

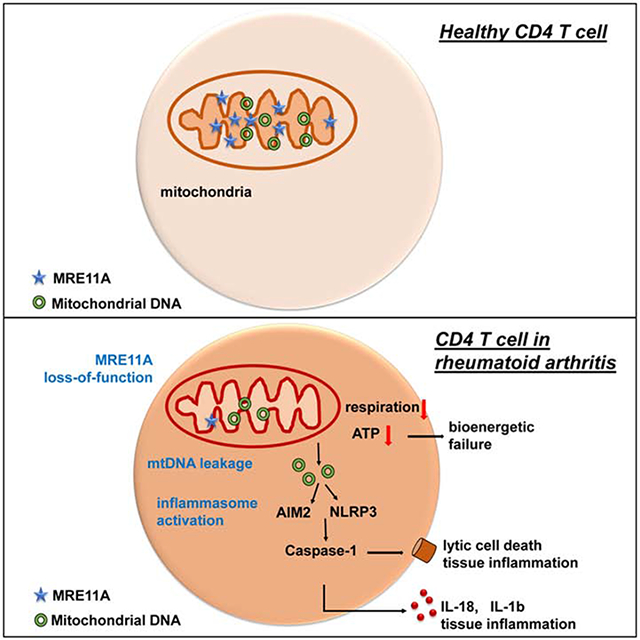
Summary
In the autoimmune disease rheumatoid arthritis (RA), CD4+ T cells promote pro-inflammatory effector functions by shunting glucose away from glycolysis and ATP production. Underlying mechanisms remain unknown and here we implicate the DNA repair nuclease MRE11A in the cells’ bioenergetic failure. MRE11A deficiency in RA T cells disrupted mitochondrial oxygen consumption and suppressed ATP generation. Also, MRE11A loss-of-function caused leakage of mitochondrial DNA (mtDNA) into the cytosol, triggering inflammasome assembly, caspase-1 activation and pyroptotic cell death. Caspase-1 activation was frequent in lymph node-residing T cells in RA patients. In vivo, pharmacologic and genetic inhibition of MRE11A resulted in tissue deposition of mtDNA, caspase-1 proteolysis and aggressive tissue inflammation. Conversely, MRE11A overexpression restored mitochondrial fitness and shielded tissue from inflammatory attack. Thus, the nuclease MRE11A regulates a mitochondrial protection program and MRE11A deficiency leads to DNA repair defects, energy production failure and loss of tissue homeostasis.
Keywords: DNA damage repair, MRE11A, mitochondrial DNA, caspase-1, pyroptosis, inflammasome, T cell aging, tissue inflammation
Graphical Abstract
eTOCblurb
In rheumatoid arthritis, T cells are deficient in the DNA repair nuclease MRE11A and age prematurely. Li et al. show that MRE11A is necessary to sustain mitochondrial fitness and ATP production. T cells deficient in MRE11A leak mtDNA into the cytoplasm, triggering pyroptosis and driving tissue inflammation.
Introduction
Innate and adaptive immune system abnormalities cause the chronic tissue inflammation typical for rheumatoid arthritis (RA).T cells represent key pathogenic drivers (Ishigaki et al., 2017) that support autoantibody production, stimulate myeloid and stromal cells and promote extranodal lymphoid organogenesis. Disease-associated genetic polymorphisms implicate abnormalities in T cell signaling and threshold setting (Saitoh et al., 2008). CD4+ T cells from RA patients have a distinct metabolic signature and age prematurely. Besides accumulating DNA breaks and telomere damage (Fujii et al., 2009), RA T cells bypass the G2/M cell cycle checkpoint (Yang et al., 2016), and differentiate into proinflammatory Th1 and Th17 effector cells. Defects in the DNA repair machinery include repression of the ataxia-telangiectasia mutated (ATM) (Shao et al., 2009), gain of the repair kinase DNA-PKcs (Shao et al., 2010), and loss-of-function of the DNA nuclease MRE11A (Li et al., 2016). In healthy individuals, MRE11A protein expression in T cells declines age-dependently; in RA patients, most naive and memory CD4+ T cells are MRE11Alow already during midlife (Li et al., 2016). MRE11A is part of the tri-molecular MRE11A/RAND50/NBS1 (MRN) complex, functions as an exonuclease and endonuclease and contributes to single- and double-strand break (DSB) repair (Syed and Tainer, 2018). Pharmacological or genetic inhibition of MRE11A causes telomeric damage and MRE11Alow T cells induce robust tissue inflammation (Li et al., 2016).
Defective DNA repair in pre-aged RA T cells is associated with a switch in bioenergetic strategies. RA T cells repress the key glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3) (Yang et al., 2013), upregulate glucose-6-phosphate dehydrogenase (G6PDH) and shunt glucose away from breakdown to the pentose phosphate pathway (PPP) (Yang et al., 2016). Such T cells have a distinct metabolic profile: ATPlow, pyruvatelow, NADPHhigh, fatty acidhigh (Weyand and Goronzy, 2017). Metabolic reprogramming changes T cell function. Excess lipogenesis leads to cytoplasmic lipid droplet accumulation, membrane ruffling, and podosome formation; imposing a tissue-invasive phenotype (Shen et al., 2017).
As the cell’s energy plant, mitochondria deliver ATP and metabolic intermediates from imported pyruvate, fatty acids, and amino acids (Weinberg et al., 2015). Oxidative phosphorylation in the electron transport chain (ETC) provides the major ATP source, and generates reactive oxygen species (ROS) (Bratic and Larsson, 2013; Yakes and VanHouten, 1997). Mitochondria house their own genome organized into DNA-protein complexes, the mitochondrial nucleoids. Lacking protective histones and being close to the ROS-producing ETC, mitochondrial DNA (mtDNA) is at risk for oxidative damage (Alexeyev et al., 2013). The organelle possesses almost all DNA repair machineries to deal with base excision repair, mismatch repair, and recombinational repair. Whether MRE11A loss-of-function affects mtDNA repair, containment and mitochondrial fitness is unknown.
Here, we have examined whether MRE11A loss-of-function in RA T cells has extranuclear functions that are of pathogenic relevance. Specifically, we explored whether transcriptional repression of MRE11A affects the metabolic conditioning of RA T cells. MRE11A knockdown in healthy T cells markedly reduced mitochondrial oxygen consumption, phenocopying the impaired mitochondrial function and ATPlow state of RA T cells. Diminished mitochondrial oxygen consumption and ATP generation was combined with cytosolic leakage of mtDNA and inflammasome activation. MRE11Alow T cells died a lytic death and were frequent in lymph nodes of RA patients; resembling the pyroptosis-prone T cells in HIV-infected lymph nodes. We exploited a chimeric mouse model to test the in vivo relevance of MRE11A in mtDNA leakage, caspase-1 activation, pyroptosis and tissue inflammation. MRE11A loss-of-function resulted in tissue deposition of mtDNA, caspase-1 proteolysis and induction of aggressive tissue inflammation. MRE11A gain-of-function was tissue protective, preventing T cell death and inflammation. The current data implicate the nuclease MRE11A in a mitochondrial stress defense program and identify MRE11A as a sensor of metabolic status that ultimately regulates T cell longevity and tissue inflammation.
Results
MRE11A regulates mitochondrial activity
MRE11A’s nucleolytic activity is critically involved in DSB repair and in telomeric maintenance (Marcomini and Gasser, 2015). CD4+ T cells in RA patients accumulate telomeric damage and age prematurely (Li et al., 2016). MRE11A protein concentrations were 50% lower in RA CD4+ T cells than in controls (Fig. S1A). We mimicked conditions in RA T cells by knocking down MRE11A mRNA in healthy T cells through siRNA technology (Fig. S1B). Alternatively, we treated T cells with Mirin, which inhibits MRE11A’s exonucleolytic activity (Dupre et al., 2008; Shibata et al., 2014). In all experiments, T cells were stimulated for 72h. We assessed metabolic competence of MRE11Alow CD4+ T cells by quantifying mitochondrial oxygen consumption rates (OCR) by Seahorse analyzer (Fig. 1A–D, Fig.S1C). Mitochondrial function was probed by serial addition of oligomycin, FCCP and antimycin A/rotenone. Dynamic tracings revealed significantly reduced OCRs in CD4+ T cells rendered MRE11Alow (Fig. 1A). Mirin treatment was similarly effective in suppressing mitochondrial function (Fig.S1C). MRE11A loss-of-function reduced basal OCRs and MRE11Alow T cells failed to uncouple respiration as efficiently as wildtype T cells (Fig. 1B–D). Accordingly, intracellular ATP concentrations declined (Fig. 1E). MRE11A overexpression normalized oxygen consumption and ATP production (Fig. 1A–E). We compared PFKFB3 and G6PD transcripts and NADPH concentrations in MRE11Alow and wildtype T cells (Fig.S1D–F) and measured extracellular acidification rates (ECARs) (Fig. 1F). Glycolysis and the pentose phosphate shunt were unaffected by MRE11A knockdown; ECARs were even higher than in controls. In RA CD4+ T cells, OCRs were depressed, basal oxygen consumptions were low, and mitochondria were unable to maximize respiration (Fig. 1G–H). As reported, ECARs and ATP concentrations were significantly reduced in RA T cells (Fig. 1I–J). Overall, mitochondrial function appeared similarly impaired in RA CD4+ T cells and healthy CD4+ T cells, in which MRE11A was genetically or pharmacologically inhibited.
Figure 1: MRE11A regulates mitochondrial function.
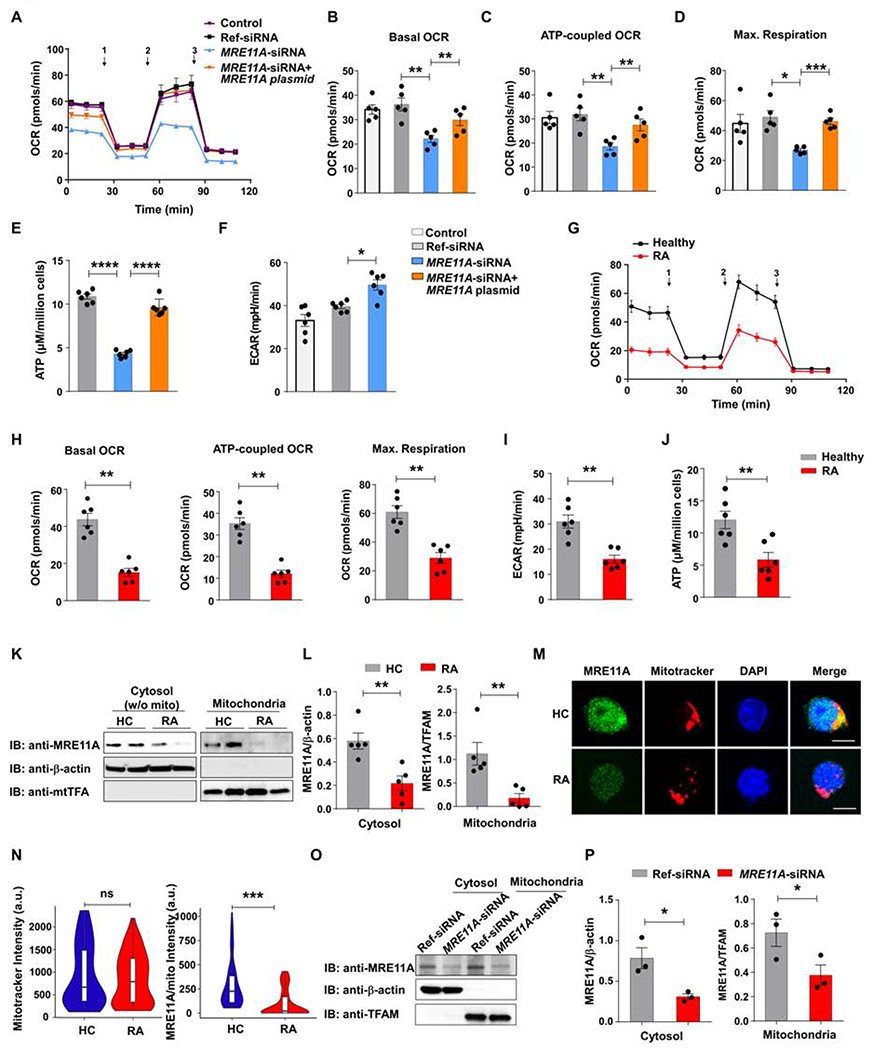
CD4+ T cells from 5 healthy controls were transfected with MRE11A siRNA plus/minus an MRE11A expression plasmid and stimulated for 72h. One aliquot was left untransfected. Oxygen consumption rates (OCR) were traced by Seahorse Bioscience XF96 analyzer and mitochondrial function was probed by serial addition of 1,oligomycin, 2,FCCP, and 3,antimycin A/rotenone. (A) Summarized OCR tracings. (B-D) Baseline respiration, ATP-coupled respiration, and maximal respiration. (E) Intracellular ATP concentrations. (F) Extracellular acidification rates (ECAR) measured by Seahorse Bioscience XF96 analyzer. (G) RA and control CD4+ T cells (n=6 each) were stimulated for 72h. OCR tracing curves collected by Seahorse analyzer. (H-J) Parameters of mitochondrial function (H), ECARs (I) and intracellular ATP (J) were determined as above. (K-L) MRE11A in cytosolic, mitochondrial, and nuclear fractions. (M, N) Cytosolic and mitochondrial MRE11A in CD4+ T cells from RA patients and controls. Immunoblots representative of 5 controls and 5 patients. (M) Dual-color immunostaining for MRE11A (green) and mitotracker (red). Nuclei marked with DAPI (blue). Scale bar; 5 μm. (N) Quantification of mitochondrial mass and mitochondrial MRE11A in control and RA CD4+ T cells. Violin plots from 5 controls and 5 patients. Median fluorescence intensities and interquartile range are indicated. (O-P) Immunoblotting of cytosolic and mitochondrial MRE11A protein after transfection of MRE11A or control siRNA. Immunoblots representative of 3 samples. All data are mean ± SEM. ANOVA with post hoc Tukey (B-F), Mann Whitney (H-J, N) ort-test (L, P).*p<0.05, **p<0.01, ***p<0.001, ****<0.0001. See also Fig.Sl
We first explored whether human T cells have detectable MRE11A outside of the nucleus. Western blotting demonstrated a cytosolic fraction and immunostaining revealed a punctuated pattern in the cytoplasmic rim (Fig.S1G–H). Further fractionation assigned MRE11A protein to both, the cytoplasm, and the mitochondria (Fig.S1I). Proteinase K treatment placed MRE11A into the mitochondrial matrix (Fig.S1J). Immunoblotting of mitochondria-free cytosol and mitochondria from RA and control T cells showed low cytosolic and barely detectable mitochondrial signals in RA CD4+ T cells (Fig. 1K–L). Mitochondrial mass was similar in control and RA T cells, whereas mitochondrial MRE11A was distinctly low in RA T cells (p<0.0001) (Fig.1M–N). MRE11A knockdown in healthy CD4+ T cells suppressed the nuclease’s cytosolic and mitochondrial fraction without affecting mitochondrial mass (Fig.1O–P, Fig.S1K–L).
These data demonstrated that the MRE11Alow status of RA T cells extended to mitochondria; resulting in low mitochondrial respiration and ATP production.
MRE11A protects mtDNA against oxidization and leakage
We analyzed whether MRE11A is physically associated with mtDNA. Immunoprecipitation of mtDNA-MRE11A complexes from Jurkat T cells with anti-MRE11A antibodies verified direct binding to mtDNA (Fig.2A).
Figure 2: MRE11A protects mtDNA against oxidization and leakage.
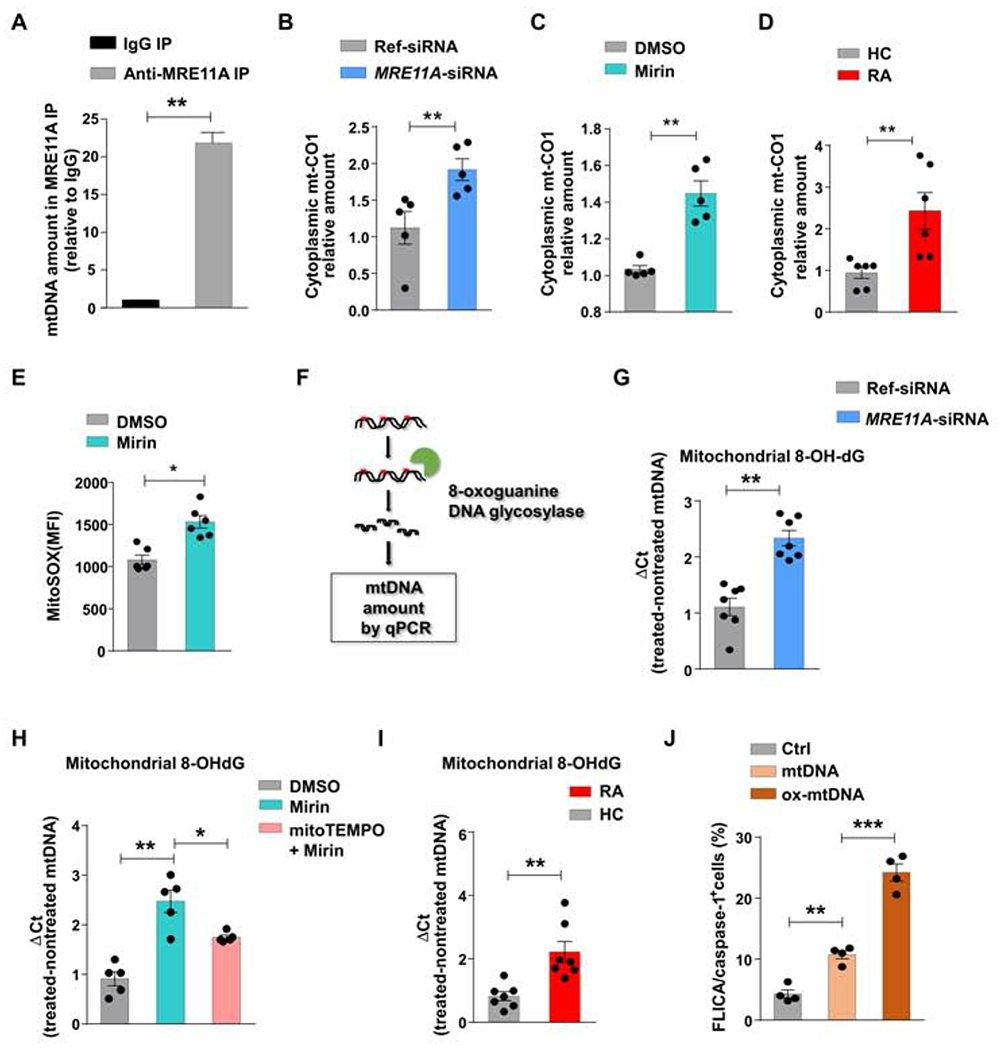
(A) Mitochondria were isolated from Jurkat T cells. mtDNA bound to MRE11A was immunoprecipitated, purified, and quantified by RT-PCR. Data from 3 experiments. (B) Healthy CD4+ T cells (n=5) were transfected with MRE11A-siRNA or reference siRNA and stimulated for 72h. Cytosolic mtDNA quantified by RT-PCR. (C) Healthy CD4+ T cells (n=5) were treated with the MRE11A inhibitor Mirin (50 μM) or vehicle. Cytosolic mtDNA measured by RT-PCR. (D) RA and control CD4+ T cells (n=6 each) were stimulated for 72h. Cytosolic mtDNA was measured by RT-PCR. (E) Flow cytometrical detection of mtROS in healthy CD4+ T cells with or without Mirin treatment. (F) Experimental scheme of measuring 8-OH-dG in mtDNA. (G) MRE11A expression in control CD4+ T cells was inhibited by MRE11A-siRNA transfection (n=7). Mitochondrial 8-OH-dG quantified by RT-PCR. (H) CD4+ T cells were pretreated with mitoTEMPO (10 μM) for 2h and then MRE11A activity was inhibited by Mirin treatment (50 μM) (n=5). Mitochondrial 8-OH-dG quantified by RT-PCR. (I) CD4+ T cells from 7 RA patients and 7 controls were stimulated for 72h. mtDNA 8-OH-dG quantified by RT-PCR. (J) Jurkat T cells were transfected with non-oxidized and oxidized mtDNA and frequencies of FLICA+ cells were measured by FACS. All data are mean ± SEM. Paired t-test (A-C), Mann Whitney (D, I), Wilcoxon signed rank test (E, G), ANOVA and pairwise comparison using Tukey’s method (H, J). *p<0.05, **p<0.01, ***p<0.001. See also Fig.S2.
To explore whether mitochondrial MRE11A contributes to mtDNA confinement, we quantified cytosolic mtDNA in healthy CD4+ T cells transfected with MRE11A siRNA (Fig.2B). MRE11A knockdown resulted in mtDNA leakage into the cytoplasmic space. MRE11A overexpression prevented mtDNA leakage (Fig.S2A). Mirin treatment yielded similar results (Fig.2C). We quantified cytosolic mtDNA in MRE11Alow RA and age-matched CD4+ T cells. Patient-derived T cells released >2-fold higher mtDNA amounts into the cytoplasm (Fig.2D).
To test whether mtDNA-bound MRE11A shields against oxidative damage, we quantified mtROS production in nuclease-intact and nuclease-inhibited CD4+ T cells (Fig.2E). Mirin treatment resulted in almost 50% higher mtROS release. We quantified mitochondrial 8-Oxo-2′-deoxyguanosine (8-OH-dG) (Fig. 2F–H), an oxidized derivative of deoxyguanosine and the most frequent oxidative lesion in mtDNA (Shimada et al., 2012). 8-OH-dG mtDNA concentrations increased by 50-100% if MRE11A activity was genetically or pharmacologically suppressed (Fig.2G–H). mitoTEMPO treatment partially protected mtDNA from oxidization (Fig.2H). Increased mtDNA oxidization was reversible with forced MRE11A overexpression (Fig.S2B). Also, oxidized mtDNA was 2.7-fold higher in RA T cells than in controls (Fig. 2I).
In macrophages, mtDNA has been implicated in inflammasome activation and caspase-1 cleavage (Shimada et al., 2012). We transfected healthy CD4+ T cells with non-oxidized and oxidized mtDNA (Fig.2J) and measured caspase-1 activation. mtDNA, but even more so the oxidized form, efficiently induced caspase-1 activation. Frequencies of FLICA+ T cells doubled after mtDNA exposure and more than tripled with oxidatively modified mtDNA. Quantification of caspase-1 activation after caspase-1 silencing validated the specificity of the FLICA probes (Fig.S2C).
Taken together, MRE11A protects mtDNA from oxidative damage and leakage into the cytosol.
MRE11A controls activation of caspase-1
Caspase-1 is autoactivated when proteolysed as part of the inflammasome complex (Martinon and Tschopp, 2004). To examine which role MRE11A plays in inflammasome activation, we determined frequencies of CD4+ T cells with activated caspase-1 and caspase-3 after T-cell receptor stimulation for 72h. In parallel, MRE11A was inhibited pharmacologically (Mirin treatment) or genetically (siRNA) (Fig.3A–B). Caspase-1 or caspase-3 activation occurred in a small fraction of healthy CD4+ T cells (2-3%). Mirin treatment more than doubled and MRE11A knockdown tripled the frequency of FLICA/caspase-1+ T cells, while caspase-3 was unaffected (Fig. 3A–B). In MRE11A siRNA-transfected CD4+ T cells, caspase-1 silencing prevented the accumulation of FLICA+ cells (Fig.S3A). We applied western blot analysis to search for pro-caspase-1 cleavage. MRE11A knockdown enhanced procaspase-1 autoproteolysis as indicated by the appearance of the p10 fragment (Fig. 3C). Reconstitution of MRE11A in MRE11A knockdown T cells prevented caspase-1 cleavage (Fig.S3B).
Figure 3. MRE11A controls activation of caspase-1.
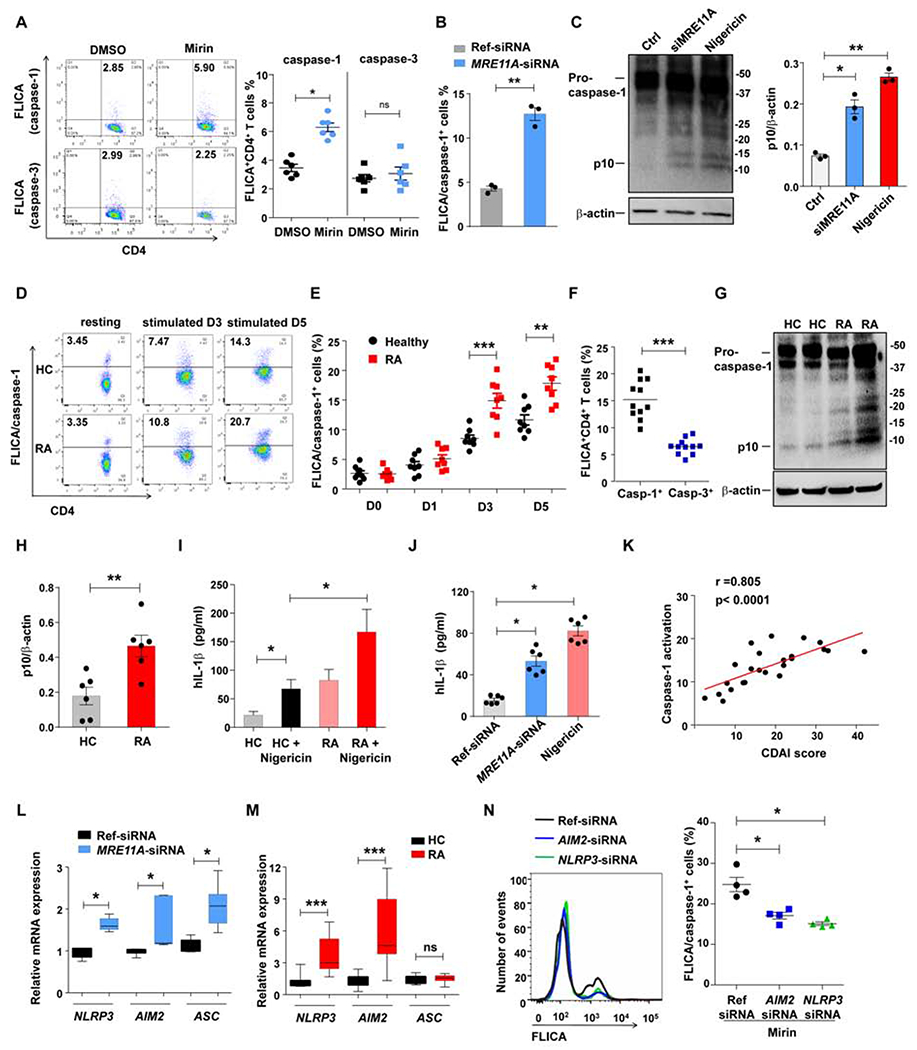
Healthy and RA CD4+ T cells were stimulated for 72h and treated as indicated. MRE11A activity was inhibited by MRE11A siRNA transfection or Mirin treatment (50 μM). The inflammasome was activated with Nigericin. Caspase-1 and caspase-3 activity was measured flow cytometrically with FLICA probes. (A) Frequencies of FLICA+CD4+ T cells after Mirin treatment. Representative scatter blots and data from 6 experiments. (B) Frequencies of FLICA/caspase-1+ CD4+ T cells after MRE11A knockdown (n=3). (C) Immunoblotting of caspase-1 cleavage in healthy CD4+ T cells treated as indicated. Quantification of p10 protein from 3 experiments. (D-F) Comparative analysis of caspase activation in control and RA CD4+ T cells. (D) Representative scatter blots of FLICA+ cells. (E) Kinetics of CD4+ FLICA/caspase-1+ T cells on day1-5 post stimulation. 8 patient-control pairs. (F) FLICA+CD4+ T cells for caspase-1 and caspase-3 measured in 11 RA patients on day 3 poststimulation. (G) Representative immunoblots for caspase-1 cleavage in post stimulation control and RA CD4+ T cells. (H) Quantification of p10 protein (n=5). (I) Concentrations of secreted IL-1β in 7 RA-control pairs. (J) Concentrations of secreted IL-1β after MRE11A knockdown. (K) Frequencies of CD4+ T cells with activated caspase-1 on day3 correlated with the clinical disease burden assessed by Clinical Disease Activity Index (CDAI). (L) NLRP3, AIM2 and ASC transcripts quantified after MRE11A knockdown in healthy CD4+ T cells. (M) NLRP3, AIM2 and ASC transcripts compared in control and RA CD4+ T cells. (N) Frequencies of FLICA/caspase-1+CD4+ T cells following NLRP3 or AIM2 knockdown and Mirin treatment. Representative histograms and data from 4 samples. All data are mean±SEM. Wilcoxon signed rank test (A, F, L), paired t-test (B, C), ANOVA and pairwise comparison using Tukey’s method (E, J, N), Mann-Whitney (H, I, M). *p<0.05, **p<0.01, ***p<0.001. See also Fig.S3.
Caspase-1 pathway activation is a typical response pattern in innate immune cells. We ruled out monocyte contamination in the T cell populations. RT-PCR-based analysis for the monocyte marker CD14 yielded a strong signal in enriched monocytes but was negative in purified T cell populations (Fig.S3C).
We tested the hypothesis that MRE11A activity in T cells is mechanistically linked to caspase-1 activation in a cohort of RA patients and age-matched controls. Resting T cells had a distinctly low signal, frequencies of T cells with activated caspase-1 peaked on day 5 (Fig. 3D–E). Higher frequencies in RA T cells were detectable after 72h and persisted till day 5. Caspase-3 activation was indistinguishable in healthy and RA T cells (Fig.S3D). A direct comparison of CD4+ T cells with activated caspase-1 versus caspase-3 revealed dominance for the caspase-1 enzyme (Fig.3F). To provide confirmatory evidence for caspase-1 autoproteolysis, we tested for the presence of the p10 fragment, a proteolytic fragment of pro-caspase-1 (Fig.3G). RA CD4+ T cells expressed substantially higher amounts of the caspase-1 cleavage product p10 (Fig. 3G–H). Also, we quantified IL-1β in the supernatant of control and RA CD4+ T cells 72h post stimulation. Patient T cells produced >3-fold more IL-1β (Fig.3I). Nigericin enhanced IL-1β secretion 4-5–fold with consistently more IL-1β released by Nigericin-stressed RA T cells. Similarly, MRE11A silencing in healthy T cells resulted in 3-fold higher IL-1β release (Fig.3J).
We questioned whether the RA disease burden, measured with the disease activity score CDAI had any impact on the frequency of CD4+ T cells with activated caspase-1. Caspase-1 activation in patients’ T cells correlated with the clinical disease activity (Fig.3K; r = 0.805).
To examine which inflammasome components contributed to the mitochondria-dependent triggering of caspase-1 proteolysis in MRE11Alow T cells, we knocked down MRE11A and measured mRNA for NLRP3, AIM2 and ASC (Fig.3L). MRE11A loss-of-function significantly enhanced transcription of all three inflammasome components. Parallel experiments testing RA T cells yielded similar results for NLRP3 and AIM2 (Fig.3M). We transfected AIM2 or NLRP3 specific siRNA into healthy CD4+ T cells before triggering inflammasome activation with the MRE11A inhibitor Mirin (Fig.S3E, Fig.3N). Blocking MRE11A’s nuclease activity enhanced caspase-1 cleavage in CD4+ T cells. Inflammasome activation was dependent on both, AIM2 as well as NLRP3 (Fig.3N).
Inflammasome and caspase-1 activation in macrophages requires two signals; the priming signal inducing pro-IL-1β and pro-IL-18 and a second danger signal promoting inflammasome aggregation/activation (Jo et al., 2016). We explored whether T cell receptor signaling is sufficient for inflammasome activation (Fig.S3F–G). CD4+ T cells stimulation with agonistic anti-CD3/CD28 beads upregulated transcription of NLRP3, AIM2 and ASC and promoted proteolytic cleavage of procaspase-1.
These data identified caspase-1 activation as a part of the T cell activation program, classified MRE11A as a physiologic protector against inflammasome assembly and connected MRE11A loss-of-function with aberrant caspase-1 activation in RA T cells.
MRE11A prevents pyroptotic T cell death
Target proteins proteolytically cleaved by activated caspase-1 promote membrane pore formation, release of intracellular content and highly inflammatory lytic cell death (Bergsbaken et al., 2009). Inhibiting MRE11A activity caused 2-3 fold higher rates of T cell death (Fig.4A, B). Similarly, MRE11A knockdown increased the propensity for T cells to die (Fig.4C). T cell viability consistently dropped among MRE11A-deficient cells (Fig.4D, Fig.S4A). MRE11Alow T cells released lactate dehydrogenase (LDH), indicative of lytic death (Fig.4E, Fig.S4B). More than 2-fold as many T cells died in RA samples compared to controls (Fig.4F). LDH release into the extracellular space was double as high in RA compared to healthy T cells (Fig.4G).
Figure 4. MRE11A prevents caspase-1-dependent pyroptotic T cell death.
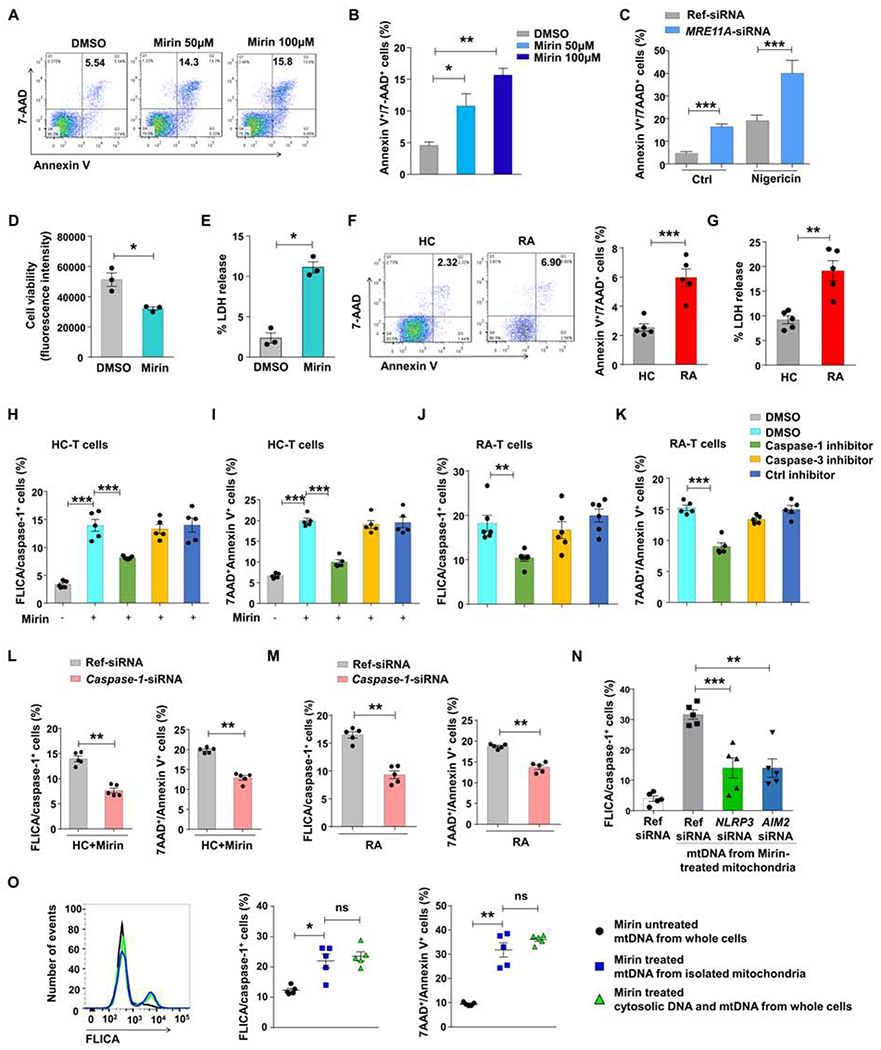
(A, B) CD4+ T cells were stimulated and treated with the MRE11A inhibitor Mirin. Cell survival was measured by flow cytometry of 7AAD+/Annexin V+ cells. Representative dot blots and data from 3 samples. (C) CD4+ T cells were transfected with MRE11A-siRNA or ref siRNA. Inflammasome activation was triggered with Nigericin (10 μM). Frequencies of 7AAD+/AnnexinV+ CD4+ T cells were measured by flow cytometry. Data from 3 samples. (D, E) Cell viability and LDH release measured in activated CD4+ T cells treated with Mirin (50 μM) (n=3). (F) T cell death quantified in control and RA CD4+ T cells day3 post stimulation. Flow cytometry for Annexin V+/7AAD+CD4+ T cells. Representative scatter blots and data from 5 patient-control pairs. (G) T cell death quantified by LDH release in 5 RA patients and 5 controls. (H, I) Healthy CD4+ T cells were pretreated with caspase inhibitors (50 μM; 48 h) before MRE11A activity was blocked with Mirin. Frequencies of FLICA/caspase-1+ cells (H) and Annexin V+/7AAD+ cells (I) measured by flow. (J, K) RA CD4+ T cells were pretreated with caspase inhibitors (50 μM). T cell death quantified by FLICA/caspase-1+ (J) and Annexin V+/7AAD+ (K) cell frequencies. (L, M) siRNA mediated knockdown of caspase-1 in control and RA CD4+ T cells prior to Mirin treatment Frequencies of FLICA/caspase-1+ and Annexin V+/7AAD+ cells measured by flow cytometry (n=5). (N) Frequencies of FLICA/caspase-1+ CD4+ T cells after NLRP3 or AIM2 knockdown and transfection of mtDNA isolated from mirin-treated mitochondria (O). Control CD4+ T cells or isolated mitochondria were treated with Mirin. mtDNA was isolated from mitochondria and mtDNA plus cytosolic DNA from the whole cells and transfected into CD4+ T cells. Control mtDNA derived from untreated whole cells. Frequencies of FLICA/caspase-1+ cells and cell death were determined by flow cytometry. All data are mean±SEM. ANOVA and pair-wise comparison using Tukey’s method (B, H-K, N, O), paired t-test (C-E, L, M), t-test (F, G). *p<0.05, **p<0.01, ***p<0.001. See also Fig.S4.
To confirm that MRE11Alow T cells undergo caspase-1 dependent cell death, we applied a caspase-1 inhibitor (Z-YVAD-FMK), a caspase-3 inhibitor (Z-DEVE-FMK) or a control inhibitor, while MRE11A activity was blocked with Mirin. Abolishing caspase-1 activation selectively reduced the frequencies of FLICA/caspase-1+ T cells and protected the cells from dying (Fig.4H–I, Fig.S4C). Similarly, caspase-1 activation and T cell death amongst RA T cells was sensitive to caspase-1, but not caspase-3 inhibition (Fig.4J–K, Fig.S4D–E). T cell death and caspase-1 activation in RA T cells and in MRE11A-deficient healthy T cells were caspase-1 dependent, as further demonstrated by silencing experiments (Fig.4L–M).
MRE11A loss-of-function affects nuclear as well as mitochondrial DNA stability. We first examined whether inhibition of mitochondrial MRE11A triggered caspase-1 activation and which inflammasome components recognized mtDNA (Fig.4N). Following knockdown of AIM2 or NLRP3 by siRNA technology, T cells were transfected with mtDNA isolated from Mirin-treated mitochondria. mtDNA induced robust caspase-1 activation, and both, AIM2 and NLRP3 participated in mtDNA-induced inflammasome activation. Also, we investigated whether cytosolic leakage of nuclear DNA contributed to inflammasome activation. Detection of cytosolic DNA by STING induces type I IFN production, which initiates NLRP3-dependent inflammasome activation (Gaidt et al., 2017). Type I interferon responses quantified by IFNB and IFIT1 transcript induction were distinctly low in RA T cells (Fig.S4F–G). We inhibited MRE11A nuclease activity with Mirin, treating either intact T cells or isolated mitochondria. mtDNA from untreated cells, mtDNA from Mirin-treated mitochondria or mtDNA plus cytosolic DNA isolated from treated whole cells were transfected into CD4+ T cells (Fig.4O). mtDNA from isolated mitochondria and the combination of mtDNA and cytosolic DNA were equally potent in inducing caspase-1 activation and cell death.
Together, these data revealed the susceptibility of MRE11Alow T cells to caspase-1–dependent death and linked mitochondrial intactness to T cell survival in RA patients.
Caspase-1 activation in tissue-residing T cells
To explore whether aberrant caspase-1 activation was a feature of tissue-residing T cells, we applied dual-color immunohistochemistry (IHC) in lymph node (LN) biopsies from RA patients and age-matched controls (Fig.5A–B) staining for cleaved caspase-1 in CD3+ T cells. Lymph node sections from patients with HIV infection served as controls (Doitsh et al., 2014). Antibody staining for cleaved caspase-1 was essentially negative in normal LN. In HIV-infected LN, CD3+ and CD3− cells were positive for cleaved caspase-1. In RA LN, about 4% of T cells stained positive for cleaved caspase-1. T cell density was lower than in healthy controls, but T cell compartmentalization was maintained. In HIV-infected specimens, the T cell density, even in the T cell zones, was visibly reduced.
Figure 5: Caspase-1 activation in tissue-residing T cells.
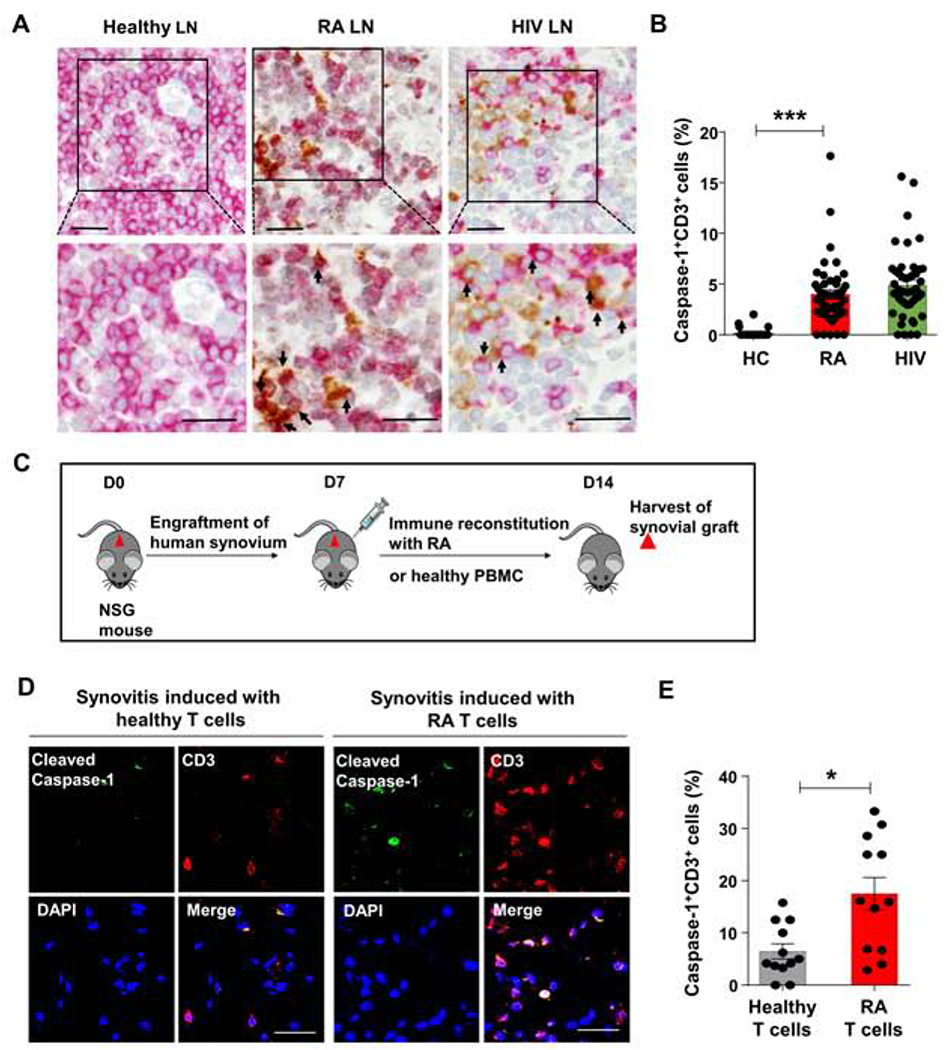
(A, B) Lymph nodes were collected from diagnostic biopsies in patients with RA or HIV infection. (A) Dual-color immunohistochemistry for cleaved caspase-1 (brown) and CD3 (red) in tissue sections from healthy, RA and HIV-infected lymph node (LN). Arrows mark double positive cells. Representative images from 3 lymph node samples each. Scale bar; 20 μm. (B) Frequencies of CD3+ T cells with cleaved caspase-1 in 10 randomly selected high-powered fields (HPF). Data are mean ± SEM. ANOVA and pairwise comparison using Tukey’s method. (C) Experimental scheme. NSG mice engrafted with human synovial tissue were reconstituted with PBMCs from healthy controls or RA patients. Synovial grafts were explanted for imaging analysis. (D) Dual-color immunohistochemistry of cleaved caspase-1 (green) and CD3 (red). Scale bar; 20 μm. (E) Percentages of CD3+ cleaved caspase-1+ T cells. Mean ± SEM compared by Wilcoxon test. *p<0.05, ***p<0.001. See also Fig.S5.
To extend these findings into synovial tissue, we made use of a humanized mouse model of RA (Li et al., 2016; Shen et al., 2017; Yang et al., 2016). Synovial tissue was engrafted into NSG mice, which were reconstituted with either healthy or RA-derived peripheral blood mononuclear cells (Fig.5C) and synovial grafts were explanted 7-10 days later. Dual-color IHC in explanted synovium visualized cleaved caspase-1+ T cells (Fig.5D–E). In mild synovitis, caspase-1 cleavage was infrequent, detected in 6% of the tissue-residing T cells. Moderate-to-severe synovitis contained dense synovial infiltrates and caspase-1 was cleaved in 17% of T cells. In contrast, caspase-3 cleavage in tissue-residing T cells occurred rarely (Fig.S5A).
Thus, RA T cells in secondary lymphoid tissues as well as in inflammatory lesions are predisposed to cleave caspase-1, shortening their survival and providing a trigger for tissue inflammation.
MRE11A regulates caspase-1 activation and mtDNA leakage in tissue-residing T cells
To investigate whether and how MRE11A regulates inflammatory T cells, we utilized a humanized NSG mouse model (Fig.S6A). To mimic the MRE11Alow state of RA T cells, healthy PMBCs were transfected with MRE11A siRNA before the adoptive transfer. MRE11A knockdown markedly increased synovial T cell density (Fig.6A). Cleaved caspase-1 became visible in cell clusters, including CD3+ T cells. MRE11A loss-of-function resulted in doubling of T cells cleaving caspase-1 (Fig.6B). Amounts of 8-OH-dG mtDNA deposited in the tissue were >3-fold higher after the transfer of MRE11A siRNA transfected cells compared to controls (Fig.6C). Lowering MRE11A broke tissue tolerance and resulted in robust tissue inflammation (Fig.6D). The inflammatory signature in tissues exposed to MRE11Alow cells included the innate cytokines TNFA, IL6 and IL1B and TNFSF11 (encoding for RANKL), a marker of bone-erosive inflammation. IL10 and TGFB1 transcripts were abundant in control tissues, but distinctly downregulated after MRE11A repression (Fig.6E).
Figure 6: MRE11A regulates caspase-1 activation and mtDNA leakage in tissue-residing T cells.
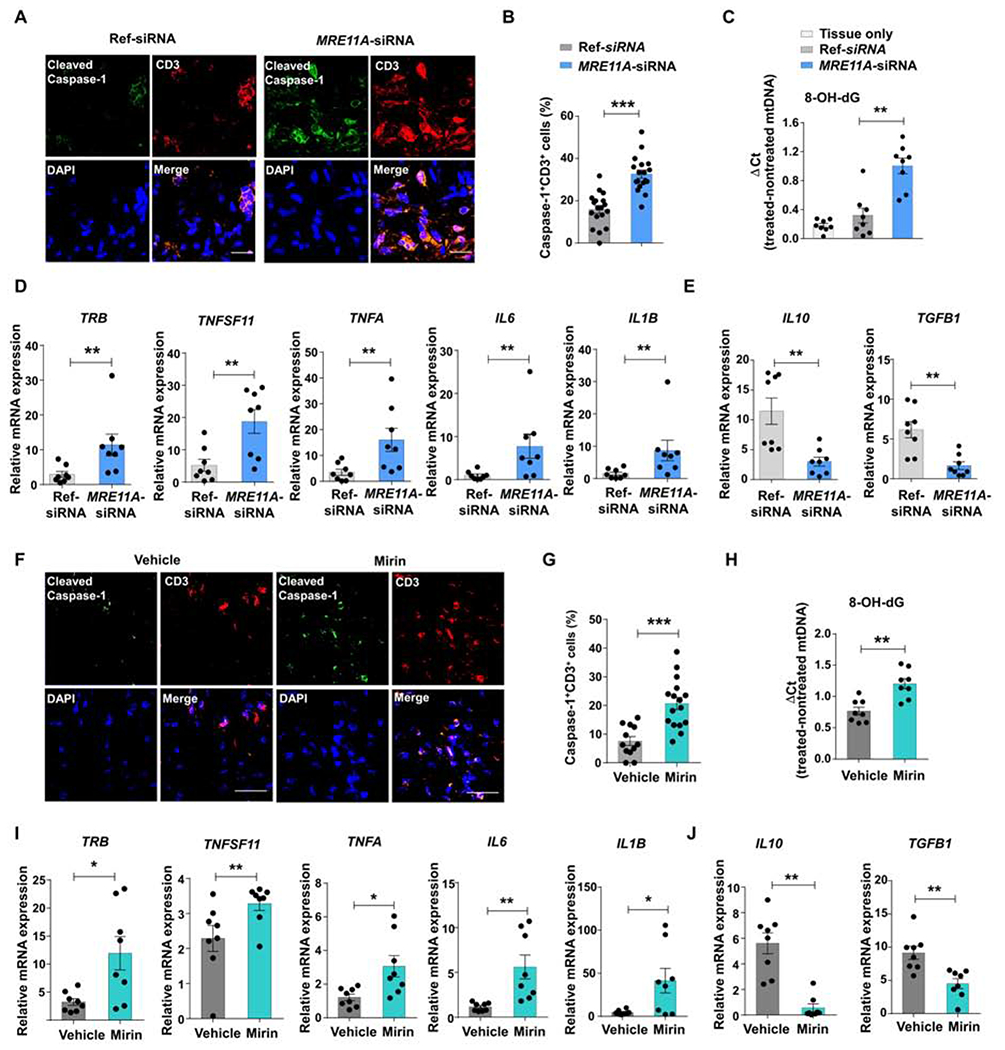
NSG mice engrafted with human synovial tissue were reconstituted with healthy PBMCs. MRE11A was silenced in PBMCs before transfer or by daily injection of the MRE11A inhibitor Mirin (or vehicle). Control mice had human synovial tissue only. (A) Dual-color immunohistochemistry for cleaved caspase-1 (green) and CD3 (red) in synovial explants. Scale bar; 20 μM. (B) Frequencies of CD3+ T cells with cleaved caspase-1+ quantified in 18 random HPF. (C) Tissue mtDNA was extracted and 8-OH-dG was determined as in Figure 2F. (D, E) Tissue transcriptome analysis for inflammation-related genes by RT-PCR. (F) Representative microphotographs from tissue IHC stained for cleaved caspase-1 (green) and CD3 (red). Scale bar; 20 μM. (G) Percentages of CD3+ T cells expressing cleaved caspase-1 (12 random HPF). (H) 8-OH-dG measured in mtDNA extracted from synovial grafts. (I, J) Tissue transcriptome analysis for inflammation-related genes by RT-PCR. Mean ± SEM compared by Wilcoxon test. *p<0.05, **p<0.01, ***p<0.001. See also Fig.S6.
Alternatively, we treated NSG chimeras reconstituted with healthy peripheral blood mononuclear cells (PBMCs) with the MRE11A inhibitor Mirin (Fig. S6A). Blocking MRE11A’s nucleolytic activity prompted T cell recruitment (Fig. 6F), >20% of synovial T cells cleaved caspase-1 (Fig.6G) and markedly higher amounts of mtDNA with oxidized 8-OH-dG sites were deposited in the tissues (Fig.6H). The tissue transcriptome signature of Mirin-treated mice resembled that from the MRE11A siRNA experiments (Fig.6D). The profile included upregulation of T cell receptor transcripts, strong induction of TNFSF11, TNFA, IL6, IL1B and suppression of IL10 and TGFB1 (Fig.6I, J).
Together, these data linked MRE11A to the protection of tissue mtDNA and the triggering of caspase-1 in tissue-infiltrating T cells. Overall, MRE11A emerged as a mitochondrial protector and upstream regulator of damage-associated molecular pattern (DAMP) sensing.
MRE11A overexpression and caspase inhibition protect synovial tissue against inflammatory attack
Placing MRE11A at the pinnacle of a mitochondrial protection program, we devised two therapeutic interventions to prevent mtDNA from triggering inflammatory disease; MRE11A gain-of-function experiments and pharmacologic blockade of caspase-1 activity.
MRE11A overexpression in RA T cells replenished the cytosolic pool of the nuclease (Fig.S7A). Restoring MRE11A protein expression enhanced OCR and ATP production (Figure 7A–C). Restoring MRE11A effectively reduced cytoplasmic mtDNA leakage (Fig. 7D) and shielded mtDNA against oxidation (Fig.7E). MRE11A gain-of-function reduced frequencies of CD4+ T cells with active caspase-1 from 45% to 18% (Fig.7F–G) and prolonged T cell survival. >20% of control plasmid-transfected T cells died (Fig.7H), while death rates fell below 10% after MRE11A restoration. We overexpressed both wildtype MRE11A and a nuclease-deficient construct in RA T cells. The construct containing mutated MRE11A left caspase-1 activation unaffected (Fig.S7B).
Figure 7. MRE11A protects synovial tissue from inflammation.
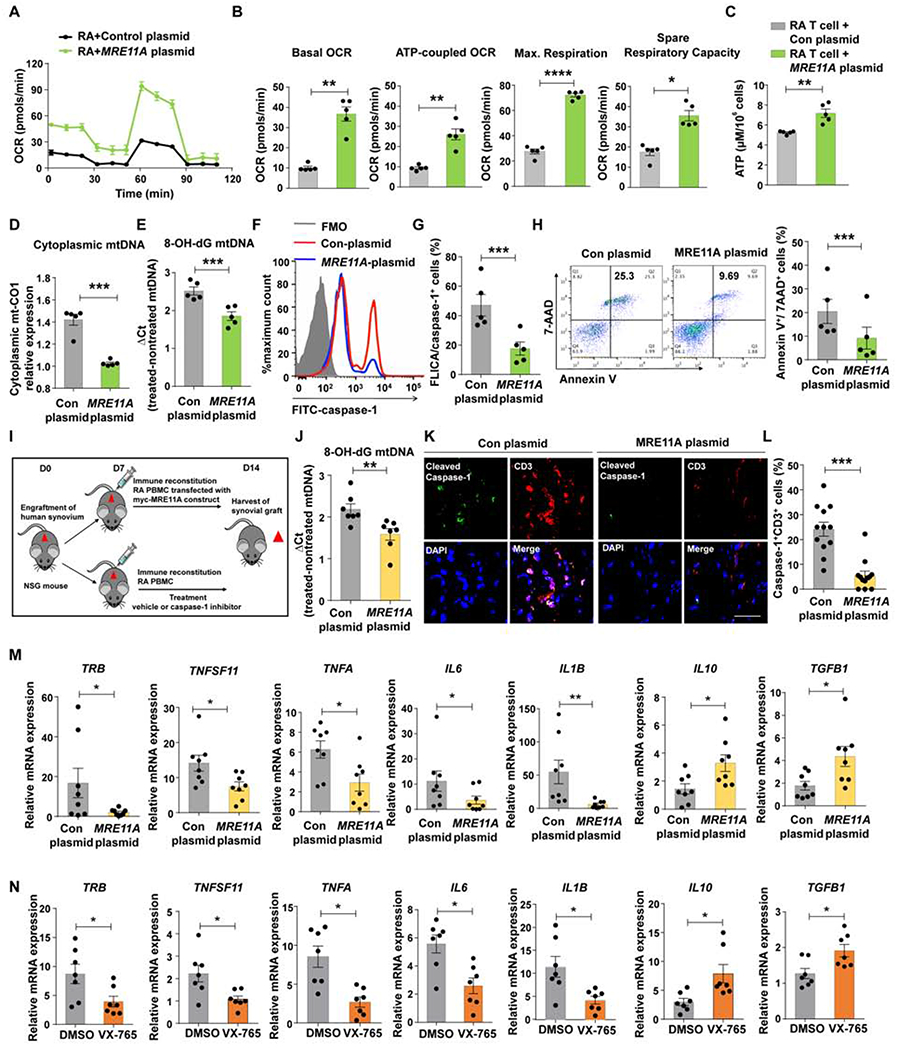
RA CD4+ T cells were transfected with MRE11A constructs to restore MRE11A expression. (A-C) Oxygen consumption rates (OCR) were traced with Seahorse Bioscience XF96 analyzer and mitochondrial function was probed as in Fig.1 (A) Summarized OCR tracings. (B) Baseline respiration, respiration coupled to ATP production, maximal respiration and spare respiratory capacity. (C) Intracellular ATP concentrations. (D) Effect of MRE11A overexpression on cytosolic mtDNA leakage. (E) Oxidized mtDNA (8-OH-dG) in RA T cells after MRE11A overexpression. (F-G) Representative histogram of FLICA/caspase-1+ CD4+ T cells after MRE11A overexpression. Results from 5 experiments. (H) Representative scatter blot of Annexin V+/7-AAD+ T cells after MRE11A overexpression. (I-N) RA PBMC were transfected with MRE11A constructs or control plasmids and adoptively transferred into NSG mice engrafted with human synovium. Alternatively, chimeric mice were treated with the caspase-1 inhibitor VX-765. Data from 7 synovial grafts in each treatment arm. (I) Experimental scheme. (J) Oxidized 8-OH-dG quantified in mtDNA extracted from synovial grafts. (K) Immunohistochemical staining of synovial tissue sections for cleaved caspase-1 (green) and CD3+ T cells (red). (L) Frequencies of CD3+ T cells with cleaved caspase-1+ in 7 synovial grafts. (M) Tissue transcriptome analysis (RT-PCR) from 8 explants harvested from MRE11A overexpression experiments. (N) Tissue transcriptome analysis (RT-PCR) of synovial explants of vehicle or caspase-1 inhibitor treated mice. All data are mean±SEM. Paired t-test (B-E, G, H), Wilcoxon test (J, L, M, N). *p<0.05, **p<0.01, ***p<0.001. See also Fig.S7.
We examined MRE11A’s protective effects in the chimeric mice (Fig. 7I). RA T cells were powerful inducers of tissue mtDNA leakage, caspase-1 cleavage and tissue inflammation (Fig. 7J–M). MRE11A overexpression suppressed tissue inflammation, reduced transcripts of innate (IL1B, IL6, TNFA) and adaptive markers (TRB, TNFSF11) and maintained tissue protective molecules (IL10, TGFB1) (Fig.7M).
Treatment with the caspase-1 inhibitor VX-765 mirrored the anti-inflammatory effects of MRE11A overexpression (Fig.7N). Gene expression of TNFA, IL6 and IL1B were significantly reduced by inhibitor treatment, while expression of IL10 and TGFB1 were preserved.
Together, these data identified MRE11A as a protector of mitochondrial DNA stability, shielding against caspase-1 induced pyroptotic death and tissue inflammation.
Discussion
A hallmark of RA T cells is their metabolic signature of suppressed glycolysis and impaired mitochondrial function combined with high biosynthetic activity, e.g., lipogenesis generating tissue-invasive membrane structures. Altered bioenergetic strategies of RA T cells directly feed their disease-promoting functions. Here, we have identified MRE11A loss-of-function as a mechanism underlying mitochondrial failure in RA T cells, mechanistically connecting genomic and metabolic instability. We have localized MRE11A to the cytoplasm and mitochondria, where the nuclease orchestrates a mitochondrial stress defense program. Our data link MRE11A’s nucleolytic activity to mitochondrial oxygen consumption and ATP generation, but also to mtDNA containment and oxidative protection. In MRE11Alow T cells, energy production failed and oxidized mtDNA leaked into the cytoplasm. Mitochondrial deprotection affected T cell function twofold: (1) metabolic malfunction with low ATP and ROS generation; (2) mtDNA instability with cytoplasmic leakage, triggering of inflammasome assembly, caspase-1 activation and mitochondria-induced pyroptotic death. Taken together, the nuclease functions as a mitochondrial protector and MRE11A deficiency turns T cells into inherently inflammatory effector cells.
Cell-intrinsic metabolic programs are now considered important risk factors in cancer and inflammation (Andrejeva and Rathmell, 2017; Huang and Perl, 2018; Weyand and Goronzy, 2017). Energy production, nutrient utilization and metabolite synthesis render hosts susceptible to aberrant and tissue-damaging immunity through multiple pathways. In innate immune cells, the unifying theme is excessive mitochondrial activity, supplying pro-inflammatory ROS (Palsson-McDermott et al., 2015; Shirai et al., 2016), Krebs cycle intermediates (Tannahill et al., 2013) and energy to fuel tissue-destructive effector functions. In RA, pro-inflammatory effector functions are suppressed by pro-oxidants, replenishing intracellular ROS (Yang et al., 2016) or by inhibiting fatty acid synthesis (Shen et al., 2017). Here, mitochondrial inactivity nurtures autoimmunity through ROS-dependent cell cycle dysregulation and failed fatty acid oxidation, supplying pathogenic T cells with biosynthetic precursors. In systemic lupus erythematosus (SLE), mitochondrial depression supports development of short-lived effector CD8 T cells (Ling et al., 2018). A mitochondrial checkpoint is critical for T cell fate decisions into short-lived effector cells (low mitochondrial activity) versus long-lived memory cells (high mitochondrial activity) (Geltink et al., 2018; Johnson et al., 2016). In SLE-prone mice, the defect was attributed to lack of complement component C1q, which binds to a mitochondrial matrix protein (Muta et al., 1997) and stimulates mitochondrial biogenesis. Thus, targeting metabolic pathways and reprogramming T cell metabolism become potential strategies to enhance immunotherapy (Sukumar et al., 2017; Sukumar et al., 2016). Current data emphasize the contribution of DNA repair processes to mitochondrial intactness and bioenergetics. MRE11A deficiency, either spontaneous in RA T cells or induced in healthy T cells, resulted in low mitochondrial respiration and poor ATP production, and rendered T cells tissue inflammatory.
How a DNA repair nuclease interferes with mitochondrial intactness and mtDNA protection is unknown. MRE11A’s exonucleolytic function was needed for mitochondrial protection, as the inhibitor Mirin specifically targets exonucleolysis (Shibata et al., 2014). Mirin treatment produced tissue-injurious T cells in vitro and in vivo. While primarily a nuclear protein, we detected cytoplasmic and mitochondrial MRE11A. Involvement in mtDNA repair processes would explain why MRE11A loss profoundly affected ATP production and mtDNA containment. Organized in DNA-protein complexes, mitochondrial nucleoids are co-packaged with multiprotein machineries that regulate mtDNA replication, transcription, and repair. A major consequence of MRE11A loss-of-function was cytoplasmic leakage of mtDNA, detected by the innate immune sensors NLRP3 and AIM2 (Zhong et al., 2018). Being histone-free and close to the ROS source, mtDNA is highly susceptible to oxidative modification (Apel and Hirt, 2004), but cytosolic misplacement of mtDNA appears sufficient to trigger inflammasome assembly and caspase activation.
The current study provides insights into how T cells monitor bioenergetic fitness and mitochondrial integrity and how ATP-low T cells are removed from the pool. Mitochondria serve as key platforms for inflammasome activation and cell death regulation (Zhou et al., 2011). Danger signals are believed to selectively trigger inflammasome activation; e.g., cell damage and K+ efflux activate the NLRP3 inflammasome, whereas the AIM2 inflammasome responds to cytosolic bacteria and DNA viruses (Jo et al., 2016; Rathinam et al., 2010). However, AIM2 engages in the activation of a noncanonical NLRP3 inflammasome (Cunha et al., 2017), compatible with cross-regulation amongst different inflammasomes. Current data suggest involvement of both the NLRP3 and AIM2 inflammasome in the monitoring of mitochondrial stress. NLRP3 inflammasome has been connected to aberrant tissue inflammation (Youm et al., 2015). Adipose tissue macrophages reduce lipolysis by degrading catecholamines; connecting metabolism, immunity and NLRP3 inflammasome-mediated disease (Camell et al., 2017).
Inflammasome and caspase-1 activation typically require two signals; the priming signal upregulating pro-IL-1β and a danger signal promoting inflammasome platform aggregation. Here, we show that T cell receptor signaling alone induces inflammasome genes and promotes procaspase-1 cleavage; confirming previous data reporting that CD4+ T cells constitutively express high levels of cytoplasmic pro-IL-1β, NLRP3 and ASC and that quiescent CD4+ T cells die by caspase-1-mediated pyroptosis (Doitsh et al., 2014). Inflammasome activity regulates T cell lineage commitment (Arbore et al., 2016; Feriotti et al., 2017) and current data point towards a role in T cell culling. When stressed bioenergetically, T cells underwent a lytic and pro-inflammatory death; likely, by engaging the membrane pore-forming caspase-1 target molecule gasdermin D (Liu et al., 2016). Protein expulsion into the extracellular space endows T cells with additional abilities beyond cytokine production or cell-to-cell signaling. Synovial mtDNA deposition suggested that mitochondria directly contribute to the intracellular proteins expulsed by pyroptotic T cells and added T cell pyroptosis to the list of pathways inducing chronic tissue inflammation. Lymph nodes collected from HIV-infected patients and RA patients suggested that caspase-1-dependent T cell death might determine T cell compartment size. Like RA patients, HIV infected individuals suffer from premature immune aging (Boulias et al., 2016), possibly a result of excessive T cell loss. The data suggest a mechanistic connection between bioenergetic fitness, inflammasome activation, T cell survival and T-cell-dependent tissue inflammation.
In summary, we report that MRE11A directs a cytoprotective program by securing energy production, repressing mitochondrial stress-induced inflammasome activation and controlling T cell longevity. The data have implications beyond the understanding of RA pathogenesis in that integrity of the mitochondrial genome is placed upstream of cellular stress sensing and programed T cell death; and identify the mitochondrion as the organelle integrating DNA repair with metabolic activity and cellular life-death decisions. Several of the molecular abnormalities seen in RA T cells are present in old T cells, albeit to a lower degree. MRE11A repression has implications for DNA double-strand repair, telomeric maintenance and mitochondrial function. Therapeutic targeting of the nuclease has the potential to restore bioenergetic competence, prolong T cell life and avoid T cell-mediated tissue injury in multiple clinical settings; including RA, the aging host, HIV infection and anti-tumor immunotherapy.
Study Limitations
Our study mechanistically connects mitochondrial fitness, DNA repair, T cell death and tissue inflammation. Pathways through which MRE11A participates in mitochondrial DNA repair and electron transport chain function remain undefined. How mitochondrial DNA reaches the cytoplasm is still unclear. In addition, further studies need to address whether the mechanisms of tissue inflammation outlined here extend beyond rheumatoid arthritis and have relevance in other chronic inflammatory diseases.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cornelia M. Weyand (cweyand@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Synovitis Induction in Chimeric Mice
Chimeric mice engrafted with human synovial tissue and reconstituted with human peripheral blood mononuclear cells were generated as previously described (Yang et al., 2016). Pieces of human synovium were transplanted into a subcutaneous pocket on the dorsal midline of NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (80 mice; 58 female and 22 male; 10 to 14 weeks old; The Jackson Laboratory). Following engraftment, the mice were immune-reconstituted with 10 million human PBMCs that either derived from RA patients or from age-matched healthy controls. A total of 30 patients were recruited for the immune reconstitution experiments; including 24 females and 6 males at an average age of 54.7 years. For interventional studies, chimeric mice from the same litter and engrafted with the same synovial tissue were randomly assigned to parallel treatment arms. In experiments testing the effect of pharmacologic inhibition of MRE11A, engrafted mice were reconstituted with PBMC and injected with vehicle control or Mirin (1 mg/kg/day) for 7 days. Alternatively, MRE11A was knocked down by transfecting PBMC with either reference siRNA or MRE11A specific siRNA. Ten million transfected cells were adoptively transferred into the mice. To explore how overexpression of MRE11A affects synovitis induction, PBMCs were isolated from RA patients and transfected with either a control plasmid or a MRE11A overexpression plasmid. Transfected cells were rested for 24h before the adoptive transfer. In the caspase-1 inhibitor trial, mice were engrafted and immune-reconstituted and then treated with daily i.p. injections of vehicle control or VX765 (50 mg/kg) for 7 consecutive days. At the end of the experiments, synovial grafts were harvested from sacrificed mice and processed for immunostaining (embedded in OCT, Tissue-Tek; Sakura Finetek) or tissue transcriptome analysis by RT-PCR (Shirai et al., 2016). Mice were maintained in a pathogen-free barrier facility under temperature-controlled (20-22°C) conditions with 12 h light/12 h dark cycles, and ad libitum access to autoclaved rodent chow and water. All animal experiments were approved by the Stanford APLAC committee.
Patients and Control Individuals
A cross-sectional cohort of 145 RA patients was recruited from Stanford Hospital and Clinics. All patients fulfilled the diagnostic criteria for RA and were positive for anti-CCP antibodies or for rheumatoid factor (Table S1). Age- and gender-matched healthy controls were enrolled through the Stanford Blood Center. The lymph node biopsies from the HIV and RA patients were done for diagnostic purposes. The control samples derived from tissue resections in patients with non-neoplastic non-infectious conditions. All samples were de-identified. The studies were approved by Stanford University Institutional Review Board and written informed consent was obtained from all participants.
T Cells Purification and Culture
Peripheral blood mononuclear cells (PBMCs) were separated from whole blood using Lymphocyte Separation Medium (Corning). Total CD4+ T cells were isolated from PBMCs using a human CD4+ T cell isolation kit (STEMCELL Technologies, # 17952). To induce effector T cells, purified CD4+ T cells were stimulated for 72h using anti-CD3/CD28 beads (Life Technologies, # 11132D) at a ratio of 1 bead per 2 cells.
METHOD DETAILS
Isolation of Cytoplasmic, Mitochondrial and Nuclear Fractions
CD4+ T cells were stimulated with anti-CD3/CD28 beads for 72h. Cytosolic, mitochondrial, and nuclear fractions were prepared using nuclear and cytoplasmic extraction reagents (78833, Thermo Fisher Scientific) followed by a mitochondrial isolation kit (89874, Thermo Fisher Scientific) according to the manufacturer’s instructions.
Measurement of Mitochondrial Oxygen Consumption Rates and Extracellular Acidification Rates
CD4+ T cells stimulated for 72h were transferred to Cell-Tak solution-coated 96-well plates (200,000 cells/175 μl/well) (Seahorse XFe96 FluxPak mini, Agilent Technologies, 102601-100). Thirty minutes before assessment, cells were incubated in a CO2-free incubator in unbuffered RPMI 1640 medium (Sigma R1383). For assessment of mitochondrial respiratory activity, cells were treated with oligomycin (1.5 μM), FCCP (1.5 μM), and Rotenone/Antimycin A (0.5 μM) respectively, at time-points indicated using a Seahorse XF Cell Mito Stress Test Kit (Agilent Technologies, 103015-100). To examine glycolytic activity, cells were treated with glucose (10 mM), oligomycin (1 μM) and 2-DG (50 mM), respectively. Mitochondrial oxygen consumption rates (OCR) and extracellular acidification rates (ECAR) were measured using an XF96 extracellular analyzer (Seahorse Bioscience).
Quantification of Intracellular ATP
CD4+ T cells were stimulated for 72h and transferred to a 96-well plate. Cellular ATP amounts were measured with a luminescent ATP Detection Assay Kit (Abcam, ab113849) according to the manufacturer’s protocol. Luminescence was measured by an Infinite M1000 Proplate Reader (Tecan).
Assessment of Activated Effector Caspases
Activated caspase-1 or caspase-3 was detected with the FAM-FLICA® Caspase-1 Assay Kit (ImmunoChemistry TECHNOLOGIES, #97) or FAM-FLICA® Caspase-3/7 Assay Kit (ImmunoChemistry TECHNOLOGIES, #93) according to the manufacturer’s instructions respectively. The fluorescent probe FAM-YVAD-FMK labels active caspase-1 and FAM-DEVD-FMK labels active caspase-3 in living cells. CD4+ T cells were incubated with FLICA solution at 37°C protected from light for 1h, and then washed with 1X Apoptosis Wash Buffer. Fluorescence-labeled cells were detected with a flow cytometer.
Immunoblot Analysis of Procaspase-1 Cleavage
CD4+ T cells were collected in lysis buffer and proteins were resolved by 4-15% SDS-PAGE (Bio-Rad, 4561083). Proteins were then transferred to PVDF membranes (Bio-Rad, 1620177) followed by incubation with 5% milk in TBST blocking buffer and caspase-1 antibody (Santa Cruz Biotechnology, sc-56036) to detect pro-caspase-1 cleavage. SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific 34094) was used to detect protein.
Transfection of T Cells
For knockdown experiments, CD4+ T cells were transfected with reference siRNA or the indicated specific siRNA using Amaxa Human T Cell Nucleofector Kits (Lonza, VPA-1002). MRE11A-specific siRNA was purchased from Thermo Fisher Scientific (HSS1429690), caspase-1 siRNA was from Santa Cruz Biotechnology (sc-29235), NLRP3 siRNA was from Dharmacon (L-017367-00-0005) and AIM2 siRNA was from Invitrogen (1299001).
For overexpression experiments, CD4+ T cells were transfected with empty pcDNA vector, shRNA resistant hMre11 WT-FLAG in pcDNA5 frt/to or shRNA resistant hMre11 H129N-FLAG in pcDNA5 frt/to (generous gift from Tanya Paull, The University of Texas at Austin) using Amaxa Human T Cell Nucleofector Kits.
Immunohistochemical Staining of Paraffin-Embedded Sections
Paraffin-embedded sections were deparaffinized using a standard technique. Antigens were retrieved in Target Retrieval Solution (pH 6.0, Dako, S1699) at 95°C for 20 min, and endogenous peroxidase was blocked in 3% H2O2 for 15 min to reduce background or unspecific staining. Deparaffinized tissues were blocked by 2% goat serum at RT for 45 min and then incubated with anti-human cleaved caspase-1 (1:50, Biorbyt, orb126550) and anti-human CD3 (1:50, Dako, M725401-2) overnight at 4°C. Immunohistochemistry was carried out using ImmPRESS™ HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit (Vector Laboratories, MP-7451), ImmPRESS™-AP Anti-Mouse IgG (alkaline phosphatase) Polymer Detection Kit (Vector Laboratories, MP-5402), VECTOR Red Alkaline Phosphatase (AP) Substrate Kit (Vector Laboratories, SK-5100) and DAB Peroxidase (HRP) Substrate Kit, 3,3′-diaminobenzidine kit (Vector Laboratories, SK-4100). Tissues were counterstained with Mayer’s Hematoxylin solution (Sigma-Aldrich, MHS16-500ML) for 45 secs, washed twice with 1x PBS, and mounted with mounting medium.
Immunohistochemical Staining of Frozen Sections
Tissues slides were placed at room temperature for 60 minutes and fixed in ice cold acetone for 20 min, followed by 2x washing in 1x PBST (0.05% Tween-20). Staining procedures followed previously published protocols (Li et al., 2016). Tissue sections were incubated with primary antibodies (anti-human CD3 (1:50, Dako, M725401-2) and anti-human cleaved caspase-1 (1:50, Biorbyt, orb126550)) for overnight at 4°C. After washing, slides were incubated with secondary goat anti-mouse IgG H&L (DyLight 488) (Abcam, ab96879) and goat anti-rabbit IgG (H+L), Alexa Fluor 594 (Invitrogen, R37117) antibodies for 1 h. Nuclei were counterstained with DAPI and stained tissues were imaged with an Olympus BX41 microscope. Stainings were quantified in a blinded manner.
Quantitative Immunostaining
Cells were fixed in 4% paraformaldehyde, permeabilized with 0.05% Triton X-100, and incubated with primary antibodies to MRE11A (Abcam, ab214) at 4°C overnight. Incubation with secondary antibodies was performed at room temperature for 1 hr using Alexa Fluor 488 labeled goat anti-mouse (Abcam, ab96879). Images were analyzed using the LSM710 microscope system with ZEN 2010 software (Carl Zeiss) and a 63× oil immersion objective (Carl Zeiss). Image analysis procedures were performed blinded.
Determination of the 8-OHdG Modifications in Mitochondrial DNA
Mitochondria were purified from cultured cells (89874, Thermo Fisher Scientific) or tissues (89801, Thermo Fisher Scientific) using appropriate kits. Mitochondrial DNA was then isolated using DNeasy Blood &Tissue Kit (69504, Qiagen). Oxidatively modified 8-hydroxy-2′-deoxyguanosine (8-OH-dG) was quantified using q-PCR. 4 μl of DNA sample (6.25 ng/μl) was treated with 8.7 μl RNase free water, 1.5 μl 10X NE buffer, 0.15 μl 100X BSA, and 0.625 μl human 8-oxoguanine glycosylase (Fpg) (treatment mixture) or RNase free water (nontreatment mix). Mixtures were incubated at 37°C for 1 hr for digestion. A fragment of mtDNA was amplified by qPCR with the following primers: forward: 5′-GTGCTATAGTGGAGGCCGGA-3′ and reverse: 5′-GGGTGGGAGTAGTTCCCTGC-3′.
Determination of Cytosolic mtDNA Amount
Mitochondria-free cytosol was prepared first using mitochondrial isolation kits (89874, Thermo Fisher Scientific). Subsequently, cytosolic DNA was collected with a DNeasy Blood &Tissue Kit (69504, Qiagen). qPCR was performed to detect mitochondrial DNA in the cytoplasmic fraction using the following primers: forward: 5′-GTGCTATAGTGGAGGCCGGA-3′ and reverse: 5′-GGGTGGGAGTAGTTCCCTGC-3′. Results were normalized with 18s rRNA as an internal control.
ChIP Assay
Mitochondria were isolated as described above. Isolated mitochondria were fixed with 1% formaldehyde for 15 min at room temperature, stopped by glycine, and spun down at 12,000 g for 10 min at 4°C, washed 2x with ice-cold PBS, and lysed for 10 min in Nuclear Lysis Buffer (pH 8.0 50 mM; Tris-HCL, 10 mM; EDTA 1% SDS). MRE11A mtDNA-immunoprecipitation was performed using chromatin immunoprecipitation kit according to the manufacturer’s instructions (cat. no. 53040; Active Motif). Purified DNA from MRE11A (ab12159, Abcam) or IgG (ab171870, Abcam) immunoprecipitated samples was quantified by real-time PCR using the following primers: forward: 5′-GTGCTATAGTGGAGGCCGGA-3′ and reverse: 5′-GGGTGGGAGTAGTTCCCTGC-3′.
Measurement of Secreted IL-1β
CD4+ T cells isolated from healthy individual or RA patients were cultured with anti-CD3/CD28 beads (2 cells per 1 bead), followed by treatment with or without Nigericin (5μM) for 5 days. Supernatant were collected and IL-1β was measured by ELISA using Human IL-1 beta/IL-1F2 Quantikine ELISA Kit (R&D Systems, #DLB50).
Cell Viability Detection
CD4+ T cells were cultured with anti-CD3/CD28 beads (2 cells per 1 bead) for 3 days, and cell viability was measured using AlamarBlue Cell Viability Reagent (Thermo Fisher Scientific, #DAL1025) according to the manufacturer’s instructions.
Measurement of LDH
CD4+ T cells were cultured with anti-CD3/CD28 beads (2 cells per 1 bead). LDH in the supernatant was measured using Pierce LDH Cytotoxicity Assay Kit according to the manufacturer’s instructions. To calculate the percentage of LDH release, the LDH activity of the spontaneous LDH release control was subtracted from the experimental sample’s LDH activity and divided by the total LDH activity (Maximum LDH release activity-Spontaneous LDH release activity).
Measurement of Mitochondrial Mass
CD4+ T cells were stimulated with anti-CD3/CD28 beads (2 cells per 1 bead) for 72h. Cells were incubated with MitoTracker Red (Invitrogen, M7512) (50 nM) at 37°C for 30 mins. After staining, cells were washed twice, and mitochondrial mass was quantified as the intensity of MitoTracker Red using the LSM710 microscope system with the ZEN 2010 software (Carl Zeiss).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics were calculated using GraphPad Prism software (GraphPad Software). The paired Wilcoxon test was used when the sample size per group was >5. Otherwise, the parametric t-test was used. To adjust for multiple testing, in addition to individual p-values, we used Hochberg’s step-down method to control for a family-wise-error rate at the 0.05 levels. Where appropriate, one-way ANOVA was used and pair-wise comparison using Tukey’s method was applied. All data points were included in the analysis and no outliers were removed. Normality testing was not performed if the sample size per group was <10. All data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 and statistical parameters can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-human cleaved caspase-1 (1:50) | Biorbyt | orb126550 |
| Anti-CD3 (1:50) | Dako | M725401-2 |
| Anti-MRE11A (1:100) | Abcam | ab214 |
| Alexa Fluor 488 labeled goat anti-mouse (1:1000) | Abcam | ab96879 |
| Anti-MRE11A-CHIP (1:100) | Abcam | ab12159 |
| IgG-CHIP (1:100) | Abcam | ab171870 |
| Caspase-1 (14F468)(1:1000) | Santa Cruz Biotechnology | sc-56036 |
| Cleaved Caspase-3 (Asp175) Antibody(1:200) | Cell Signaling Technology | 9661S |
| Goat anti-mouse IgG H&L (DyLight 488)(1:500) | Abcam | ab96879 |
| Goat anti-rabbit IgG (H+L), Alexa Fluor 594 (1:500) | Invitrogen | R37117 |
| Biological Samples | ||
| Human synovial tissue | Stanford University | N/A |
| Human lymph node | Stanford University | N/A |
| PBMC from healthy donors | Stanford Blood Center | N/A |
| PBMC from patients | Stanford University | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Mirin | Sigma-Aldrich | M9948-5MG |
| VX765 | MedChem Express | HY-13205 |
| Anti-CD3/CD28 beads | Life Technologies | #11132D |
| Lymphorep | Cosmo Bio | AXS-1115758 |
| RPMI 1640 Medium | ThermoFisher Scientific | 11875135 |
| glucose | Sigma-Aldrich | 50-99-7 |
| 4-15% SDS-PAGE | Bio-Rad | 4561083 |
| PVDF membranes | Bio-Rad | 1620177 |
| TBST | Cell Signaling Technology | 9997S |
| PBS | Boston BioProducts | BM-220 |
| Tris-Glycine-SDS Running Buffer (10X) | Boston BioProducts | NBP-150 |
| Tris-Glycine Transfer Buffer (10X) #12539 | Cell Signaling Technology | 12539S |
| SuperSignal West Femto Maximum Sensitivity Substrate | ThermoFisher Scientific | 34094 |
| Target Retrieval Solution | Dako | S1699 |
| ImmPRESS™ HRP Anti-Rabbit IgG (Peroxidase) Polymer Detection Kit | Vector Laboratories | MP-7451 |
| ImmPRESS™-AP Anti-Mouse IgG (alkaline phosphatase) Polymer Detection Kit | Vector Laboratories | MP-5402 |
| VECTOR Red Alkaline Phosphatase (AP) Substrate Kit | Vector Laboratories | SK-5100 |
| DAB Peroxidase (HRP) Substrate Kit | Vector Laboratories | SK-4100 |
| Mayer’s Hematoxylin solution | Sigma-Aldrich | MHS16-500ML |
| Direct-zol RNA MiniPrep | Genesee Scientific | 11-331 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368813 |
| Ac-YVAD-cmk | Sigma-Aldrich | SML0429-1MG |
| Z-DEVD-FMK | Cayman Chemical Company | 210344-95-9 |
| Z-FA-FMK | Cayman Chemical Company | 197855-65-5 |
| Critical Commercial Assays | ||
| human CD4+ T cell isolation kit | STEMCELL Technologies | #17952 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | ThermoFisher Scientific | 78833 |
| Mitochondria Isolation Kit for Cultured Cells | ThermoFisher Scientific | 89874 |
| Mitochondria Isolation Kit for Tissue | ThermoFisher Scientific | 89801 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | 103015-100 |
| luminescent ATP Detection Assay Kit | Abcam | ab113849 |
| FAM-FLICA® Caspase-1 Assay Kit | ImmunoChemistry TECHNOLOGIES | #97 |
| FAM-FLICA® Caspase-3/7 Assay Kit | ImmunoChemistry TECHNOLOGIES | #93 |
| Amaxa Human T Cell Nucleofector Kits | Lonza | VPA-1002 |
| DNeasy Blood &Tissue Kit | Qiagen | 69504 |
| ChIP-IT High Sensitivity® | Active Motif | 53040 |
| Human IL-1 beta/IL-1F2 Quantikine ELISA Kit | R&D Systems | #DLB50 |
| Pierce LDH Cytotoxicity Assay Kit | ThermoFisher Scientific | 88953 |
| 4% paraformaldehyde | Santa Cruz Biotechnology | sc-281692 |
| Fpg | New England Biolabs (NEB) | M0240S |
| MitoTracker Red | Invitrogen | M7512 |
| Experimental Models: Cell Lines | ||
| Jurkat | ATCC | TIB-152™ |
| Experimental Models: Organisms/Strains | ||
| NSG mice | The Jackson Laboratory | Stock No:005557 |
| Oligonucleotides | ||
| MRE11-siRNA | Thermo Fisher Scientific | HSS1429690 |
| Caspase-1-siRNA | Santa Cruz Biotechnology | sc-29235 |
| NLRP3-siRNA | Dharmacon | L-017367-00-0005 |
| AIM2-siRNA | Invitrogen | 1299001 |
| Primer for human TRB: forward: CCTTCAACAACAGCATTATTCCAG; reverse: CGAGGGAGCACAGGCTGTCTT | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human TNFA: forward CGCATCGCCGTCTCCTACCA; reverse: AGGGCAATGATCCCAAAGTA | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human TNFSF11: forward: GGTGGATGGCTCATGGTTAGA; reverse: CATGTTGGAGATCTTGGCCC | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human IL6: forward: CTTCGGTCCAGTTGCCTTCT; reverse: GTGCCTCTTTGCTGCTTTCA | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human IL1B: forward: ATCCAGCTACGAATCTCCGA; reverse: CCACTTGTTGCTCCATATCC | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human AIM2: forward: TCAAGCTGAAATGAGTCCTGC; reverse: CTTGGGTCTCAAACGTGAAGG | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human ASC: forward: GACGGGGCCAATACCACAC; reverse: TCTGTAACAAAAGTCGTGCTTCT | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human NLRP3: forward: CGTGAGTCCCATTAAGATGGAGT; reverse: CCCGACAGTGGATATAGAACAGA | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human PFKFB3: forward: TGTGAGGAGCTGACCTACGA; reverse: CCATGATCACTGGCTCCAAG | Protein and Nuclei acid Facility at Stanford University | N/A |
| Primer for human G6PD: forward: GGCCGTCACCAAGAACATTC; reverse: TGGTCGATGCGGTAGATCTG | Protein and Nuclei acid Facility at Stanford University | N/A |
| Recombinant DNA | ||
| hMre11 WT-FLAG | The University of Texas at Austin | gift from Tanya Paull |
| hMre11 H129N-FLAG | The University of Texas at Austin | gift from Tanya Paull |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| FlowJo | BD | https://www.flowjo.com/ |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ZEN 2010 software | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| Seahorse Wave Controller software | Seahorse/Agilent | https://www.agilent.com/en/products/cell-analysis/cell-analysis-software/instrument-software/wave-controller-2-4 |
Highlights:
The DNA repair nuclease MRE11A is located in mitochondria.
Mitochondrial MRE11A protects mtDNA from oxidation and cytoplasmic leakage.
MRE11Alow T cells fail to produce ATP and undergo caspase-1-dependent pyroptosis.
MRE11A loss-of-function results in tissue inflammation.
Context and Significance.
In rheumatoid arthritis, immune cells promote chronic tissue inflammation. Molecularly, the immune cells known as T cells have a defect in DNA repair, age prematurely and adapt to a distinct metabolic program. Here, researchers at Stanford University show that in rheumatoid arthritis, the loss of a molecule used for DNA repair directly impacts mitochondrial DNA and function, leading to both energetic failure and pro-inflammatory behavior of T cells. Due to a deficiency in the DNA repair enzyme known as MRE11A in mitochondria, rheumatoid arthritis T cells have low production of the fuel ATP and leak mitochondrial DNA into the cytoplasm, where it triggers an inflammatory series of events and cell death. These findings indicate that targeting the DNA repair machinery to protect mitochondria could be a promising addition to therapeutic interventions aimed at blocking chronic tissue inflammation in rheumatoid arthritis.
Acknowledgment
This work was supported by the NIH (R01AR042527, R01HL117913, R01AI108906, P01HL129941, R01AI108891, R01AG045779, R01AI129191). Tanya T. Paull (U. of Texas, Austin) generously provided MRE11A wild type and nuclease-deficient constructs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Alexeyev M, Shokolenko I, Wilson G, and LeDoux S (2013). The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harbor perspectives in biology 5, a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva G, and Rathmell JC (2017). Similarities and Distinctions of Cancer and Immune Metabolism in Inflammation and Tumors. Cell metabolism 26, 49–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apel K, and Hirt H (2004). Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annual review of plant biology 55, 373–399. [DOI] [PubMed] [Google Scholar]
- Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, Dutow P, Woodruff TM, Yu ZX, O’Neill LA, et al. (2016). T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 352, aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T, Fink SL, and Cookson BT (2009). Pyroptosis: host cell death and inflammation. Nature reviews. Microbiology 7, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulias K, Lieberman J, and Greer EL (2016). An epigenetic clock measures accelerated aging in treated HIV infection. Molecular cell 62, 153–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratic A, and Larsson NG (2013). The role of mitochondria in aging. The Journal of clinical investigation 123, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camell CD, Sander J, Spadaro O, Lee A, Nguyen KY, Wing A, Goldberg EL, Youm YH, Brown CW, Elsworth J, et al. (2017). Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 550, 119–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL, Flavell RA, and Zamboni DS (2017). AIM2 Engages Active but Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell Rep 20, 794–805. [DOI] [PubMed] [Google Scholar]
- Doitsh G, Galloway NL, Geng X, Yang Z, Monroe KM, Zepeda O, Hunt PW, Hatano H, Sowinski S, Munoz-Arias I, et al. (2014). Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 505, 509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre A, Boyer-Chatenet L, Sattler RM, Modi AP, Lee JH, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT, et al. (2008). A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nature chemical biology 4, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feriotti C, de Araujo EF, Loures FV, da Costa TA, Galdino NAL, Zamboni DS, and Calich VLG (2017). NOD-Like Receptor P3 Inflammasome Controls Protective Th1/Th17 Immunity against Pulmonary Paracoccidioidomycosis. Frontiers in immunology 8, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Shao L, Colmegna I, Goronzy JJ, and Weyand CM (2009). Telomerase insufficiency in rheumatoid arthritis. Proceedings of the National Academy of Sciences of the United States of America 106, 4360–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. (2017). The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124 e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geltink RIK, Kyle RL, and Pearce EL (2018). Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annual review of immunology 36, 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, and Perl A (2018). Metabolism as a Target for Modulation in Autoimmune Diseases. Trends in immunology 39, 562–576. [DOI] [PubMed] [Google Scholar]
- Ishigaki K, Kochi Y, Suzuki A, Tsuchida Y, Tsuchiya H, Sumitomo S, Yamaguchi K, Nagafuchi Y, Nakachi S, Kato R, et al. (2017). Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nature genetics 49, 1120–1125. [DOI] [PubMed] [Google Scholar]
- Jo EK, Kim JK, Shin DM, and Sasakawa C (2016). Molecular mechanisms regulating NLRP3 inflammasome activation. Cell Mol Immunol 13, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MO, Siska PJ, Contreras DC, and Rathmell JC (2016). Nutrients and the microenvironment to feed a T cell army. Seminars in immunology 28, 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Shen Y, Hohensinner P, Ju J, Wen Z, Goodman SB, Zhang H, Goronzy JJ, and Weyand CM (2016). Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity 45, 903–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling GS, Crawford G, Buang N, Bartok I, Tian K, Thielens NM, Bally I, Harker JA, Ashton-Rickardt PG, Rutschmann S, et al. (2018). C1q restrains autoimmunity and viral infection by regulating CD8(+) T cell metabolism. Science 360, 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcomini I, and Gasser SM (2015). Nuclear organization in DNA end processing: Telomeres vs double-strand breaks. DNA repair 32, 134–140. [DOI] [PubMed] [Google Scholar]
- Martinon F, and Tschopp J (2004). Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 117, 561–574. [DOI] [PubMed] [Google Scholar]
- Muta T, Kang D, Kitajima S, Fujiwara T, and Hamasaki N (1997). p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. The Journal of biological chemistry 272, 24363–24370. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR, Domingo-Fernandez R, Johnston DGW, et al. (2015). Pyruvate Kinase M2 Regulates Hif-1alpha Activity and IL-1beta Induction and Is a Critical Determinant of the Warburg Effect in LPS-Activated Macrophages. Cell metabolism 21, 347. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nature immunology 11, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. (2008). Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268. [DOI] [PubMed] [Google Scholar]
- Shao L, Fujii H, Colmegna I, Oishi H, Goronzy JJ, and Weyand CM (2009). Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. The Journal of experimental medicine 206, 1435–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Goronzy JJ, and Weyand CM (2010). DNA-dependent protein kinase catalytic subunit mediates T-cell loss in rheumatoid arthritis. EMBO molecular medicine 2, 415–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Wen Z, Li Y, Matteson EL, Hong J, Goronzy JJ, and Weyand CM (2017). Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nature immunology 18, 1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata A, Moiani D, Arvai AS, Perry J, Harding SM, Genois MM, Maity R, van Rossum-Fikkert S, Kertokalio A, Romoli F, et al. (2014). DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Molecular cell 53, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et al. (2012). Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, et al. (2016). The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. The Journal of experimental medicine 213, 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Kishton RJ, and Restifo NP (2017). Metabolic reprograming of anti-tumor immunity. Current opinion in immunology 46, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Mehta GU, Patel SJ, Roychoudhuri R, Crompton JG, Klebanoff CA, Ji Y, Li P, Yu Z, et al. (2016). Mitochondrial Membrane Potential Identifies Cells with Enhanced Sternness for Cellular Therapy. Cell metabolism 23, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed A, and Tainer JA (2018). The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annual review of biochemistry 87, 263–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1 beta through HIF-1 alpha. Nature 496, 238-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, and Chandel NS (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand CM, and Goronzy JJ (2017). Immunometabolism in early and late stages of rheumatoid arthritis. Nature reviews. Rheumatology 13, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, and VanHouten B (1997). Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proceedings of the National Academy of Sciences of the United States of America 94, 514–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Fujii H, Mohan SV, Goronzy JJ, and Weyand CM (2013). Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. The Journal of experimental medicine 210, 2119–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Shen Y, Oishi H, Matteson EL, Tian L, Goronzy JJ, and Weyand CM (2016). Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Science translational medicine 8, 331ra338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, D’Agostino D, Planavsky N, Lupfer C, Kanneganti TD, et al. (2015). The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nature medicine 21, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B, et al. (2018). New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, and Tschopp J (2011). A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.