
Fig. 4. Phylogenetic analysis of SARSr-CoVs and MERSr-CoVs.
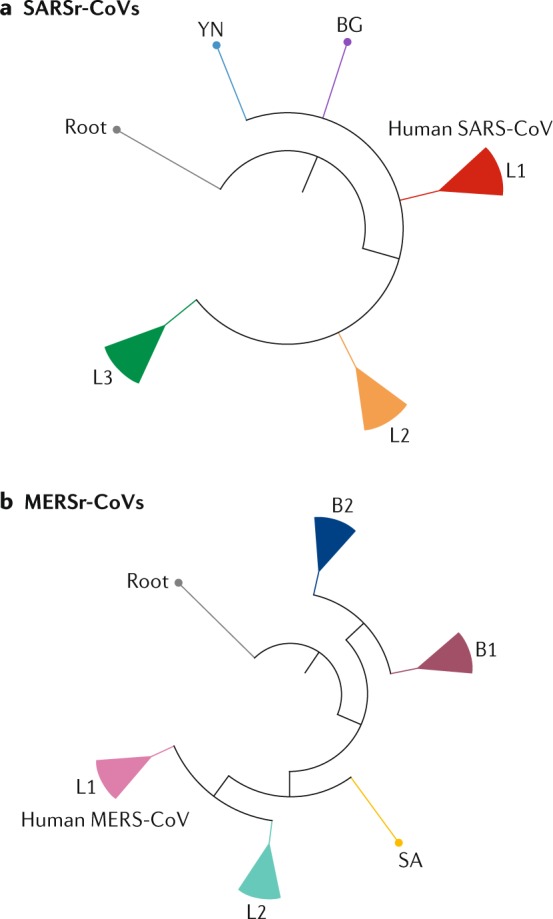
a | The figure shows a simplified phylogenetic tree of severe acute respiratory syndrome-related coronaviruses (SARSr-CoVs) from bats. SARSr-CoVs cluster into three lineages, L1–L3, and human severe acute respiratory syndrome coronaviruses (SARS-CoVs) embed in L1. Two individual SARSr-CoVs do not cluster into these lineages: YN, a virus isolated from Yunnan province, China, and BG, a virus from Bulgaria, Europe. The tree is based on published trees20,138 and reconstructed using sequences of the complete RNA-dependent RNA polymerase-coding region (maximum likelihood method under the GTR + I + Γ model of nucleotide substitution as implemented in PhyML, version 3.1 (ref.137)).The strain Zhejiang2013 (GenBank No. KF636752) was used as a root. b | By contrast, Middle East respiratory syndrome-related coronaviruses (MERSr-CoVs) form two major viral lineages, L1 and L2. L1 is found in humans and camels, and L2 is found only in camels. Two small clusters, B1 (bat 1) and B2, and one single virus, SA, from South Africa, were found in bats. The phylogenetic tree of MERSr-CoVs is based on a published trees94,139 and reconstructed using full-genome alignment of all coding regions using the same method as above. HKU4-1 (EF065505) and HKU5-1 (EF065509), two 2c betacoronaviruses, served as the root of the tree. Detailed phylogenetic trees and grouping information can be found in Supplementary Fig. S1. MERS-CoV, Middle East respiratory syndrome coronavirus.