
Figure 2. Pathways for the development of safer immunomodulatory biologics.
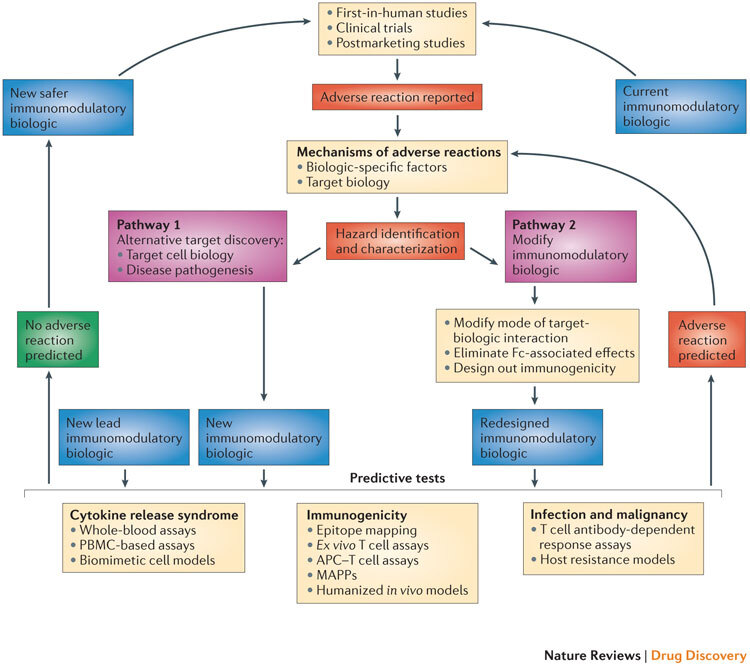
The iterative cycle for the development of safer immunomodulatory biologics incorporates two key pathways and depends on the collective knowledge obtained from adverse reactions observed in first-in-human studies, clinical trials and postmarketing pharmacovigilance analyses. These data provide the basis for understanding the frequency and nature of the adverse reactions associated with immunomodulatory biologic therapy, as well as the potential mechanisms by which these adverse reactions are induced. The next step in this process is the identification and characterization of hazards associated with the immunomodulatory biologic (for example, characterizing excessive Fc-mediated effector functions that result in cytokine release syndrome (CRS), overt or global immunosuppression leading to serious infections or the presence of immunogenic structures within the biologic that trigger immunogenicity). Pathways 1 and 2 are two different trajectories that utilize the understanding of the mechanism of adverse reactions to inform the design of safer and potentially more effective immunomodulatory biologics. Pathway 1 is followed when the adverse reaction cannot be dissociated from the target biology, and involves generating a biologic that engages an alternative target or mechanism to produce the desired pharmacodynamic effect without the associated adverse reaction. Pathway 2 involves redesigning the biologic to engineer out components within the biologic structure that trigger adverse effects, or to alter the nature of the target–biologic interactions. New immunomodulatory biologics from both of these pathways (and any other lead drug candidates) need to go through a panel of predictive tests until a safer biologic emerges. The selection of the predictive tests to be used is on a case-by-case basis, as it should take into account the nature of the target biology, the effector mechanisms that are engaged and any other factor or factors that influence the intended pharmacological effect and the risk of adverse reactions. Currently available tests that are suitable for determining the CRS-inducing risk of a new biologic with immunostimulatory properties could include whole-blood assays, peripheral blood mononuclear cell (PBMC)-based assays and biomimetic cell models. Predicting the risk of serious infections and malignancies for a new immunomodulatory biologic still remains a challenge, and limitations in T cell-dependent antibody response (TDAR) assays and host resistance models are due to species-specific variations both in target biology and in exposure to risk factors. Preclinical tools for predicting the risk of immunogenicity involve the use of in silico models (immunogenic epitope mapping), in vitro or ex vivo models (T cell assays, antigen-presenting cell (APC)–T cell assays and major histocompatibility complex (MHC)-associated peptide proteomics (MAPPs)) and in vivo models (humanized animals). The integration of in silico and in vitro or ex vivo assays increases the predictive value of these preclinical tools. A new or redesigned immunomodulatory biologic that is considered to be safe based on predictive preclinical assessments enters the cycle of testing in first-in-human studies and clinical trials, and then moves to the clinic with necessary safety precautions in place and with continuous monitoring for any potential adverse reaction. By contrast, any new immunomodulatory biologic that is flagged using predictive tests for its potential to cause adverse reactions (or observed to cause adverse reactions in clinical studies) will be passed through this iterative cycle again for the design and development of a safer immunomodulatory biologic.