S4D and S4I); SOX2 expression was high only in the early nasal lesions of RP mice (Figures S4E and S4J).
Nasal tumors were found in 71% of RP-Fgfr1 mice (19 out of 27) upon Ad5-CGRP-Cre injection (Tables S3 and S4) and not in RP mice (n = 20). However, nasal lesions originating from CGRPPOS cells showed a later onset (Figure S4P), permitting development of lung tumors within the same time window. In line with Ad5-CMV-Cre-injected mice, nasal tumors originating from Ad5-CGRP-Cre-injected RP-Fgfr1 mice appeared morphologically very similar (Figures S4K and S4L) showing NE differentiation as indicated by the consistent expression of SYN (Figure S4M), FGFR1K656E expression (Figure S4N), and marginal SOX2 expression (Figure S4O).
RP-Fgfr1 mice injected with Ad5-K14-Cre developed nasal tumors with a penetrance of 67%. The number of nasal samples collected from Ad5-K14-Cre-injected RP mice do not allow for a reliable estimation of the % of nasal tumors, which we believe is overestimated due to the low number of samples (2 out of 7) (Tables S5 and S6). Nasal tumors showed consistent NE differentiation: they expressed high levels of CGRP and SYN (data not shown).
Taken together, our data indicate that FGFR1K656E expression promotes the transformation of nasal epithelium cells into neoplastic lesions with NE differentiation.
FGFR1K656E Is a Context-Dependent Oncogene
RP mice injected with recombinant adenoviruses driving Cre expression, either ubiquitously (Ad5-CMV-Cre) or specifically in CGRPPOS, K14POS, SPCPOS, and CC10POS lung cells, consistently and exclusively developed NE lesions with the typical central SCLC as the most pronounced lesion, although the latency, penetrance, and number of lesions per mouse ranged from high (CMV) to low (CC10) (Figure 7; Tables S2, S4, S6, S8, S10, and S11). Data obtained by targeting CGRPPOS, SPCPOS, and CC10POS cells are in line with previous studies (Table S13). Expression of constitutively active FGFR1 dramatically affected the tumor phenotype of RP mice causing CGRPPOS cells to be much more refractory in developing SCLC, whereas K14POS cells emerged as effective SCLC cells of origin of RP-Fgfr1 mice (Figure 7; Tables S1, S3, S5, S7, S9, and S11).
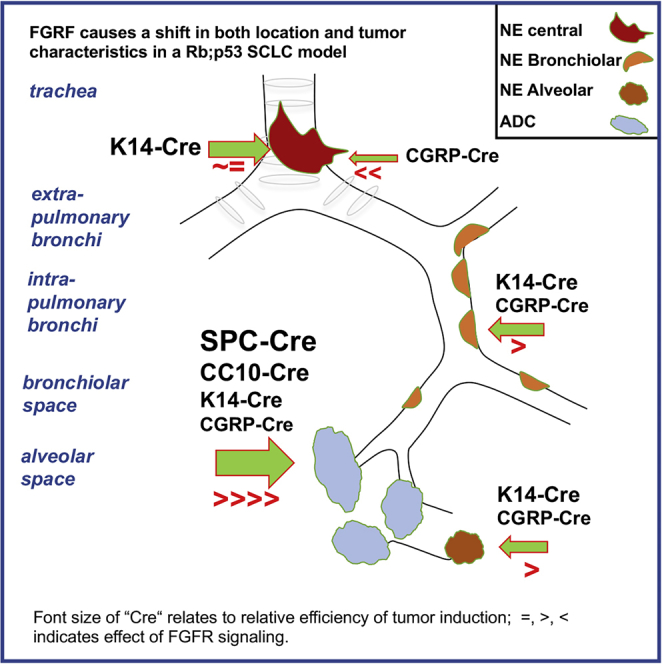
Figure 7.

FGFR1 Is a Context-Dependent Oncogene
Schematic representation of neoplastic lesions developed according to the targeted cell type by RP mice and RP-Fgfr1 mice: RP mice develop primarily typical central SCLC when CGRPPOS cells are targeted; RP mice injected with Ad5-CMV-Cre also develop BLs and ALs, but their contribution to the tumor burden is very marginal. In RP-Fgfr1 mice injected with any virus except Ad5-K14-Cre virus, the development of SCLC is inhibited. Peripheral NE lesions are promoted, especially when mice are injected with Ad5-CMV-Cre. RP-Fgfr1 mice also develop ADC. Ad5-SPC-Cre- and Ad5-CC10-Cre-injected mice develop ADC exclusively, whereas ADC is quite marginal in Ad5-CGRP-Cre- and Ad5-K14-Cre-injected mice.
RP-Fgfr1 mice developed peripheral low-grade bronchial and alveolar NE lesions most predominantly when Ad5-CMV-Cre was used to switch the conditional alleles and, to a lesser extent, when CGRPPOS or K14POS cells were targeted (Figure 7). In contrast, RP-Fgr1 mice developed LADC from any of the targeted cells: Ad5-SPC-Cre- or Ad5-CC10-Cre-injected mice developed LADC exclusively, whereas in Ad5-K14-Cre-injected mice, the LADC compartment was rather marginal, giving predominant rise to central and peripheral NE lesions (Figure 7).
Taken together, our data show that FGFR1 activation can promote different degrees of NE lesions and LADC depending on the cell of origin. Most importantly, they indicate that FGFR1 activation can inhibit SCLC when it originates from CGRPPOS cells whereas it promotes SCLC when initiated from K14POS cells.
Discussion
Here, we have studied how FGFR signaling, often observed as a result of amplification of the FGFR1 gene in human SCLC, is associated with a distinct tumor phenotype and cell of origin. Therefore, we have generated a conditional dominant active allele of Fgfr1 inserted in the Col1A1 locus. Side-by-side comparison of mice carrying conditional Rb1 and Trp53 alleles (RP) with mice carrying on top the conditional mutant Fgfr1 allele (RP-Fgfr1) have shown how FGFR1 signaling can influence tumor development in a Rb1;Trp53-deficient background. By using a set of previously described Ad5-Cre viruses, we have targeted these genetic lesions to specific subsets of lung cells and monitored tumor development.
Neuroendocrine Lung Tumors Can Originate from Different Cells in Lung
Previously, we and others have reported that SCLC in mice can be induced by Cre-mediated inactivation of Rb1 and Trp53 in a variety of lung cells (Table S13). Targeting lung cells by using the ubiquitous CMV promoter appeared the most efficient approach, followed by switching CGRPPOS NE cells and SPCPOS cells, respectively (Sutherland et al., 2011). This, together with data of other investigators, who did not observe SCLC upon switching SPCPOS cells (Gazdar et al., 2017), has led to the view that lung NE cells represent the predominant cell of origin of SCLC.
Our results reveal that SCLC can originate from more cell lineages than previously assumed (Figure 7) e.g., inactivation of Rb1 and Trp53 in K14POS cells enables development of SCLC with a similar penetrance as found for CGRPPOS cells.
Moreover, we observe a diversity of NE lesions when tumors are induced by a generally expressed CMV-Cre virus. This results in “typical” central lesions, most reminiscent of human SCLC and peripheral lesions that are more benign and found in the BL and AL space. BLs and ALs appear less aggressive with no evidence of local dissemination, although this might change as a result of additional lesions (Yang et al., 2018). These peripheral lesions are much more efficiently induced by the CMV-Cre virus than by any of the other cell-type-specific Cre viruses. These observations align with those made for RP mice overexpressing Mycl (RPM) (Böttger et al., 2019) and RP mice with the additional inactivation of Rbl2 (Yang et al., 2018).
Finally, comparing the expression profiles of these peripheral lesions that are more abundant in RPM mice with typical central SCLC tumors showed distinct differences as illustrated by principal component analysis (Figure 3A). We conclude that NE lesions induced in the bronchiolar and alveolar space represent closely related entities that significantly differ from more centrally located SCLC. Concomitant Fgfr1 signaling enhances the penetrance and the number per mouse of these NE peripheral lesions while retaining their more benign in situ character. Apparently, it requires additional driver lesions such as p130 loss or Nfib overexpression to transform these peripheral tumors in more aggressive metastasizing tumors (Yang et al., 2018).
Overexpression of FGFR K656E Promotes Low-Grade NE Lesions
We observe more BLs and ALs when RP mice express FGFR1K656E although there appears selection against its continued expression. The observation that these peripheral lesions are most predominant when CMV-Cre virus is used while virtually absent when Cre is delivered in AT2 or Club cells indicates that cells not previously identified serve as the cells of origin of these tumors.
As indicated by the gene expression analysis, the unique features of these tumors are further substantiated by distinct expression markers that distinguish them from the typical central SCLC lesions. For instance, both BLs and ALs of RPM and RP-Fgfr1 mice express high levels of ALDH1 and EGFR. A high level of ALDH1 has been previously related to stem cell properties in LADC (Huang et al., 2013, Ji et al., 2014, Jiang et al., 2009). In line with this observation, RP-Fgfr1 mice showed expression of ALDH1, also in LADC (Figure 3F), whereas even higher levels of ALDH1 are seen in BLs and ALs (Figure 3F), suggesting that an “ALDH1high cancer stem cell population” likely gives rise to these low-grade NE lesions. We also observed high levels of EGFR in ALs and BLs: EGFR has been reported as amplified in human bronchial carcinoids (Voortman et al., 2010), suggesting that NE peripheral lesions we describe here resemble human carcinoids.
The observation that these peripheral lesions gradually lose FGFR1K656E expression suggests that FGFR1 plays a role during initiation of these tumors, but subsequently, its expression is poorly tolerated. This is reminiscent of the situation observed in human LADC carrying EGFR mutations in which treatment with EGFR inhibitors can result in transdifferentiation to SCLC that shows impaired MAPK signaling (Niederst et al., 2015). We have previously observed that forced expression of mutant KRAS is poorly tolerated by SCLC cells, resulting in loss of NE marker profile and acquisition of NSCLC phenotype (Calbo et al., 2011). The apparent mutual exclusiveness of sustained MAPK signaling with NE features is particularly intriguing in view of the notion that a fraction of the SCLC show amplification of the FGFR1 gene. As we will argue below, this appears dictated by the cell of origin and both applies to SCLC as well as LADC.
Overexpression of FGFR K656E Impairs SCLC Initiated from NE CGRPPOS while Promoting SCLC Initiated from K14POS Epithelial Cells
Whereas FGFR1K656E promoted the initiation of peripheral lesions, it strongly impaired typical central SCLC development when induced by either CMV-Cre or CGRP-Cre viruses. Because we used time-matched mice for these comparisons, this cannot be ascribed to differences in mice survival. As observed for SCLC cell lines, which show intolerance to constitutive MAPK signaling, CGRPPOS cells apparently do not tolerate FGFR1 signaling well. Central lesions arising following Ad5-CGRP-Cre instillation of RP-Fgfr1 mice did not show expression of FGFR1K656E (Figures 6B and 6C). Intriguingly, when tumors were induced by K14-Cre in either RP or RP-Fgfr1 mice, SCLC was found in a substantial percentage of the mice. In this case, many of the tumors in RP-Fgfr1 mice, but not in RP mice, expressed FGFR1K656E, indicating that K14POS cells of origin of SCLC might actually benefit from FGFR1K656E expression. Therefore, central lesions can have distinct cells of origin with a differential tolerance to FGFR1 signaling. This might imply that human SCLC samples showing FGFR1 overexpression preferentially originate from a K14POS, CGRPNEG cell.
FGFR1 Catalyzes Development of LADC from a Wide Variety of Lung Cells
Given the differential effect of FGFR1K656E expression on the induction of SCLC, we asked whether similar preferences also apply to the cell of origin of LADC. Therefore, we monitored LADC development in RP and RP-Fgfr1 mice using the complete set of Cre viruses. In RP mice, LADC was virtually absent. Even Ad5-SPC-Cre virus failed to induce LADC. In contrast, LADC was found at high incidence in RP-Fgfr1 mice irrespective of the virus used, although CMV, SPC, and CC10-Cre viruses were the most effective. Interestingly, CGRP-Cre and K14-Cre delivery also gave rise to LADC. LADC induced by CGRP-Cre virus did not express FGFR1K656E, similarly to what we observed for SCLC, although its activation was clearly required for LADC development. Apparently, CGRPPOS cells do not tolerate continuous FGFR1K656E expression, this in contrast to LADC induced by SPC, CC10, and K14-Cre viruses. A principal component analysis (PC1, Figure 6A) shows close resemblance of LADCs induced by either SPC-Cre or CGRP-Cre (PC1), whereas CGRP-Cre-induced LADC shared a number of markers specifically with CGRP-Cre-induced SCLC (PC2). This indicates that the cell of origin of LADC also imposes distinct expression requirements for tumor outgrowth.
RP and RP-Fgfr1 Mice Develop Tumors in the Nasal Cavity
All viruses except SPC-Cre and CC10-Cre induced NE tumors in the nasal cavity. This phenotype was much more pronounced in RP-Fgfr1 mice than in RP mice using either CMV-Cre, CGRP-Cre, or K14-Cre viruses, indicating that FGFR1K656E acts as a collaborating oncogenic lesion in the induction of NE lesions in the nasal cavity. Because this was a confounding factor in our analysis, we took care to always use age- and infection time-matched mice in comparing lung tumor incidence and phenotype to exclude the modifying influence of independent tumors arising in these mice as much as possible. This was also done for mice in which both SCLC and LADC were found concurrently.
Implications for Human SCLC and LADC
Our study illustrates that a larger variety of lung cell types can give rise to both NE tumors as well as LADC (Figure 7). Earlier, we have shown that lung squamous cell carcinoma can arise from most cell types in lung, resulting in tumors that are indistinguishable with respect to their expression pattern (Ferone et al., 2016). The most remarkable observation reported here is the differential effect of FGFR1 signaling on tumor development: depending on the cell of origin, it can either promote or impair SCLC development. Even when promoting the initiation of NE lesions, it appears to have an adverse effect in later phases of tumor development. This effect was observed for the peripheral NE BLs and ALs and was likely also responsible for the impairment of typical central SCLC originating from CGRPPOS cells. In contrast, SCLC induced by K14-Cre did not show any adverse effect of continued FGFR1 expression. Interestingly, LADC induced in RP-Fgfr1 mice by CGRP-Cre virus did show very marginal FGFR1K656E expression, although FGFR1K656E was evidently required for its induction. This suggests that a subset of lung cells do not tolerate well sustained mutant FGFR1 expression at specific stages of tumor development. Because we used a constitutive active form of FGFR1, we cannot exclude that, upon FGFR1 amplification as seen in a subset of human SCLC, FGFR signaling is less pronounced and therefore not associated with some of the adverse effects of FGFR1K656E expression we observed here. However, our observations indicate that the effects of inhibiting FGFR1 signaling in both SCLC and LADC are hard to predict, because the selective advantage conferred by aberrant FGFR signaling might be confined to the early stage of tumorigenesis. It will therefore be important to explore whether some of the markers that can distinguish these tumor subsets in the mouse have a counterpart in human SCLC and LADC and can be used in the future to assess whether they can serve as predictors for response to therapies.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CGRP | Sigma-Aldrich | Cat# C8198; RRID: AB_259091 |
| Rabbit monoclonal anti-CDH1 | Cell Signaling Technology | Cat# 3195; RRID: AB_2291471 |
| Rabbit monoclonal anti-FGFR1 | Cell Signaling Technology | Cat# 9740; RRID: AB_11178519 |
| Rabbit polyclonal anti-GFP | Abcam | Cat# ab6556; RRID: AB_305564 |
| Rabbit polyclonal anti-ALDH1A1 | Abcam | Cat# ab23375; RRID: AB_2224009 |
| Rabbit monoclonal anti-EGFR | Abcam | Cat# ab52894; RRID: AB_869579 |
| Rabbit polyclonal anti-SOX2 | Millipore | Cat# AB5603; RRID: AB_2286686 |
| Rabbit polyclonal anti-SOX9 | Millipore | Cat# AB5535; RRID: AB_2239761 |
| Mouse monoclonal anti-TTF1 | Agilent | Cat# M357501-2; RRID: AB_2801260 |
| Rabbit monoclonal anti-SYP | Abcam | Cat# ab32127; RRID: AB_2286949 |
| Bacterial and virus strains | ||
| Ad5-CMV-Cre | Viral Vector Core Facility, University of IOWA Health Care | N/A |
| Ad5-CGRP-Cre | Viral Vector Core Facility, University of IOWA Health Care | N/A |
| Ad5-K14-Cre | Viral Vector Core Facility, University of IOWA Health Care | N/A |
| Ad5-SPC-Cre | Viral Vector Core Facility, University of IOWA Health Care | N/A |
| Ad5-CC10-Cre | Viral Vector Core Facility, University of IOWA Health Care | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cyclosporin A | Sigma-Aldrich | Cat# 30024 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE132759 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Rb1flox/flox;Trp53flox/flox | Meuwissen et al., 2003 | N/A |
| Mouse: Rb1flox/flox;Trp53flox/flox; CAG < Lox66Mycl-LucLox71 > | Huijbers et al., 2014 | N/A |
| Mouse: Rb1flox/flox;Trp53flox/flox; LSL-Fgfr1K656E | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: LSL-FGFR1K656E | Ferone et al., 2016 | N/A |
| Software and Algorithms | ||
| STAR (STAR_2.5.0c) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq-count | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| DESeq2 | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R (version 3.6.0) | https://www.r-project.org/contributors.html | https://www.r-project.org |
| AxioVision 4 software | Carl Zeiss Vision | N/A |
| GraphPad Prism, versions 7 | GraphPad Software | https://www.graphpad.com |
| Other | ||
| Zeiss Axioskop2 Plus microscope | Carl Zeiss Microscopy | N/A |
| Zeiss AxioCam HRc digital camera | Carl Zeiss Vision | N/A |
| HiSeq 2000 Sequencing System | Illumina | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anton Berns (a.berns@nki.nl).
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Experimental Model and Subject Details
Mice
All experiments involving animals were performed in accordance with Dutch and European regulations on care and protection of laboratory animals and have been approved by the local animal experiment committee at Netherlands Cancer Institute, DEC NKI (OZP ID 14016). All mice were maintained on an FVB background (backcrossed from strains generated from 129 Ola ESCs) and housed under standard condition of feeding, light and temperature with free access to food and water. Mice aged eight to ten weeks were used for experiments. Male and female mice were represented equally in the experimental cohorts and were group-housed (maximum 4 mice per cage) in individually ventilated cages (IVC) with standard enrichment.
We introduced LSL-Fgfr1K656E allele directly into ES cells derived from Rb1flox/flox;Trp53flox/flox mice, previously generated in our lab (Meuwissen et al., 2003), by performing Flp-recombinase mediated cassette exchange in the Col1A1 locus.
Method Details
Intratracheal Adenovirus instillation
Eight to ten weeks old mice were first treated with Cyclosporine A in drinking water (100 μg/ml) for immunosuppression, 1 week prior and 2 weeks following adenoviral injection. The day of virus injection mice were anesthetized with ketamine/sedazine and intratracheally injected with 20 μL of 1 × 1010 pfu/mL of purified adenovirus (Ad5-CMV-Cre, Ad5-CGRP-Cre, Ad5-K14-Cre, Ad5-SPC-Cre and Ad5-CC10-Cre; Viral Vector Core Facility, University of IOWA Health Care). Mice were monitored daily for signs of illness and culled upon respiratory distress or excessive weight loss (> 20% of initial weight).
Histology and Immunohistochemistry
For histological analysis, lungs were inflated and fixed for 24 h with ethanol–acetic acid–formalin (EAF). Fixed tissues were subsequently dehydrated, embedded in paraffin and sections of 2-4 μm were prepared, and stained with hematoxylin and eosin (H&E) for subsequent histopathological analyses. For IHC, tissue sections were rehydrated, blocked in BSA containing PBS, and sequentially incubated with specific primary antibodies and with biotinylated secondary antibodies (DAKO).
The following primary antibodies were applied: CGRP (Sigma, C8198),
E-cadherin/CDH1 (Cell signaling, 3195), FGFR1 (Cell signaling, 9740), GFP (Abcam, ab6556), ALDH1A1 (Abcam, ab23375), EGFR (Abcam, ab52894), SOX2 (Millipore, AB5603), SOX9 (Millipore AB5535), TTF1 (Agilent M357501-2), Synaptophysin/SYP (Abcam, ab32127).
The sections were reviewed with a Zeiss Axioskop2 Plus microscope (Carl Zeiss Microscopy, Jena, Germany) and images were captured with a Zeiss AxioCam HRc digital camera and processed with AxioVision 4 software (both from Carl Zeiss Vision, München, Germany).
RNA sequencing
RNA was isolated from laser capture micro-dissected paraffin sections of distinct lung lesions. 75bp paired-end RNA sequencing was performed on an Illumina HiSeq2000. Sequence reads were mapped to the mouse reference genome (Mus musculus, GRCm38) using STAR (STAR_2.5.0c) (Dobin et al., 2013). Mapped sequence reads were counted using HTSeq-count (Anders et al., 2015). Genes with False Discovery Rate (FDR) for differential expression lower than 0.05 were considered significant. Principal component analysis was performed using all genes in the dataset. To generate heatmaps related to the principle components the genes with an absolute rotation values four times higher than the standard deviation were selected.
Quantification and Statistical Analysis
Normalization and statistical analyses
Statistical analysis of the differential expression of genes was performed using DESeq2 (Love et al., 2014). All downstream analyses were performed in R (version 3.6.0) using the Bioconductor framework. The statistical methods used as well as the p values defining significance are stated in all figure legends referencing this data.
Survival analysis
Kaplan-Meier survival curves were analyzed using the log-rank test. All p values were calculated using a nonparametric Mann-Whitney test (statistical analyses were performed by GraphPad Prism, version 7).
Data visualization
Heatmaps were generated using the R-function heatmap.3 (https://github.com/obigriffith/biostar-tutorials/blob/master/Heatmaps/heatmap.3.R) using the z-score generated from RNA sequence read count data. Clustering was based on euclidean distance and complete linkage. PCA plots, GSEA-plots and volcano plots were generated in R (version 3.6.0). MAplots were generated using the ‘plotMA’ function from the DESeq2 R-package.
Expression plots were made using GraphPad Prism (version 7).
Data and Code Availability
The accession number for the sequencing data reported in this paper is Gene Expression Omnibus (GEO): GSE132759.
Acknowledgments
We thank the division of animal pathology Ellen Riem, Joost van Ooij, Lex de Vrije, and Jelrik van der Meer for producing high-quality histopathologic material and the animal facility for its support in maintaining the mice. This study was supported by European Research Council (319661 COMBATCANCER) and the Queen Wilhelmina Prize from the Dutch Cancer Society to A.B.
Author Contributions
G.F. conceived the experiments and conducted most of them, coordinated the project, and wrote the paper. J.-Y.S. analyzed tumors and scored IHC staining. O.K. performed RNA-seq data analysis. J.v.d.V. and M.C. gave technical support for intratracheal injections. E.A.S. scored IHC staining. D.J.A. coordinated RNA sequencing. D.P. provided input in manuscript writing. A.B. supervised the whole project.
Declaration of Interests
The authors declare no competing interests.
Published: March 17, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.052.
Supplemental Information
References
- Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni M.G., Woolner L.B., Bernatz P.E. Atypical carcinoid tumors of the lung. J. Thorac. Cardiovasc. Surg. 1972;64:413–421. [PubMed] [Google Scholar]
- Böttger F., Semenova E.A., Song J.Y., Ferone G., van der Vliet J., Cozijnsen M., Bhaskaran R., Bombardelli L., Piersma S.R., Pham T.V. Tumor heterogeneity underlies differential cisplatin sensitivity in mouse models of Small-Cell Lung Cancer. Cell Rep. 2019;27:3345–3358.e4. doi: 10.1016/j.celrep.2019.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calbo J., van Montfort E., Proost N., van Drunen E., Beverloo H.B., Meuwissen R., Berns A. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19:244–256. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- Cui M., Augert A., Rongione M., Conkrite K., Parazzoli S., Nikitin A.Y., Ingolia N., MacPherson D. PTEN is a potent suppressor of small cell lung cancer. Mol. Cancer Res. 2014;12:654–659. doi: 10.1158/1541-7786.MCR-13-0554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny S.K., Yang D., Chuang C.H., Brady J.J., Lim J.S., Grüner B.M., Chiou S.H., Schep A.N., Baral J., Hamard C. Nfib Promotes Metastasis through a Widespread Increase in Chromatin Accessibility. Cell. 2016;166:328–342. doi: 10.1016/j.cell.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferone G., Song J.Y., Sutherland K.D., Bhaskaran R., Monkhorst K., Lambooij J.P., Proost N., Gargiulo G., Berns A. SOX2 Is the Determining Oncogenic Switch in Promoting Lung Squamous Cell Carcinoma from Different Cells of Origin. Cancer Cell. 2016;30:519–532. doi: 10.1016/j.ccell.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A.F., Brambilla E. Preneoplasia of lung cancer. Cancer Biomark. 2010;9:385–396. doi: 10.3233/CBM-2011-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar A.F., Bunn P.A., Minna J.D. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat. Rev. Cancer. 2017;17:765. doi: 10.1038/nrc.2017.106. [DOI] [PubMed] [Google Scholar]
- George J., Lim J.S., Jang S.J., Cun Y., Ozretić L., Kong G., Leenders F., Lu X., Fernández-Cuesta L., Bosco G. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C.P., Tsai M.F., Chang T.H., Tang W.C., Chen S.Y., Lai H.H., Lin T.Y., Yang J.C., Yang P.C., Shih J.Y., Lin S.B. ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 2013;328:144–151. doi: 10.1016/j.canlet.2012.08.021. [DOI] [PubMed] [Google Scholar]
- Huijbers I.J., Bin Ali R., Pritchard C., Cozijnsen M., Kwon M.C., Proost N., Song J.Y., de Vries H., Badhai J., Sutherland K. Rapid target gene validation in complex cancer mouse models using re-derived embryonic stem cells. EMBO Mol. Med. 2014;6:212–225. doi: 10.1002/emmm.201303297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakawa R., Takenaka M., Kohno T., Shimada Y., Totoki Y., Shibata T., Tsuta K., Nishikawa R., Noguchi M., Sato-Otsubo A. Genome-wide identification of genes with amplification and/or fusion in small cell lung cancer. Genes Chromosomes Cancer. 2013;52:802–816. doi: 10.1002/gcc.22076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y., Zheng M., Ye S., Chen J., Chen Y. PTEN and Ki67 expression is associated with clinicopathologic features of non-small cell lung cancer. J. Biomed. Res. 2014;28:462–467. doi: 10.7555/JBR.27.20130084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F., Qiu Q., Khanna A., Todd N.W., Deepak J., Xing L., Wang H., Liu Z., Su Y., Stass S.A., Katz R.L. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009;7:330–338. doi: 10.1158/1541-7786.MCR-08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazanjian A., Wallis D., Au N., Nigam R., Venken K.J., Cagle P.T., Dickey B.F., Bellen H.J., Gilks C.B., Grimes H.L. Growth factor independence-1 is expressed in primary human neuroendocrine lung carcinomas and mediates the differentiation of murine pulmonary neuroendocrine cells. Cancer Res. 2004;64:6874–6882. doi: 10.1158/0008-5472.CAN-04-0633. [DOI] [PubMed] [Google Scholar]
- Koutsami M.K., Doussis-Anagnostopoulou I., Papavassiliou A.G., Gorgoulis V.G. Genetic and molecular coordinates of neuroendocrine lung tumors, with emphasis on small-cell lung carcinomas. Mol. Med. 2002;8:419–436. [PMC free article] [PubMed] [Google Scholar]
- Kwon M.C., Proost N., Song J.Y., Sutherland K.D., Zevenhoven J., Berns A. Paracrine signaling between tumor subclones of mouse SCLC: a critical role of ETS transcription factor Pea3 in facilitating metastasis. Genes Dev. 2015;29:1587–1592. doi: 10.1101/gad.262998.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden D.G., Papagiannakopoulos T., Taylor-Weiner A., Stewart C., Carter S.L., Cibulskis K., Bhutkar A., McKenna A., Dooley A., Vernon A. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014;156:1298–1311. doi: 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R., Linn S.C., Linnoila R.I., Zevenhoven J., Mooi W.J., Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Mills S.E., Cooper P.H., Walker A.N., Kron I.L. Atypical carcinoid tumor of the lung. A clinicopathologic study of 17 cases. Am. J. Surg. Pathol. 1982;6:643–654. doi: 10.1097/00000478-198210000-00006. [DOI] [PubMed] [Google Scholar]
- Niederst M.J., Sequist L.V., Poirier J.T., Mermel C.H., Lockerman E.L., Garcia A.R., Katayama R., Costa C., Ross K.N., Moran T. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo O.E., Arcaro A., Salerno G., Tetley T.D., Valovka T., Gout I., Seckl M.J. Novel cross talk between MEK and S6K2 in FGF-2 induced proliferation of SCLC cells. Oncogene. 2001;20:7658–7667. doi: 10.1038/sj.onc.1204994. [DOI] [PubMed] [Google Scholar]
- Pardo O.E., Lesay A., Arcaro A., Lopes R., Ng B.L., Warne P.H., McNeish I.A., Tetley T.D., Lemoine N.R., Mehmet H. Fibroblast growth factor 2-mediated translational control of IAPs blocks mitochondrial release of Smac/DIABLO and apoptosis in small cell lung cancer cells. Mol. Cell. Biol. 2003;23:7600–7610. doi: 10.1128/MCB.23.21.7600-7610.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo O.E., Wellbrock C., Khanzada U.K., Aubert M., Arozarena I., Davidson S., Bowen F., Parker P.J., Filonenko V.V., Gout I.T. FGF-2 protects small cell lung cancer cells from apoptosis through a complex involving PKCepsilon, B-Raf and S6K2. EMBO J. 2006;25:3078–3088. doi: 10.1038/sj.emboj.7601198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo O.E., Latigo J., Jeffery R.E., Nye E., Poulsom R., Spencer-Dene B., Lemoine N.R., Stamp G.W., Aboagye E.O., Seckl M.J. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009;69:8645–8651. doi: 10.1158/0008-5472.CAN-09-1576. [DOI] [PubMed] [Google Scholar]
- Park K.S., Liang M.C., Raiser D.M., Zamponi R., Roach R.R., Curtis S.J., Walton Z., Schaffer B.E., Roake C.M., Zmoos A.F. Characterization of the cell of origin for small cell lung cancer. Cell Cycle. 2011;10:2806–2815. doi: 10.4161/cc.10.16.17012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peifer M., Fernández-Cuesta L., Sos M.L., George J., Seidel D., Kasper L.H., Plenker D., Leenders F., Sun R., Zander T. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012;44:1104–1110. doi: 10.1038/ng.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi R.K., Weber E., McMahon M., Williams J.R., Baylin S., Mal A., Harter M.L., Dillehay L.E., Claudio P.P., Giordano A. Activated Raf-1 causes growth arrest in human small cell lung cancer cells. J. Clin. Invest. 1998;101:153–159. doi: 10.1172/JCI831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi R.K., Thiagalingam A., Weber E., McMahon M., Nelkin B.D., Mabry M. Raf-1 causes growth suppression and alteration of neuroendocrine markers in DMS53 human small-cell lung cancer cells. Am. J. Respir. Cell Mol. Biol. 1999;20:543–549. doi: 10.1165/ajrcmb.20.4.3406. [DOI] [PubMed] [Google Scholar]
- Rizvi S.M., Goodwill J., Lim E., Yap Y.K., Wells A.U., Hansell D.M., Davis P., Selim A.G., Goldstraw P., Nicholson A.G. The frequency of neuroendocrine cell hyperplasia in patients with pulmonary neuroendocrine tumours and non-neuroendocrine cell carcinomas. Histopathology. 2009;55:332–337. doi: 10.1111/j.1365-2559.2009.03371.x. [DOI] [PubMed] [Google Scholar]
- Rudin C.M., Durinck S., Stawiski E.W., Poirier J.T., Modrusan Z., Shames D.S., Bergbower E.A., Guan Y., Shin J., Guillory J. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012;44:1111–1116. doi: 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer B.E., Park K.S., Yiu G., Conklin J.F., Lin C., Burkhart D.L., Karnezis A.N., Sweet-Cordero E.A., Sage J. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010;70:3877–3883. doi: 10.1158/0008-5472.CAN-09-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E.A., Kwon M.C., Monkhorst K., Song J.Y., Bhaskaran R., Krijgsman O., Kuilman T., Peters D., Buikhuisen W.A., Smit E.F. Transcription Factor NFIB Is a Driver of Small Cell Lung Cancer Progression in Mice and Marks Metastatic Disease in Patients. Cell Rep. 2016;16:631–643. doi: 10.1016/j.celrep.2016.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriuranpong V., Borges M.W., Ravi R.K., Arnold D.R., Nelkin B.D., Baylin S.B., Ball D.W. Notch signaling induces cell cycle arrest in small cell lung cancer cells. Cancer Res. 2001;61:3200–3205. [PubMed] [Google Scholar]
- Sutherland K.D., Berns A. Cell of origin of lung cancer. Mol. Oncol. 2010;4:397–403. doi: 10.1016/j.molonc.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland K.D., Proost N., Brouns I., Adriaensen D., Song J.Y., Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–764. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- Tatematsu A., Shimizu J., Murakami Y., Horio Y., Nakamura S., Hida T., Mitsudomi T., Yatabe Y. Epidermal growth factor receptor mutations in small cell lung cancer. Clin. Cancer Res. 2008;14:6092–6096. doi: 10.1158/1078-0432.CCR-08-0332. [DOI] [PubMed] [Google Scholar]
- Travis W.D. Advances in neuroendocrine lung tumors. Ann. Oncol. 2010;21(Suppl 7):vii65–vii71. doi: 10.1093/annonc/mdq380. [DOI] [PubMed] [Google Scholar]
- Voortman J., Lee J.H., Killian J.K., Suuriniemi M., Wang Y., Lucchi M., Smith W.I., Jr., Meltzer P., Wang Y., Giaccone G. Array comparative genomic hybridization-based characterization of genetic alterations in pulmonary neuroendocrine tumors. Proc. Natl. Acad. Sci. USA. 2010;107:13040–13045. doi: 10.1073/pnas.1008132107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Denny S.K., Greenside P.G., Chaikovsky A.C., Brady J.J., Ouadah Y., Granja J.M., Jahchan N.S., Lim J.S., Kwok S. Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov. 2018;8:1316–1331. doi: 10.1158/2159-8290.CD-17-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported in this paper is Gene Expression Omnibus (GEO): GSE132759.