

Graphical abstract
Abbreviations: PRR, pattern recognition receptor; PAMP, pathogen-associated molecular pattern; DAMP, danger-associated molecular pattern; NLRP3, NOD-, LRR- and pyrin domain-containing 3; TLR, toll-like receptor; CLR, C-type lectin receptor; NLR, NOD-like receptor; RIG-I, retinoic acid-inducible gene 1; RLR, retinoic acid-inducible gene 1-like receptor; CARD, caspase activation and recruitment domain; MAVS, mitochondrial antiviral signaling protein; TNFα, tumor necrosis factor α; TRAF, TNF receptor-associated factor; NF-κB, nuclear factor-kappaB; MAPK, mitogen-activated protein kinase; IRF, interferon regulatory factor; IFN, interferon; DENV, dengue virus; HCV, hepatitis C virus; mtDNA, mitochondrial DNA; ROS, reactive oxygen species; cGAS, cyclic GMP–AMP synthase; STING, stimulator of interferon genes; Drp 1, dynamin-related protein 1; Mfn, mitofusin; OPA1, optic atrophy 1; vMIA, viral mitochondrion-localized inhibitor of apoptosis; SARS-CoV, severe acute respiratory syndrome-coronavirus; ORF, open reading frame; HPIV3, human parainfluenza virus type 3; RSV, respiratory syncytial virus; RIPK2, cytosolic receptor interacting serine/threonine kinase 2; KSHV, kaposi’s sarcoma-associated herpesvirus; JEV, Japanese encephalitis virus; CO, cytochrome C oxidase; PINK1, phosphatase and tensin homolog (PTEN)-induced kinase 1
Keywords: Virus, Mitochondria, Therapy, Immune
Abstract
Mitochondria have been recognized as ancient bacteria that contain evolutionary endosymbionts. Metabolic pathways and inflammatory signals interact within mitochondria in response to different stresses, such as viral infections. In this commentary, we address several interesting questions, including (1) how do mitochondrial machineries participate in immune responses; (2) how do mitochondria mediate antiviral immunity; (3) what mechanisms involved in mitochondrial machinery, including the downregulation of mitochondrial DNA (mtDNA), disturbances of mitochondrial dynamics, and the induction of mitophagy and regulation of apoptosis, have been adopted by viruses to evade antiviral immunity; (4) what mechanisms involve the regulation of mitochondrial machineries in antiviral therapeutics; and (5) what are the potential challenges and perspectives in developing mitochondria-targeting antiviral treatments? This commentary provides a comprehensive review of the roles and mechanisms of mitochondrial machineries in immunity, viral infections and related antiviral therapeutics.
1. Introduction
Viral infections induce antiviral immunity in human bodies. Many organelles in virus-infected cells operate in conjunction with activated immune effector cells to trigger optimal antiviral immune responses. While facing immune defenses, viruses develop mechanisms to antagonize the immune machinery to ensure the successful production of viral progenies. Diverse and complex mechanisms are involved in antiviral immunity in host cells and immune evasion by viruses. Among the organelles in virus-infected cells, mitochondria are crucial for triggering antiviral immunity and represent critical targets that can be manipulated by viruses to escape immune protection. In the following section, we address the roles and mechanisms of mitochondria in innate immunity against viral infection and discuss the strategies adopted by viruses to fight against antiviral immunity by modifying the function of the mitochondrial machinery. Evidence supporting the benefit of correcting dysfunctional mitochondria is based on the antiviral effects of some medications. The challenges and perspectives of targeting virus-modulated mitochondrial machineries are also discussed.
2. Viral infection and innate immunity
Viruses are obligatory intracellular organisms that require cellular organelles and machineries to replicate and produce viral progeny. However, the invasion of viruses alerts the immune system and elicits a response. To detect the signals of invading pathogens and sense the danger from intracellular damage, cells adopt an alarm system using a group of molecules called pattern recognition receptors (PRRs) [1]. PRRs are localized on the cell surface, in the endosome, or in the cytosol and function to detect pathogen-associated molecular patterns (PAMPs) present in microbial invaders and danger-associated molecular patterns (DAMPs) exposed upon the occurrence of damage to cells [1]. The compartmentalization of these PRRs provides spatial information for better microbial detection. The accumulation of these receptor-mediated signals leads to the development of optimal immunity against pathogens. PAMPs are important structural components necessary for the survival of microorganisms and appear to have been highly conserved along the course of evolution. For example, in bacteria, PAMPs are mainly found in the components of the cell wall. The recognition of PAMPs mainly depends on 20–40 PRR proteins in the innate immune system in mammals. Most opportunistic pathogens contain at least one PAMP within their structural range, and evolutionary pressure drives the human immune system to selectively recognize only the most prevalent and conserved PAMPs. Nevertheless, the recognition of only a limited number of PAMPs bears risks because some pathogens can evade immune detection through mechanisms such as losing PAMPs or structural modification of PAMPs [2]. To overcome these challenges, such defects can be compensated for by the development of more sophisticated recognition systems by the innate immune system [3]. Recently, a recognition system of homeostasis-altering molecular processes involving inflammasome components, such as NLRP3 (NOD-, LRR- and pyrin domain-containing 3) and pyrin that integrate signals from perturbations in cytoplasmic homeostasis has been shown to promote great flexibility in the innate immune system, enabling this system to sense evolutionarily novel infections [4].
There are four major groups of PRR protein families, including the Toll-like receptor (TLR), C-type lectin receptor (CLR), NOD-like receptor (NLR) and retinoic acid-inducible gene 1 (RIG-I)-like receptor (RLR) families. The importance of PRRs in viral infections has been demonstrated in RIG-I-deficient mice in which viral infections fail to induce a type I interferon (IFN) reaction [5]. RIG-I preserves several critical regions, including the N-terminal caspase activation and recruitment domains (CARDs), a DECH helicase, and a C-terminal domain. Together, these subunits sense signals and mediate downstream signal transduction. The crystal structures of RIG-I reveal that the CARD domain, which mediates CARD-CARD interactions, is sequestered by a helical domain inserted between the two helicase moieties in the inactive status. Once RIG-I binds the 5′-triphosphate double-stranded RNA (5′-ppp-dsRNA) of viruses through its C-terminal domain, the CARD domain is exposed leading to its release from the helicase domain and triggers downstream signaling [6].
Through the CARD-CARD interaction, RIG-I delivers a signal to a critical adaptor molecule residing in mitochondria. This adaptor is known as the mitochondrial antiviral signaling protein (MAVS), interferon-beta promoter stimulator 1 (IPS-1), virus-induced signaling adaptor (VISA) or CARDIF that has been identified by several groups using different strategies [7]. In the resting status, MAVS exists in the monomeric form, and there are multiple N-terminal truncated isoforms of MAVS required to prevent full-length MAVS from spontaneous aggregation and assembly into active prion-like filaments that propagate signaling cascades [7], [8]. Once activated by the RIG-I signal, MAVS recruits several ubiquitin E3 ligase tumor necrosis factor (TNF) receptor-associated factor (TRAF) proteins, including TRAF2, TRAF5, and TRAF6, via TRAF binding motifs. Then, the TRAF proteins recruit the nuclear factor-kappaB (NF-κB) essential modulator to the MAVS signaling complex, which induces the activation of inhibitor of kappaB (IκB) kinase and TNF receptor-associated factor family member-associated NF-κB-binding kinase 1 [9]. The posttranslational phosphorylation of MAVS at the conserved serine and threonine clusters in the active domains by IκB kinase and/or TNF receptor-associated factor family member-associated NF-κB-binding kinase 1 is required for MAVS to serve as a binding site for downstream signaling molecules [10]. The stimulation of MAVS results in the activation of downstream signaling pathways, such as NF-κB, mitogen-activated protein kinases (MAPKs) and interferon regulatory factors (IRFs), leading to intense immune responses and the production of IFNs, IFN-stimulated genes, proinflammatory cytokines, chemokines, and many inflammation-associated mediators.
3. Mitochondria regulate metabolism and immunity
Mitochondria have their own genome, i.e., circular mitochondrial DNA (mtDNA), which contains nonmethylated CpG motifs. The mitochondrial genome shares similarities with bacterial DNA, and thus, mitochondria exhibit many different features of bacteria. Mitochondria are ubiquitously present in eukaryotic cells, and the number of mitochondria in a single cell ranges from one to a few thousand. Mitochondria are constantly in a dynamic status, and fission and fusion processes (discussed below) continually occur throughout the cell cycle, during the processes of generating young mitochondria and removing old mitochondria and even during the process of cell death. As predicted, mitochondria may change their shape, size and number throughout these processes. In addition, mitochondria can be transported from one location to a different location via microtubules. Accordingly, mitochondria perform both anabolic and catabolic processes requiring precise biogenesis, coordinated protein transportation, and protein expression originating from both the mitochondria and nuclear genomes. Expectedly, well-organized communication among mitochondria, the nucleus and the cytosol is necessary to maintain intact mitochondrial function and homeostasis. The breakdown of the balance in the mitochondrial machinery is highly related to the development of many human diseases, such as atherosclerosis, pulmonary hypertension, cancer, and autoinflammatory disorders [11], [12]. Mitochondrial dysfunction leads to the development of autoinflammatory diseases in humans through several mechanisms, such as the generation of reactive oxygen species (ROS) in mitochondria, the release of mtDNA, and the induction of autophagy, resulting in the activation of the inflammasome pathway [12].
Accumulating studies have indicated that in addition to being efficient cell powerhouses due to their roles in ATP production through oxidative phosphorylation and the synthesis of critical cellular components, such as nucleotides and amino acids, mitochondria can regulate the activation of the NLRP3 and NLRP6 inflammasomes, microbial- and host-derived metabolites, the glycolysis process and metabolism to affect immune responses [13]. Importantly, the activation of PRR programing sends signals to mitochondria, which, in turn, undergo a metabolic switch from oxidative phosphorylation to glycolysis to prepare the cells for defense against pathogens [14]. This effect also places mitochondria in a frontier position in microbial-triggered PRR activation and subsequent signal transduction. An earlier study demonstrated that when cocultured with nonactivated Kupffer cells, AH70 cells derived from a rat hepatoma cell line reduce mitochondrial energization, which is likely a secondary mechanism to the release of Kupffer cell-derived nitric oxide through membrane-to-membrane attachment between Kupffer cells and tumor cells [15]. Enzymes involved in metabolism are also in close approximation with immune regulators. According to Cao et al., methylcrotonyl-CoA carboxylase 1, which is an enzyme located in mitochondria, is constitutively associated with TRAF6 and interacts with MAVS to induce antiviral immunity by increasing type I IFN production [16]. Furthermore, many intermediates involved in the Krebs cycle, such as succinate, fumarate and citrate, participate in different processes that are highly related to immunity and inflammation in both innate and adaptive immune cells [17]. Specifically, the individual immune effector cells, such as granulocytes, dendritic cells, macrophages, T lymphocytes and B lymphocytes, all play distinctive roles in interactions with metabolic signaling and pathways [18]. Moreover, IFNs induce changes in the cellular metabolism with increased fatty acid oxidation and oxidative phosphorylation through an autocrine effect on the type 1 IFN receptor-dependent pathway [19].
4. Viruses mediate immune evasion by regulating the mitochondrial machinery
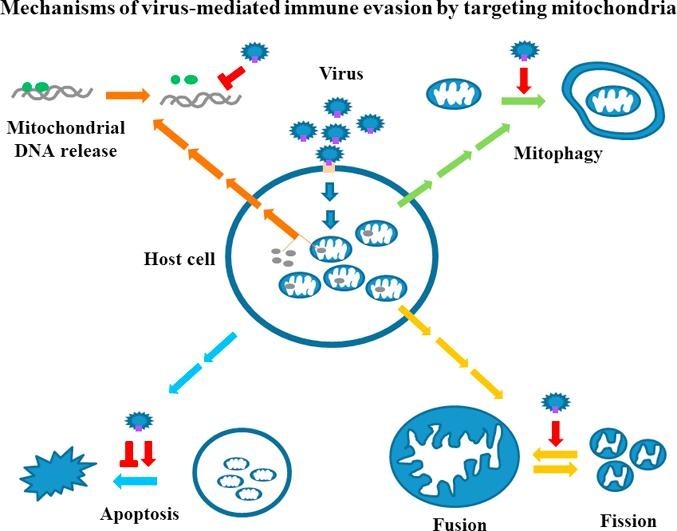
Due to the intensive immune response of host cells, viruses have developed many different strategies to evade suppression, such as targeting the antiviral cytokine IFN signaling pathways. A recent review listed the ten most common mechanisms adopted by viruses to antagonize IFN effects and achieve the goals of immune evasion [20]. The activation of the mitochondrial machinery may not necessarily be a viral target and may be a side effect of triggering antiviral immunity. In contrast, given the immune-regulatory roles of mitochondria in the defense against viral infections, targeting mitochondrial machineries is expected to be a strategy used by viruses to circumvent host immunity. In the following sections, we discuss several major categories of events occurring in the mitochondrial machinery in viral infections and address how viruses have developed mechanisms, such as targeting these events, to regulate mitochondria-associated antiviral immunity (Fig. 1 ).
Fig. 1.
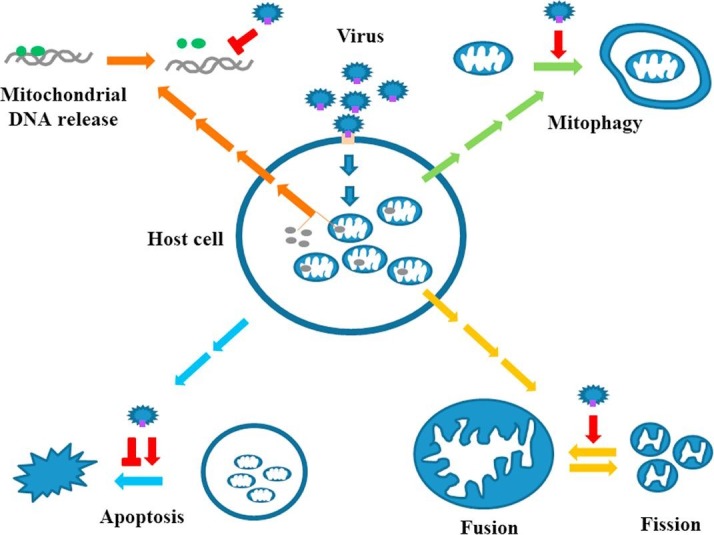
Regulation of mitochondrial machineries by viruses to antagonize immune defense. Viral infection triggers antiviral immunity in host cells leading to the production of antiviral IFNs. Mitochondria play crucial roles in antiviral immunity. In order to evade antiviral immunity, viruses target interfering mitochondrial function or causing mitochondrial elimination (mitophagy). Several major strategies are developed by viruses to achieve the goals. These include at least the disturbance of mitochondrial dynamics, the induction of mitophagy, the suppression of mtDNA copy number or interfering mtDNA-mediated effects as well as the regulation of apoptotic processes.  stands for inducing and
stands for inducing and  stands for suppressing.
stands for suppressing.
4.1. Downregulation of mitochondrial DNA (mtDNA)
The sources of DAMPs are most likely derived from the plasma membrane, nucleus, endoplasmic reticulum and other cytosolic organelles, such as mitochondria. Accordingly, the release of molecules or components from mitochondria, including mtDNA, into the cytosol or the extracellular milieu can potentially serve as potent DAMPs [17]. Certain PRRs are involved in the recognition of mtDNA in cell-type- and context-dependent manners. By engaging PRRs expressed in different cell types, mtDNA functions as a potent inducer of the antimicrobial immune response, IFN production and possibly consequential immunopathologies [21]. Many clinical disorders, such as HIV infection [22], trauma [23], sepsis and acute respiratory distress syndrome [24], [25], are associated with increased plasma mtDNA levels. In addition, the serum level of mtDNA can serve as a predictor of outcome in severe sepsis patients in the emergency room [24] and a useful biomarker of mortality in intensive care unit patients [25].
Dengue virus (DENV) infection induces the release of mtDNA into the cytosol, which upregulates TLR9 expression, binds TLR9 and translocates together with TLR9 into lysosomes to trigger downstream signaling events [26]. Interestingly, in response to the proinflammatory characteristics of mtDNA and to prevent further damage to tissues or organs, such as the lung, red blood cells can bind mtDNA through TLR9 to maintain homeostasis [27]. Another commonly recognized sensor of mtDNA is cyclic GMP–AMP synthase (cGAS), which shares similarities with the antiviral cytosolic double-stranded RNA sensor 2′-5′-oligoadenylate synthase (OAS1) [28]. The recognition of cytosolic mtDNA by cGAS leads to the production of the noncanonical cyclic dinucleotide 2′,5′ cGAMP, the activation of the stimulator of interferon genes (STING) and IRF3, and the induction of type I IFN [28]. The manipulation of the release of mtDNA or the mtDNA response has been generally adopted by invading viruses in the defense against antiviral immunity. Aguirre et al. demonstrated that the DENV non-structural (NS)2B protein recognizes and guides cGAS to the lysosome for degradation to suppress the mtDNA-mediated immune response [29]. In addition, DENV nonstructural proteins adopt different mechanisms to affect the mitochondrion-ER interaction and prevent the accumulation of DENV proteins in that organelle and attenuate mtDNA leakage into the cytoplasm [30]. Using a different strategy, Epstein-Barr virus (EBV)-encoded latent membrane protein 1 (EBV-LMP1) induces DNA methyltransferase 1 expression and promotes its mitochondrial translocation. Then, activated DNA methyltransferase 1 causes the activation of AKT signaling and the hypermethylation of the mtDNA D-loop region, leading to a reduction in oxidative phosphorylation complexes [31].
The mitochondrial single-stranded DNA binding protein (mtSSB) is critical for facilitating mtDNA replication in the dynamic status. The EBV B-ZIP transcription factor Zta, which is an immediate-early protein of the virus, binds many DNA sequences containing methylated CG dinucleotides with either 5-methylcytosine or 5-hydroxymethylcytosine present in one strand of the DNA sequence [32]. Zta can stimulate viral gene transcription and inhibit cell cycle proliferation. Weidmer et al. showed that Zta interacts with mtSSB, causing the translocation of mtSSB from the mitochondria to the nuclear compartment, attenuating mtDNA synthesis and reducing mtDNA genome copy number [33]. Full-length herpes simplex virus type 1 (HSV-1) UL12 localizes to the nucleus and is responsible for promoting viral genome generation. UL12.5 preserves alkaline DNase activity and is mainly localized to the mitochondrial matrix. UL12.5 can degrade the mitochondrial genome via its exonuclease activity, resulting in mtDNA depletion and the suppression of antiviral immunity [34]. Interestingly, Moren et al. observed significant mtDNA depletion and decrease of mitochondrial complex IV activity in human immunodeficiency virus (HIV-1)-infected U1 promonocytic cells but not HIV-1-infected lymphoid cells, although the mitochondrial apoptotic events measured by the voltage-dependent-anion-channel-1 content and caspase-9 levels were equally affected in these two cell lines [35]. Duguay et al. showed that although infection by HSV-1 results in the rapid elimination of mtDNA from host cells, this process does not appear to be required for the effective replication of viruses in cultured Vero kidney epithelial cells [36].
4.2. Disturbance of mitochondrial dynamics
Dynamic processes, namely, mitochondrial fission and mitochondrial fusion, occur constantly to maintain mitochondrial homeostasis. Mitochondrial fission is a process guided by dysfunctional daughter mitochondria to lysosomal digestion in order to properly adjust distribution of mitochondria and energy supply to daughter cells in mitosis and to maintain energetic state of mitochondria. Mitochondrial fusion requires the reorganization by mitochondrial exchange of mitochondrial components, mtDNA copies and matrix components of partially impaired mitochondrion from two individual mitochondria to form healthy and intact mitochondria. All events involved in both fission and fusion play roles in many aspects of immune-cell activation and are tightly regulated by critical molecules [17]. The GTPase dynamin-related protein 1 (Drp1) plays critical roles in the mitochondrial fission process. Drp1 is mainly localized to the cytosol and is recruited to the mitochondria to regulate the mitochondrial fission of both the outer and inner mitochondrial membranes when the appropriate signal is delivered.
The mitofusins (Mfn1 and Mfn2) and optic atrophy 1 (OPA1) proteins are three major players mediating the mitochondrial fusion process. Mitofusins localized in the outer mitochondrial membrane coordinate the fusion of the outer mitochondrial membrane. OPA1, which is a GTPase localized on the inner mitochondrial membrane, is responsible for the fusion of the inner mitochondrial membrane. A deficiency in either Mfn1 or Mfn2 causes cells to retain low levels of mitochondrial fusion, although the major cellular functions remain intact [37]. Cells lacking both Mfn1 and Mfn2 cannot undergo mitochondrial fusion and manifest severe cellular dysfunction, such as poor cell growth, widespread mitochondrial membrane potential heterogeneity, and decreased cellular respiration [37]. The introduction of OPA1 RNA interference results in an interruption of mitochondrial fusion and similar cellular defects [37]. Metabolic programming of the mitochondria can indeed regulate T-cell fate. Buck et al. demonstrated that by modulating mitochondrial cristae morphology, fusion in memory T lymphocytes configures electron transport chain complex associations toward oxidative phosphorylation and fatty acid oxidation; however, fission in activated effector T cells causes cristae expansion, reduces electron transport chain efficacy and promotes aerobic glycolysis [38].
To facilitate virus production by interfering with mitochondrial function, viruses target mitochondrial dynamics. Recent studies have suggested that the dysregulation of mitochondrial dynamics by the upregulation of Drp1 may favor viral production and play a role in the viral persistence of HCV infection [39]. Similarly, HBV infection induces Drp1 phosphorylation, causes mitochondrial translocation of Drp1 and modulates mitochondrial dynamics toward fission [40]. While both the full-length HBV genome and HBx alone are capable of achieving these effects, the deletion of HBx from the viral genome fails to show such effects [40]. The EBV latent membrane protein 2A activates the Notch signaling pathway, upregulates Drp1 expression and elevates mitochondrial fission in both gastric and breast cancer cells [41]. Studies with human cytomegalovirus infection reveal that disruption of the normal and well-organized reticular mitochondrial network induces dispersed and punctate mitochondria [42]. Molecular analyses have revealed that a 68-amino-acid antiapoptotic derivative of the immediate-early (alpha) antiapoptotic UL37x1 gene product viral mitochondrion-localized inhibitor of apoptosis (vMIA) localizes to the mitochondria after viral infection and mediates the observed effects [42]. The open reading frame 9b (ORF-9b) of severe acute respiratory syndrome-coronavirus (SARS-CoV) activates the ubiquitin pathway and induces Drp1 degradation in a proteasome-dependent manner. In addition, ORF-9b targets the MAVS signalosome and causes the degradation of MAVS, TRAF3, and TRAF6. Altogether, these effects suppress the IFN response [43]. The DENV NS2B3 protease complex cleaves Mfn1 and Mfn2 and jeopardizes antiviral responses and cell survival [44]. Furthermore, the DENV NS4B protein induces mitochondrial elongation that physically interacts with convoluted membranes at the ER-mitochondria interface, inhibiting IFN responses and promoting viral replication [45]. These examples illustrate the mechanisms by which a viral infection dysregulates mitochondrial dynamics and, thus, may jeopardize mitochondrial function, leading to reduced antiviral immunity and enhanced viral production.
4.3. Induction of mitophagy
Autophagy is a highly conserved and coordinated catabolic process that occurs in response to energy or nutrient shortages or following exposure to cytotoxic stimuli. Triggering the autophagic program helps prevent cell damage and provides a protective strategy for promoting survival and maintaining homeostasis in eukaryotes [46]. During the autophagic process in the cytosol, double-membraned structures form and transport targeted content to the lysosome for degradation. Autophagy can occur in a nonselective manner, such as through cell starvation, to remove cytoplasmic material and supply the cell with nutrients or in a selective manner, such as viral infections, in which specific harmful targets are chosen to be degraded. The targeted substrates of selective autophagy can be diverse, and depending on the nature of the organelles, proteins or lipids can be used as targets [47]. Therapeutic approaches modulating autophagic processes (either inhibiting or inducing these processing) are gaining interest and have been proposed as strategies for the treatment of various hematological disorders [48].
Mitophagy is a specific autophagic process used to remove dysfunctional or damaged mitochondria to maintain cellular homeostasis. The induction of mitophagy differs from the induction of cell death by viruses because mitochondria are not the major organelles used by viruses to produce viral progeny. Inducing mitophagy is a mechanism that also helps viruses escape from mitochondria-triggered antiviral immunity and apoptotic signaling. Human parainfluenza virus type 3 (HPIV3) infection induces the translocation of the HPIV3 matrix protein to mitochondria, where it interacts with mitochondrial Tu translation elongation factor. Subsequently, the interaction between the matrix protein, mitochondrial Tu translation elongation factor and LC3 proteins regulates the formation of the autophagosome, the induction of mitophagy and the inhibition of the type I IFN response [49]. Respiratory syncytial virus (RSV) activates the autophagocytic program and induces mitophagy through a mechanism that decreases the release of cytochrome C and blocks the apoptotic mechanism [50]. Measles virus infection activates SQSTM1/p62-mediated mitophagy and decreases mitochondrion-tethered MAVS, leading to the suppression of the innate immune response in non-small-cell lung cancer [51]. Mice with deficiencies in cytosolic receptor interacting serine/threonine kinase 2 (RIPK2) are susceptible to the virulent influenza A/PR/8/34 (PR8) H1N1 virus infection. Ripk2(−/−) cells are defective in mitophagy, resulting in an increased production of superoxide, the accumulation of damaged mitochondria, the activation of the NLRP3 inflammasome and the increased production of interleukin-18 [52]. Molecular investigations suggest that RIPK2 regulates mitophagy by phosphorylating the mitophagy inducer ULK1. The results also demonstrated that compared to wild-type animals, the defective mitophagy program in Ripk2(−/−) mice does not cause concomitant increases in virus production in the lungs. Interestingly, coxsackievirus B infection induces mitophagy and the viruses are released from the cell and disseminate through the formation of an autophagosome-bound mitochondrion-virus complex [53]. This mechanism serves as a new evasive manner for viruses to escape antiviral immunity.
4.4. Regulation of apoptosis
Apoptosis is a naturally occurring process in which organisms and tissues execute the cell death program without inciting overwhelming inflammatory reactions causing unwanted damage. Viral infections have both positive and opposite effects and outcomes on the apoptotic process. Turning on cellular apoptosis can shut down antiviral immunity, such as IFN production. However, premature apoptosis in infected cells may limit the production of viral progeny. To prevent overwhelming immune responses leading to damage to infected hosts, the appropriate regulation of nucleases and apoptosis-associated caspases is also needed in the course of viral infections. Mitochondria are capable of releasing cytochrome C to mediate apoptosis through a process that is tightly regulated by the permeabilization of the outer mitochondrial membrane and the Bcl-2 family proteins.
The HPV type 16 E1 empty set E4 protein binds mitochondria and induces the detachment of mitochondria from microtubules. The subsequent events result in a reduction of the mitochondrial membrane potential and the induction of apoptosis [54]. By inhibiting Mfn2 expression, HIV Vpr interferes with the Mfn2-mediated ER-mitochondria interaction and structural integrity of the outer mitochondrial membrane, causing host cell death [55]. HIV Vpr also binds adenine nucleotide translocase, which is critical for forming the inner membrane channel of the mitochondrial permeability transition pore and converts it into a proapoptotic pore [56]. The HIV gp120 protein binds CXCR4, which is a G protein coupled receptor that mediates viral infection in CD4+ T cells, to form the complex gp120-CXCR4. This complex binds mitochondria and causes mitochondrial membrane depolarization and cytochrome C release. Subsequently, caspases are activated, and the apoptotic program is turned on [57]. The human T-cell leukemia virus type 1 p13(II) protein preserves mitochondrial targeting sequence targets, accumulates in the mitochondrial inner membrane and induces the influx of K(+). Then, the mitochondrial matrix volume expands and becomes fragmented, and apoptosis induced by ceramide and the Fas ligand is promoted [58].
For long-term benefit, establishing and maintaining latency is also critical for viral survival in an infected cell. Therefore, viruses may target the mitochondrial machinery to inhibit proapoptotic pathways and promote prosurvival signals to facilitate viral production. The vaccinia virus F1L protein localizes to the mitochondria and prevents cellular apoptosis by inhibiting inner mitochondrial membrane potential loss and cytochrome cytochrome c release [59]. Cytomegalovirus encodes the cell death suppressor vMIA, which binds Bax, sequesters Bax at the mitochondria as a vMIA–Bax complex and prevents Bax-mediated mitochondrial membrane permeabilization and cellular apoptosis [60]. To maintain cell survival, Kaposi’s sarcoma-associated herpesvirus (KSHV) upregulates the expression of the antiapoptotic proteins Bcl-2 and Mcl-1 to promote the survival of virus-infected B cells. KSHV also encodes the viral Bcl-2 homolog KsBcl-2, which inhibits mitochondria-mediated apoptosis and promotes cell survical [61]. Epstein-Barr nuclear antigen 3C binds Gemin3, which is an RNA helicase regulating DNA transcription, recombination and repair, and RNA metabolism, to increase its stability and promote its interaction with p53, causing antiapoptosis and cell proliferation [62]. The antiapoptotic mechanisms mediated by the antiapoptotic gene UL36 in cytomegalovirus infection also play critical roles in cytomegalovirus transmission across primate species barriers [63]. These examples suggest a bi-directional mechanisms (both apoptotic and antiapoptotic) operated by viruses to ensure viral production whereas host cells are undergoing apoptotic program.
5. Viral targeting of specific mitochondrial molecules
Viral infection results in mitochondrial dysfunction in various aspects; the identification of the specific mitochondrial targets of the virus may help narrow the therapeutic options and effectively restore mitochondrial function. There are four mitochondrial subcompartments, the outer membrane, the intermembrane space, the inner membrane, and the matrix. The molecules in these different subcompartments may be up- or down-regulated, translocate to different subcompartments and participate in virus-induced antiviral immunity, virus-mediated immune evasion and the prevention of overwhelming immune responses. The mitochondrial outer membrane is enriched in phospholipids, and the inner membrane is enriched in protein content. The phospholipid content regulates mitochondrial function, affects biogenesis and modulates protein translocation. Among the negatively charged phospholipids, including cardiolipin, phosphatidylethanolamine, and phosphatidylglycerol, cardiolipin plays a crucial role in resuming the mitochondrial membrane architecture and recruiting soluble components to the membrane to maintain mitochondrial dynamics [64], [65]. Cardiolipin also interacts with several mitochondrial fusion- and division-associated molecules, including OPA1, Mfn, and Drp1, to regulate the function of these dynamin-related GTPases. In addition, different with the general concept that ROS is required for the inflammasome activators to induce inflammasome signaling, Lyer et al. found that the oxazolidinone antibiotic linezolid, a NLRP3 agonist, activates the NLRP3 inflammasome independently of ROS, whereas mitochondrial cardiolipin is required for NLRP3 inflammasome activation [66]. Under cardiolipin deficiency, mitophagy is defective likely due to the impairment of MAPK signaling and the reduced activation of the protein kinase C pathway [67].
Viral interferon regulatory factor 1 (vIRF-1) is encoded by human herpesvirus 8 (HHV-8), which through direct interactions with cardiolipin targets the mitochondrial detergent-resistant membrane fraction. vIRF-1 downregulates MAVS-mediated antiviral responses, promotes the productive replication of the virus and inhibits the development of cellular apoptosis [68]. By analyzing clinical samples, Martinez et al. demonstrated that a significant correlation exists among the level of anti-cardiolipin antibodies, the plasma HIV load and the cell-associated DNA level in HIV-infected patients presenting with slow disease progression [69]. This study highlights the possibility of direct or indirect involvement of cardiolipin in HIV infection-mediated pathogenesis.
Given the crucial roles of MAVS in antiviral immunity, this adaptor has been a common target of many viruses to antagonize the immune defense mechanism. Through association with the N-terminal CARD-like domain and the C-terminal transmembrane domain of MAVS, the DENV NS4A protein prevents MAVS binding to RIG-I and suppresses the downstream activation of IRF3 and NF-κB and IFN production [70]. By translocating to the mitochondrial inner membrane subcompartment, binding to MAVS and decreasing the mitochondrial membrane potential, the influenza A virus protein PB1-F2, which is a 90-amino-acid protein expressed by the PB1 gene of some influenza A viruses, induces mitochondrial fragmentation, inhibits IFN production and mediates immune evasion [71], [72]. Hepatitis C viral NS3-4A protease is capable of targeting MAVS from the outer mitochondrial membrane for cleavage and inhibiting MAVS signaling complex formation and IFN production to counteract antiviral immunity [73], [74]. The Borna disease virus X protein colocalizes and interacts with MAVS in the mitochondria and exerts antiapoptotic effects [75]. Interestingly, this apoptosis blockage process is independent of type I IFN production and NF-κB activity. In response to viral infection, receptor for globular head domain of complement component 1q (gC1qR), which is an RNA-binding protein critical for mitochondrial translation, translocates to the outer mitochondrial membrane, interacts with MAVS and prevents the interaction between MAVS and RIG-I/MDA5 to block antiviral immunity and promote viral replication [76]. Poly(C)-binding protein 2 (PCBP2), which is encoded by the PCBP2 gene, functions as a translational regulator of poliovirus, human papillomavirus and hepatitis A virus RNA. PCBP2 is induced after viral infection and interacts with MAVS to recruit the HECT domain-containing E3 ligase atrophin-1-interacting protein 4 (AIP4) to polyubiquitinate and degrade MAVS. Subsequent experimental results have suggested that the stabilization of MAVS in AIP4-deficient (Itch(−/−)) mouse embryonic fibroblasts results in exaggerated and prolonged antiviral responses [77]. Another molecule, i.e., PCBP1, uses similar mechanisms as PCBP2 to ubiquitinate and cause the degradation of MAVS. The synergistic effect on MAVS is observed when the combined effects of both PCBP1 and PCBP2 are simultaneously blocked [78]. Furthermore, by enhancing the ubiquitination of MAVS, HBx attenuates the antiviral response [79]. Collectively, these examples illustrate the mechanisms by which different viruses target and inhibit MAVS-mediated effects to antagonize antiviral immunity.
6. Antiviral therapeutics that preserve the effects of mitochondrial function regulation
Over the past few decades, significant progress has been achieved in the treatment of viral infections, and many therapeutic options are continually emerging. These achievements include the development of antiviral small molecules, vaccines, and protease inhibitors and epigenetic therapeutic approaches [80], [81]. However, scientists still face great challenges regarding many viral infections. The presence of complicated mechanisms, such as persistent and recurrent viral infections, are even more challenging [82]. Given that mitochondria govern many different aspects of antiviral immunity, small-molecule carnitinoids have been suggested to offer a promising opportunity for epigenetic treatment of persistent viral infections [83]. Carnitinoids are mitochondria-localizing novel acylcarnitine esters that regulate antioxidant and cytoprotective genes and mitophagic pathways while inhibiting histone deacetylase activity [83]. A phase II study investigating the mitochondria-targeted antioxidant mitoquinone has demonstrated benefits in patients with hepatitis C virus infection [84].
By examining limited postmortem brain samples from patients with herpes simplex encephalitis, Wnek et al. observed a preferential reduction in mitochondrial genome encoded transcripts in the genome-wide transcriptomic study [85]. There is also a preferential loss of mitochondrial function among mtDNA encoded components in primary human astrocytes infected by HSV-1 [85]. Strikingly, the results show that the dysfunction of the mitochondrial enzyme cytochrome C oxidase (CO, composed predominantly of mtDNA encoded subunits) precedes that of succinate dehydrogenase (composed entirely of nuclear encoded subunits). Treatment with minocycline leads to increases in the CO1 transcript abundance, sustained CO activity and cell viability [85]. This study highlights the significance of mitochondrial damage that occurs during the very early stages of HSV-1 infection and demonstrates the usefulness of minocycline as a potential therapeutic agent for patients with herpes simplex encephalitis.
Many commercially available drugs preserve mitochondria, and by protecting the function of mitochondria or improving mitochondrial dysfunction, their therapeutic effects are achieved. For example, alisporivir, which is an analog of cyclosporine, functions as a nonimmunosuppressive cyclophilin inhibitor, inhibiting HCV replication. Alisporivir also inhibits mitochondrial permeability transition by binding cyclophilin D. Furthermore, alisporivir blocks several HCV protein-mediated detrimental effects on mitochondria, such as reduction in cell respiration, collapse of mitochondrial membrane potential, overproduction of ROS and overloaded calcium in mitochondria [86]. The antiviral drug amantadine has been evaluated in combination therapies among HCV-infected patients. Quarato et al. demonstrated that amantadine prevents and rescues HCV protein-mediated mitochondrial dysfunction through several different mechanisms, including the correction of mitochondrial Ca2+ overload, disturbed respiratory chain activity and oxidative phosphorylation, reduced mitochondrial membrane potential and overproduced ROS [87]. To overcome the disadvantage of direct-acting antiviral treatment with sofosbuvir, which may lead to symptoms such as fatigue and migraine, Kim et al. screened several ginsenoside compounds exhibiting antiviral activity in a cell culture system and identified the compound ginsenoside Rg3 (G-Rg3), which shows promising anti-HCV effects. The treatment rescues HCV-induced mitophagy by restoring the HCV-induced Drp1-mediated aberrant mitochondrial fission process, achieving antiviral activity and suppressing persistent HCV infection [88].
Targeting the regulation of mitochondrial calcium fluxes may serve as another therapeutic approach. The regulation of calcium uptake into the mitochondrial matrix is important for cellular function, and this process may also affect energy production and trigger cell death. Mitochondria do not necessarily play a role as a dynamic buffer of cytosolic calcium under physiological conditions. Nevertheless, a prolonged increase in intracellular calcium levels may stimulate mitochondrial calcium uptake by up to 10- to 1000-fold and affect the intracellular calcium dynamics [89]. The mitochondrial calcium uniporter, which is a mitochondrial luminal redox sensor, is the major transporter of calcium into mitochondria. The overload of calcium in mitochondria affects redox homeostasis [90]. The HBV HBx protein activates cytosolic calcium-dependent proline-rich tyrosine kinase-2 by targeting mitochondrial calcium channels to elevate the cytosolic calcium levels and viral replication [91]. Compounds, such as cyclosporine A and SDZ NIM811, block the mitochondrial permeability transition pore and reduce cytosolic calcium signaling to inhibit HBV replication. In contrast, reagents that increase cytosolic calcium levels rescue the replication of an HBx-deficient HBV mutant in human hepatoblastoma cell line HepG2 cells [92]. Altogether, these examples help evaluate the weight of the restoration of dysregulated mitochondrial function in the overall benefits of antiviral therapeutics.
7. Challenges and perspectives
Tight interactions exist among different machineries and molecules in mitochondria after a viral infection [93]. For example, the mitochondrial fission process can be associated with mitophagy based on the concomitant observation that the decreased length of mitochondria helps in the engulfment by autophagosomes and avoidance of the fusion between damaged mitochondria and healthy mitochondria, which could jeopardize their function [94]. The regulation of mitochondrial dynamics, such as enhancing mitophagy and inhibiting apoptosis, may modify the fate of the mitochondrial machinery. HCV infection induces Drp1 expression and phosphorylation at Ser616 [95]. Then, phosphorylated Drp1 translocates to mitochondria, interferes with the mitochondrial dynamics and promotes mitochondrial fission. Subsequently, the mitophagy program is triggered, HCV-induced apoptosis is inhibited and viral production is increased [95]. Silencing Drp1 enhances antiviral immunity and inhibits viral production; concomitantly, cellular glycolysis and the ATP levels decrease. The results suggest that the virus triggers mitochondrial fission and the mitophagy program, which can help inhibit the apoptotic process, resulting in persistent HCV infection [95]. These examples illustrate the close interaction among mitochondrial machineries such as dynamics, mitophagy and apoptosis in viral infections. In addition, autophagy-related proteins can affect innate immune responses by reducing mtDNA release mediated by the NALP3 inflammasome [96]. Furthermore, mitochondrial apoptotic signaling can activate the NLRP3 inflammasome, and, in turn, NLRP3 secondary signal activator ATP induces mitochondrial dysfunction and apoptosis, leading to the release of oxidized mtDNA into the cytosol, where it binds and activates the NLRP3 inflammasome [97]. Accordingly, the interaction among different mitochondrial machineries may complicate the therapeutic strategies and the final outcomes.
The interaction between mitophagy and apoptosis is of special interest. The accumulated studies suggest that virus may modulate mitophagy to block cellular apoptosis [93]. Huang et al. showed that HBx can enhance phosphatase and tensin homolog (PTEN)-induced kinase 1 (PINK1)-Parkin signaling and mediate mitophagy in starvation status and this effect concurrently reduces mitochondrial apoptosis in starved hepatoma cells [98]. Studies suggest that the triggering of mitochondrial damage after virus infection may result in the release of a great volume of ROS into the cytosol. Subsequently, the integrity of healthy mitochondria is threatened and a vicious cycle is established finally leading to cell death [93], [99]. Thus, initiating a rapid clearance mechanism to get rid of toxic factors like ROS is very critical to prevent cell death after mitochondrial injury to maintain cell survival and to establish the status of viral persistency. Accordingly, the inhibition of mitophagy results in a surge in mitochondrial apoptotic signaling followed by death of HBV-infected hepatocytes [40]. In supportive, in the model of heart failure, the induction of mitophagy by activating AMPKalpha2-PINK1-Parkin signaling pathway results in mitochondrial depolarization, removal of damaged mitochondria, reduction of ROS production, improvement in mitochondrial function as well as decrease of apoptosis of cardiomyocytes [100]. Nevertheless, the consequences of mitophagy or apoptosis may be complicated. For example, the process of mitochondrial depolarization triggers polyubiquitination of many mitochondrial outer membrane proteins in a Parkin- and PINK1-dependent manner leading to mitophagy. However, Parkin, a ubiquitin E3 ligase, can sensitize cells toward apoptosis induced by mitochondrial depolarization but not by proapoptotic stimuli that cannot activate Parkin [101]. Molecular analysis reveals that Parkin-dependent apoptosis induced by mitochondrial depolarization is through promoting degradation of Bcl-2 family protein Mcl-1 and can be inhibited by knockdown of Bax and Bak [101]. The results suggest that Parkin may function as cytoprotective (mitophagy) or cytotoxic (apoptosis) depending on the degree of mitochondrial damage.
Given the roles of mitochondria in immune activation and viral immune evasion, the benefits/risks of the correction of dysfunctional mitochondrial machineries after viral infection are also complicated. The dysfunction of mitochondria can be explained as an evasion mechanism by which the virus counteracts antiviral activity; however, the dysfunctional mitochondria can also be regarded as a self-saving mechanism in virus-infected cells to prevent the development of overwhelming immune responses. Therefore, treatment targeting mitochondria serves two main purposes. First, this treatment can restore the dysregulated function of mitochondria after viral infection and prevent the deterioration of the dysfunctional mitochondria. Second, this treatment can prevent the sustainable activation of the mitochondrial system to avoid possibly overwhelming immune activation after viral infection.
Thus far, no medications targeting mitochondrial molecules or mitochondrial function-related machineries are market available or currently undergoing clinical investigation. Nevertheless, some observations may bring some hopes in this regard. Given that mitochondria fragmentation induced by cisplatin treatment reduces renal levels of sirtuin 3, a soluble protein located in the mitochondrial matrix, and results in acute kidney injury, Morigi et al. demonstrated that restoration of sirtuin 3 with the AMP-activated protein kinase agonist AICAR recovers mitochondrial function in cultured human tubular cells and improves renal function in cisplatin-treated animals [102]. A hypoglycemic agent metformin, also a mitochondrial complex I inhibitor, exerts antiatherosclerotic effects through inhibiting Drp1-mediated mitochondrial fission in an AMP-activated protein kinase-dependent manner [103]. Meanwhile, metformin treatments reduce mitochondrial fragmentation, mitigate mitochondrial-derived superoxide release and inhibit atherosclerosis in streptozotocin-induced diabetic ApoE(−/−) mice [103]. These examples, although not investigated as antiviral therapeutics, suggest the benefit of targeting mitochondrial specific component in restoration of mitochondrial function. Uncovering the crucial characteristics of mitochondrial machineries in both antiviral immunity and viral evasion from immune defense has a high potential for opening a new door for designing and manufacturing effective antiviral therapeutic agents. A clearer understanding of the mechanisms involving mitochondria in viral infections is needed.
Funding
This work was supported in part by grants from the Ministry of Science and Technology (MOST 107-2314-B-182A-132-MY3) and Chang Gung Memorial Hospital (CMRPG3F0573 and CMRPG3E2163), Taiwan, R.O.C.
Acknowledgments
Acknowledgements
We are grateful for the help provided by the members of Dr. Ho’s and Dr. Lai’s laboratories.
Conflicts of Interest
The authors have no conflicts of interests related to this study.
References
- 1.Brubaker S.W., Bonham K.S., Zanoni I., Kagan J.C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bardoel B.W., Strijp J.A. Molecular battle between host and bacterium: recognition in innate immunity. J. Mol. Recognit. 2011;24:1077–1086. doi: 10.1002/jmr.1156. [DOI] [PubMed] [Google Scholar]
- 3.Bayne C.J. Origins and evolutionary relationships between the innate and adaptive arms of immune systems. Integr. Comp. Biol. 2003;43:293–299. doi: 10.1093/icb/43.2.293. [DOI] [PubMed] [Google Scholar]
- 4.Liston A., Masters S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017;17:208–214. doi: 10.1038/nri.2016.151. [DOI] [PubMed] [Google Scholar]
- 5.Kato H., Sato S., Yoneyama M., Yamamoto M., Uematsu S., Matsui K. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23:19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 6.Kowalinski E., Lunardi T., McCarthy A.A., Louber J., Brunel J., Grigorov B. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. doi: 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 7.Hou F., Sun L., Zheng H., Skaug B., Jiang Q.X., Chen Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qi N., Shi Y., Zhang R., Zhu W., Yuan B., Li X. Multiple truncated isoforms of MAVS prevent its spontaneous aggregation in antiviral innate immune signalling. Nat. Commun. 2017;8:15676. doi: 10.1038/ncomms15676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heaton S.M., Borg N.A., Dixit V.M. Ubiquitin in the activation and attenuation of innate antiviral immunity. J. Exp. Med. 2016;213:1–13. doi: 10.1084/jem.20151531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S., Cai X., Wu J., Cong Q., Chen X., Li T. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. 2015;347 doi: 10.1126/science.aaa2630. aaa2630. [DOI] [PubMed] [Google Scholar]
- 11.Dromparis P., Michelakis E.D. Mitochondria in vascular health and disease. Annu. Rev. Physiol. 2013;75:95–126. doi: 10.1146/annurev-physiol-030212-183804. [DOI] [PubMed] [Google Scholar]
- 12.van der Burgh R., Boes M. Mitochondria in autoinflammation: cause, mediator or bystander? Trends Endocrinol. Metab. 2015;26:263–271. doi: 10.1016/j.tem.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 13.Prochnicki T., Latz E. Inflammasomes on the crossroads of innate immune recognition and metabolic control. Cell Metab. 2017;26:71–93. doi: 10.1016/j.cmet.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Monlun M., Hyernard C., Blanco P., Lartigue L., Faustin B. Mitochondria as molecular platforms integrating multiple innate immune signalings. J. Mol. Biol. 2017;429:1–13. doi: 10.1016/j.jmb.2016.10.028. [DOI] [PubMed] [Google Scholar]
- 15.Kurose I., Miura S., Fukumura D., Yonei Y., Saito H., Tada S. Nitric oxide mediates Kupffer cell-induced reduction of mitochondrial energization in hepatoma cells: a comparison with oxidative burst. Cancer Res. 1993;53:2676–2682. [PubMed] [Google Scholar]
- 16.Cao Z., Xia Z., Zhou Y., Yang X., Hao H., Peng N. Methylcrotonoyl-CoA carboxylase 1 potentiates RLR-induced NF-kappaB signaling by targeting MAVS complex. Sci. Rep. 2016;6:33557. doi: 10.1038/srep33557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills E.L., Kelly B., O'Neill L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017;18:488–498. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 18.Pearce E.L., Pearce E.J. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu D., Sanin D.E., Everts B., Chen Q., Qiu J., Buck M.D. Type 1 interferons induce changes in core metabolism that are critical for immune function. Immunity. 2016;44:1325–1336. doi: 10.1016/j.immuni.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ten Garcia-Sastre A. Strategies of interferon evasion by viruses. Cell Host Microbe. 2017;22:176–184. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West A.P., Shadel G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017;17:363–375. doi: 10.1038/nri.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cossarizza A., Pinti M., Nasi M., Gibellini L., Manzini S., Roat E. Increased plasma levels of extracellular mitochondrial DNA during HIV infection: a new role for mitochondrial damage-associated molecular patterns during inflammation. Mitochondrion. 2011;11:750–755. doi: 10.1016/j.mito.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kung C.T., Hsiao S.Y., Tsai T.C., Su C.M., Chang W.N., Huang C.R. Plasma nuclear and mitochondrial DNA levels as predictors of outcome in severe sepsis patients in the emergency room. J. Transl. Med. 2012;10:130. doi: 10.1186/1479-5876-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakahira K., Kyung S.Y., Rogers A.J., Gazourian L., Youn S., Massaro A.F. Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med. 2013;10 doi: 10.1371/journal.pmed.1001577. e1001577; discussion e1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai J.H., Wang M.Y., Huang C.Y., Wu C.H., Hung L.F., Yang C.Y. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018;19 doi: 10.15252/embr.201846182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hotz M.J., Qing D., Shashaty M.G.S., Zhang P., Faust H., Sondheimer N. Red blood cells homeostatically bind mitochondrial DNA through TLR9 to maintain quiescence and to prevent lung injury. Am. J. Respir. Crit. Care Med. 2018;197:470–480. doi: 10.1164/rccm.201706-1161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dhanwani R., Takahashi M., Sharma S. Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr. Opin. Immunol. 2018;50:82–87. doi: 10.1016/j.coi.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aguirre S., Luthra P., Sanchez-Aparicio M.T., Maestre A.M., Patel J., Lamothe F. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017;2:17037. doi: 10.1038/nmicrobiol.2017.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aguirre S., Fernandez-Sesma A. Collateral damage during dengue virus infection: making sense of DNA by cGAS. J. Virol. 2017;91(14) doi: 10.1128/JVI.01081-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo X., Hong L., Cheng C., Li N., Zhao X., Shi F. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018;9:619. doi: 10.1038/s41419-018-0662-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tillo D., Ray S., Syed K.S., Gaylor M.R., He X., Wang J. The Epstein-Barr virus B-ZIP protein Zta recognizes specific DNA sequences containing 5-methylcytosine and 5-hydroxymethylcytosine. Biochemistry. 2017;56:6200–6210. doi: 10.1021/acs.biochem.7b00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiedmer A., Wang P., Zhou J., Rennekamp A.J., Tiranti V., Zeviani M. Epstein-Barr virus immediate-early protein Zta co-opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. J. Virol. 2008;82:4647–4655. doi: 10.1128/JVI.02198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corcoran J.A., Saffran H.A., Duguay B.A., Smiley J.R. Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol. 2009;83:2601–2610. doi: 10.1128/JVI.02087-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moren C., Gonzalez-Casacuberta I., Alvarez-Fernandez C., Bano M., Catalan-Garcia M., Guitart-Mampel M. HIV-1 promonocytic and lymphoid cell lines: an in vitro model of in vivo mitochondrial and apoptotic lesion. J. Cell Mol. Med. 2017;21:402–409. doi: 10.1111/jcmm.12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duguay B.A., Saffran H.A., Ponomarev A., Duley S.A., Eaton H.E., Smiley J.R. Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J. Virol. 2014;88:2967–2976. doi: 10.1128/JVI.03129-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H., Chomyn A., Chan D.C. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 2005;280:26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 38.Buck M.D., O'Sullivan D., Klein Geltink R.I., Curtis J.D., Chang C.H., Sanin D.E. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khan M., Syed G.H., Kim S.J., Siddiqui A. Mitochondrial dynamics and viral infections: a close nexus. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids. 2015;1853:2822–2833. doi: 10.1016/j.bbamcr.2014.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S.J., Khan M., Quan J., Till A., Subramani S., Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pal A.D., Basak N.P., Banerjee A.S., Banerjee S. Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis. 2014;35:1592–1601. doi: 10.1093/carcin/bgu069. [DOI] [PubMed] [Google Scholar]
- 42.McCormick A.L., Smith V.L., Chow D., Mocarski E.S. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 2003;77:631–641. doi: 10.1128/JVI.77.1.631-641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi C.S., Qi H.Y., Boularan C., Huang N.N., Abu-Asab M., Shelhamer J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014;193:3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu C.Y., Liang J.J., Li J.K., Lee Y.L., Chang B.L., Su C.I. Dengue virus impairs mitochondrial fusion by cleaving mitofusins. PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1005350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chatel-Chaix L., Cortese M., Romero-Brey I., Bender S., Neufeldt C.J., Fischl W. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe. 2016;20:342–356. doi: 10.1016/j.chom.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dikic I., Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018;19(6):349–364. doi: 10.1038/s41580-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 47.Stolz A., Ernst A., Dikic I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014;16:495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 48.Orsini M., Morceau F., Dicato M., Diederich M. Autophagy as a pharmacological target in hematopoiesis and hematological disorders. Biochem. Pharmacol. 2018;152:347–361. doi: 10.1016/j.bcp.2018.04.007. [DOI] [PubMed] [Google Scholar]
- 49.Ding B., Zhang L., Li Z., Zhong Y., Tang Q., Qin Y. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21(538–547) doi: 10.1016/j.chom.2017.03.004. [DOI] [PubMed] [Google Scholar]
- 50.Li M., Li J., Yang J., Liu J., Zhang Z., Song X. Respiratory syncytial virus replication is promoted by autophagy-mediated inhibition of apoptosis. J. Virol. 2018;92(8) doi: 10.1128/JVI.02193-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xia M., Gonzalez P., Li C., Meng G., Jiang A., Wang H. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J. Virol. 2014;88:5152–5164. doi: 10.1128/JVI.03851-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lupfer C., Thomas P.G., Anand P.K., Vogel P., Milasta S., Martinez J. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nat. Immunol. 2013;14:480–488. doi: 10.1038/ni.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sin J., McIntyre L., Stotland A., Feuer R., Gottlieb R.A. Coxsackievirus B escapes the infected cell in ejected mitophagosomes. J. Virol. 2017;91(24) doi: 10.1128/JVI.01347-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raj K., Berguerand S., Southern S., Doorbar J., Beard P. E1 empty set E4 protein of human papillomavirus type 16 associates with mitochondria. J. Virol. 2004;78:7199–7207. doi: 10.1128/JVI.78.13.7199-7207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang C.Y., Chiang S.F., Lin T.Y., Chiou S.H., Chow K.C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0033657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halestrap A.P., Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 57.Roggero R., Robert-Hebmann V., Harrington S., Roland J., Vergne L., Jaleco S. Binding of human immunodeficiency virus type 1 gp120 to CXCR4 induces mitochondrial transmembrane depolarization and cytochrome c-mediated apoptosis independently of Fas signaling. J. Virol. 2001;75:7637–7650. doi: 10.1128/JVI.75.16.7637-7650.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D'Agostino D.M., Silic-Benussi M., Hiraragi H., Lairmore M.D., Ciminale V. The human T-cell leukemia virus type 1 p13II protein: effects on mitochondrial function and cell growth. Cell Death Differ. 2005;12(Suppl. 1):905–915. doi: 10.1038/sj.cdd.4401576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wasilenko S.T., Stewart T.L., Meyers A.F., Barry M. Vaccinia virus encodes a previously uncharacterized mitochondrial-associated inhibitor of apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:14345–14350. doi: 10.1073/pnas.2235583100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Arnoult D., Bartle L.M., Skaletskaya A., Poncet D., Zamzami N., Park P.U. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarid R., Sato T., Bohenzky R.A., Russo J.J., Chang Y. Kaposi’s sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat. Med. 1997;3:293–298. doi: 10.1038/nm0397-293. [DOI] [PubMed] [Google Scholar]
- 62.Cai Q., Guo Y., Xiao B., Banerjee S., Saha A., Lu J. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002418. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Burwitz B.J., Malouli D., Bimber B.N., Reed J.S., Ventura A.B., Hancock M.H. Cross-species rhesus cytomegalovirus infection of cynomolgus macaques. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1006014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martensson C.U., Doan K.N., Becker T. Effects of lipids on mitochondrial functions. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids. 2017;1862:102–113. doi: 10.1016/j.bbalip.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 65.Kameoka S., Adachi Y., Okamoto K., Iijima M., Sesaki H. Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 2018;28:67–76. doi: 10.1016/j.tcb.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iyer S.S., He Q., Janczy J.R., Elliott E.I., Zhong Z., Olivier A.K. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity. 2013;39:311–323. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shen Z., Li Y., Gasparski A.N., Abeliovich H., Greenberg M.L. Cardiolipin regulates mitophagy through the protein kinase C pathway. J. Biol. Chem. 2017;292:2916–2923. doi: 10.1074/jbc.M116.753574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hwang K.Y., Choi Y.B. Modulation of mitochondrial antiviral signaling by human herpesvirus 8 interferon regulatory factor 1. J. Virol. 2016;90:506–520. doi: 10.1128/JVI.01903-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Martinez V., Diemert M.C., Braibant M., Potard V., Charuel J.L., Barin F. Anticardiolipin antibodies in HIV infection are independently associated with antibodies to the membrane proximal external region of gp41 and with cell-associated HIV DNA and immune activation. Clin. Infect. Dis. 2009;48:123–132. doi: 10.1086/595013. [DOI] [PubMed] [Google Scholar]
- 70.He Z., Zhu X., Wen W., Yuan J., Hu Y., Chen J. Dengue virus subverts host innate immunity by targeting adaptor protein MAVS. J. Virol. 2016;90:7219–7230. doi: 10.1128/JVI.00221-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Varga Z.T., Grant A., Manicassamy B., Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012;86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshizumi T., Ichinohe T., Sasaki O., Otera H., Kawabata S., Mihara K. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014;5:4713. doi: 10.1038/ncomms5713. [DOI] [PubMed] [Google Scholar]
- 73.Li X.D., Sun L., Seth R.B., Pineda G., Chen Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hiscott J., Lacoste J., Lin R. Recruitment of an interferon molecular signaling complex to the mitochondrial membrane: disruption by hepatitis C virus NS3-4A protease. Biochem. Pharmacol. 2006;72:1477–1484. doi: 10.1016/j.bcp.2006.06.030. [DOI] [PubMed] [Google Scholar]
- 75.Li Y., Song W., Wu J., Zhang Q., He J., Li A. MAVS-mediated host cell defense is inhibited by Borna disease virus. Int. J. Biochem. Cell Biol. 2013;45:1546–1555. doi: 10.1016/j.biocel.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 76.Xu L., Xiao N., Liu F., Ren H., Gu J. Inhibition of RIG-I and MDA5-dependent antiviral response by gC1qR at mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2009;106:1530–1535. doi: 10.1073/pnas.0811029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.You F., Sun H., Zhou X., Sun W., Liang S., Zhai Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009;10:1300–1308. doi: 10.1038/ni.1815. [DOI] [PubMed] [Google Scholar]
- 78.Zhou X., You F., Chen H., Jiang Z. Poly(C)-binding protein 1 (PCBP1) mediates housekeeping degradation of mitochondrial antiviral signaling (MAVS) Cell Res. 2012;22:717–727. doi: 10.1038/cr.2011.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei C., Ni C., Song T., Liu Y., Yang X., Zheng Z. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010;185:1158–1168. doi: 10.4049/jimmunol.0903874. [DOI] [PubMed] [Google Scholar]
- 80.Lai J.H., Lin Y.L., Hsieh S.L. Pharmacological intervention for dengue virus infection. Biochem. Pharmacol. 2017;129:14–25. doi: 10.1016/j.bcp.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 81.Hong X., Kim E.S., Guo H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: implications for epigenetic therapy against chronic hepatitis B. Hepatology. 2017;66:2066–2077. doi: 10.1002/hep.29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsai K.N., Kuo C.F., Ou J.J. Mechanisms of hepatitis B virus persistence. Trends Microbiol. 2018;26:33–42. doi: 10.1016/j.tim.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Moos W.H., Pinkert C.A., Irwin M.H., Faller D.V., Kodukula K., Glavas I.P. Epigenetic treatment of persistent viral infections. Drug Dev. Res. 2017;78:24–36. doi: 10.1002/ddr.21366. [DOI] [PubMed] [Google Scholar]
- 84.Gane E.J., Weilert F., Orr D.W., Keogh G.F., Gibson M., Lockhart M.M. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010;30:1019–1026. doi: 10.1111/j.1478-3231.2010.02250.x. [DOI] [PubMed] [Google Scholar]
- 85.Wnek M., Ressel L., Ricci E., Rodriguez-Martinez C., Guerrero J.C., Ismail Z. Herpes simplex encephalitis is linked with selective mitochondrial damage; a post-mortem and in vitro study. Acta Neuropathol. 2016;132:433–451. doi: 10.1007/s00401-016-1597-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Quarato G., D'Aprile A., Gavillet B., Vuagniaux G., Moradpour D., Capitanio N. The cyclophilin inhibitor alisporivir prevents hepatitis C virus-mediated mitochondrial dysfunction. Hepatology. 2012;55:1333–1343. doi: 10.1002/hep.25514. [DOI] [PubMed] [Google Scholar]
- 87.Quarato G., Scrima R., Ripoli M., Agriesti F., Moradpour D., Capitanio N. Protective role of amantadine in mitochondrial dysfunction and oxidative stress mediated by hepatitis C virus protein expression. Biochem. Pharmacol. 2014;89:545–556. doi: 10.1016/j.bcp.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 88.Kim S.J., Jang J.Y., Kim E.J., Cho E.K., Ahn D.G., Kim C. Ginsenoside Rg3 restores hepatitis C virus-induced aberrant mitochondrial dynamics and inhibits virus propagation. Hepatology. 2017;66:758–771. doi: 10.1002/hep.29177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Williams G.S., Boyman L., Chikando A.C., Khairallah R.J., Lederer W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U.S.A. 2013;110:10479–10486. doi: 10.1073/pnas.1300410110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dong Z., Shanmughapriya S., Tomar D., Siddiqui N., Lynch S., Nemani N. Mitochondrial Ca(2+) uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell. 2017;65(6):1014–1028.e7. doi: 10.1016/j.molcel.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bouchard M.J., Wang L.H., Schneider R.J. Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science. 2001;294:2376–2378. doi: 10.1126/science.294.5550.2376. [DOI] [PubMed] [Google Scholar]
- 92.Bouchard M.J., Puro R.J., Wang L., Schneider R.J. Activation and inhibition of cellular calcium and tyrosine kinase signaling pathways identify targets of the HBx protein involved in hepatitis B virus replication. J. Virol. 2003;77:7713–7719. doi: 10.1128/JVI.77.14.7713-7719.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kim S.J., Ahn D.G., Syed G.H., Siddiqui A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion. 2018;41:21–27. doi: 10.1016/j.mito.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Twig G., Elorza A., Molina A.J., Mohamed H., Wikstrom J.D., Walzer G. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim S.J., Syed G.H., Khan M., Chiu W.W., Sohail M.A., Gish R.G. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. U.S.A. 2014;111:6413–6418. doi: 10.1073/pnas.1321114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakahira K., Haspel J.A., Rathinam V.A., Lee S.J., Dolinay T., Lam H.C. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shimada K., Crother T.R., Karlin J., Dagvadorj J., Chiba N., Chen S. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang X.Y., Li D., Chen Z.X., Huang Y.H., Gao W.Y., Zheng B.Y. Hepatitis B Virus X protein elevates Parkin-mediated mitophagy through Lon Peptidase in starvation. Exp. Cell Res. 2018;368:75–83. doi: 10.1016/j.yexcr.2018.04.016. [DOI] [PubMed] [Google Scholar]
- 99.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 100.Wang B., Nie J., Wu L., Hu Y., Wen Z., Dong L. AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ. Res. 2018;122:712–729. doi: 10.1161/CIRCRESAHA.117.312317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Carroll R.G., Hollville E., Martin S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014;9:1538–1553. doi: 10.1016/j.celrep.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 102.Morigi M., Perico L., Rota C., Longaretti L., Conti S., Rottoli D. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Invest. 2015;125:715–726. doi: 10.1172/JCI77632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Q., Zhang M., Torres G., Wu S., Ouyang C., Xie Z. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-mediated mitochondrial fission. Diabetes. 2017;66:193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]