

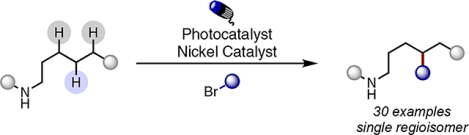
Abstract
The functionalization of unactivated C(sp3)-H bonds poses a significant challenge due to their ubiquity and relative similarity in most organic frameworks. Herein, we describe the use of a combined photoredox and nickel catalytic system for the regioselective C(sp3)-C(sp3) coupling of unactivated C(sp3)-H bonds and alkyl bromides. Positional selectivity is dictated by a 1,5-hydrogen atom transfer (HAT) reaction by a pendent amide. Interception of this radical by a Ni catalyst allows distal alkylation to occur in good yield and excellent selectivity.
Graphical Abstract
The ubiquity of C-H bonds decorating organic molecules presents itself as the ultimate handle of chemical diversification. The ability to selectively and efficiently manipulate these C-H functionalities can greatly expedite and simplify synthetic routes through a greater diversity of potential bond disconnections. However, this ubiquity also poses a considerable selectivity challenge, as the differentiation between unbiased reactive sites can be marginal to largely indistinguishable.1 Metal-catalyzed C-H activation has revolutionized our ability to quickly generate molecular complexity through the installment of new C-C, C-O, C-N, and C-X bonds.2 While metal-catalyzed C-H activation has thrived on the functionalization of C(sp2)-H bonds, the selective functionalization of C(sp3)-H bonds poses an ongoing challenge.3
Techniques involving hydrogen atom transfer (HAT) have proven useful for the functionalization of intrinsically weak C(sp3)-H bonds.4 Photoredox strategies that facilitate the generation of radical intermediates capable of HAT have expedited the growth of these avenues.5 However, these radical intermediates target the weakest C-H bond in solution capable of polarity matched transition state, which typically lie α to an existing functionality or heteroatom. In the absence of electronic positional bias, functionalization through these methods often occurs indiscriminately at chemically indistinguishable sites.6 The coupling of a transition metal with these strategies has broadly expanded the breadth of bond disconnections through single-electron transmetallation.7 This merger has culminated in the ability to cross-couple alkyl radicals generated from C-H bonds with metal-activated electrophiles to forge new C(sp3)-C(sp2) and C(sp3)-C(sp3) bonds.8 Recent methodology has extended this reactivity to unactivated positions, although it requires a large excess of substrate and may generate mixtures of regioisomers.9
Our group and the Knowles group has previously demonstrated selective functionalization of distal C(sp3) sites with new C-C bonds through a 1,5-HAT strategy through a photoredox- catalyzed oxidation of a pendent amide moiety.10 The remote carbon radical, generated through a six-membered transition state, is sufficiently long-lived to trap exogenous Michael acceptors to furnish a δ-functionalized amide. This strategy showcases the ability to regioselectively mono-functionalize an unactivated distal methine or methylene in the presence of more electronically or sterically activated C-H bonds.
We sought to unify the selectivity imparted through 1,5-HAT with the versatility offered through nickel catalysis to selectively alkylate distal bonds. In doing so, one of numerous otherwise indistinguishable methylene sites can be coupled selectively to the wealth of commercially available or easily accessible alkyl bromides.
We first examined whether the distal alkyl radical generated through 1,5-HAT could be captured by a metal catalyst. Due to the well-established ability of nickel bipyridine complexes to participate in metallophotoredox cross-couplings,11 we chose to screen
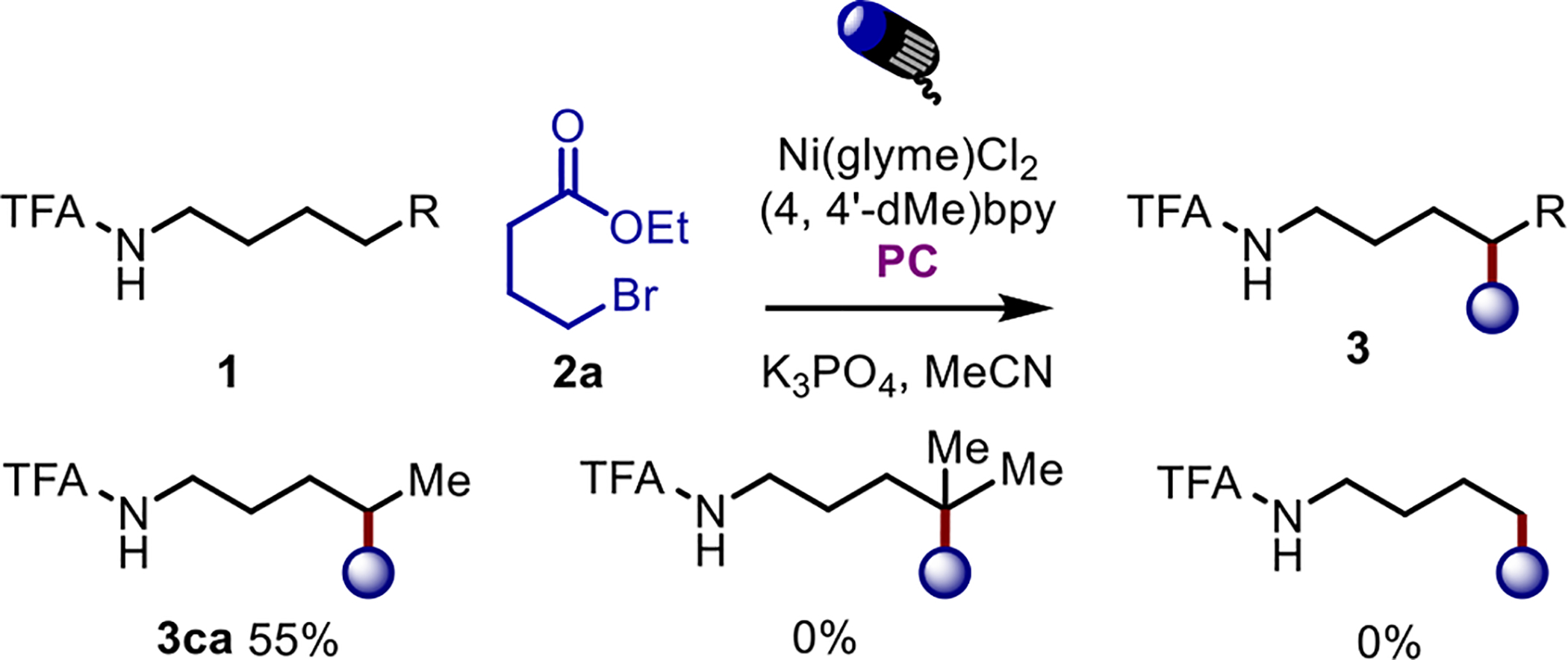
these species in conjunction with our previously established methods10a for forming a distal alkyl radical (Table 1).12 Alkyl trifluoroacetamides are particularly well-suited to directing the 1,5-HAT in the presence of a sufficiently strong base. Interestingly, the reaction only proceeds at remote sites bearing secondary C(sp3)-H bonds (Scheme 2). Other directing groups which decrease the acidity of the N-H proton proved unreactive, including when these systems were run using conditions developed by Knowles10b relying on proton-coupled electron transfer (PCET; Table 1, entries 4–6). Additionally, all of the reactive components are necessary, as removal of light, the photocatalyst, Ni, ligand, or base all result in no product formation (Table 1, entries 7–11).
Table 1. Optimization and Control Studies.
| |||
|---|---|---|---|
| Entry | R Source | Deviation from Standard Conditionsa | Yield (%) |
| 1 | CF3 | none | 79 |
| 2 | CHF2 | none | 0 |
| 3 | CF2CF3 | none | 61 |
| 4 | Ph | PC-2, NBu4OP(O)(OBu)2 | 0 |
| 5 | Ph | none | 0 |
| 6 | CF3 | PC-2, NBu4OP(O)(OBu)2 | 0 |
| 7 | CF3 | Without Ni | 0 |
| 8 | CF3 | Without Ligand | 0 |
| 9 | CF3 | Without PC | 0 |
| 10 | CF3 | Without Light | 0 |
| 11 | CF3 | Without Base | 0 |

Optimizations were performed on 0.1 mmol scale using 1a (1 equiv), 2j (2 equiv) Ni source (10 mol%), ligand (12 mol%), base (4 equiv), and PC (1 mol%) over a period of 36 hours. PC = [lr(dF(CF3)ppy)2(dtbbpy)]PF6 PC-2 = [lr(dF(CF3)ppy)2 (5,5’-dCF3bpy)]PF6
Scheme 2. Effect of Substitution at δ-position.
Having established that this strategy effectively forges a new C-C bond regioselectively at a distal C-H, we examined its scope. Aliphatic linear amides participate well and are monofunctionalized at the δ-methylene site (Scheme 3). Interestingly, even with the generation of a tertiary site at the δ-position, no second cross-coupling occurs at that site, which differs from the related capture of Michael acceptors.10a In the case of multiple functionalizable sites, secondary positions are selectively functionalized in preference over primary positions, 3fa. In cyclic systems, cross-coupling tends to occur in a trans relationship, probably due to the preference of radical capture of the active Ni catalyst on the sterically less hindered side of the ring (see SI, S5). This allows for alkylation of five and six-membered ring systems with high diastereoselectivity. In substrates featuring more activated C(sp3)-H bonds in positions other than those δ to the amide moiety, functionalization occurs solely at the unactivated, directed site in preference over the weaker, more reactive C(sp3)-H bonds. This allows for the tolerance of sulfonamides (3ja), ethers (3ia and 3la), arenes and alkenes (3oa and 3pa), as well as their adjacent activated C-H bonds, albeit with occasionally diminished yields.
Scheme 3. Reaction Scope (Amides).
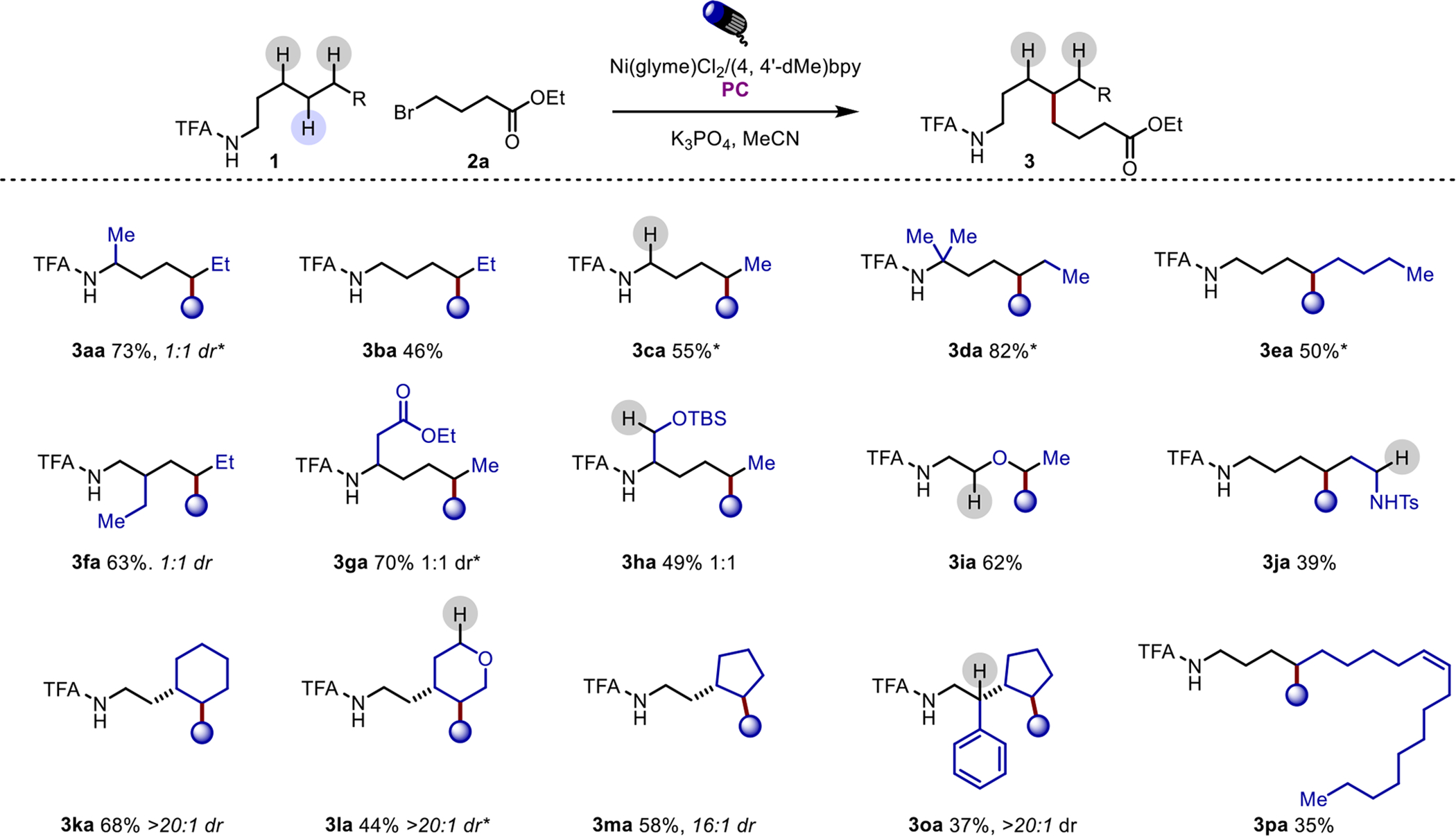
Reactions were run on 0.1 mmol scale with 2 equivalents of 2a, 10 mol % Ni(glyme)Cl2, 12 mol % 4,4’-dimethyl-2,2’-bipyridine, 1 mol% PC ([Ir(dF-CF3ppy)2dtbbpy]PF6) and 4 equivalents K3PO4 in 0.75 mL of MeCN for 36 hours. *Product assay purity 93–96% (contains 4%−7% inseparable impurity of product-derived olefin). (See SI)
The scope of the alkyl bromide coupling partner was also explored (Scheme 4). In nearly all cases, the alkyl halide is used in excess to the aliphatic amine, as the alkyl bromide is consumed in a competing reductive homo-coupling. This reductive homo-coupling outcompetes desired cross-coupling in low concentrations of amide, resulting in incomplete conversion of the amide but complete consumption of the alkyl bromide. Most aliphatic alkyl bromides, including ethyl, are tolerated under the reaction conditions (3kb). Other functionalities, including protected alcohols, 3kf, nitriles, 3kg, and alkenes, 3kh, also participate well in the reaction. Interestingly, benzylic bromides are not tolerated, presumably due to the competing off-cycle reduction by the photocatalyst which generates free benzyl radicals. Other heterocycles are tolerated, including dioxolanes, 3ki, phthalimides, 3kk, and pyrroles, 3kl. Aryl bromides are tolerated, 3km, as the primary alkyl bromide is considerably more reactive. Alkyl bromides bearing electronically diverse arene rings are well tolerated under the standard conditions (3kj −3ko). Currently, alkyl bromides are the only productive alkyl halides as chlorides, iodides, and pseudo-halides such as tosylates and triflates are either unreactive or lead to unproductive byproducts. Secondary and tertiary alkyl bromides show diminished yields under the optimized conditions and have thus far only shown modest levels of synthetic efficiency.14
Scheme 4. Representative examples of alkyl bromides.
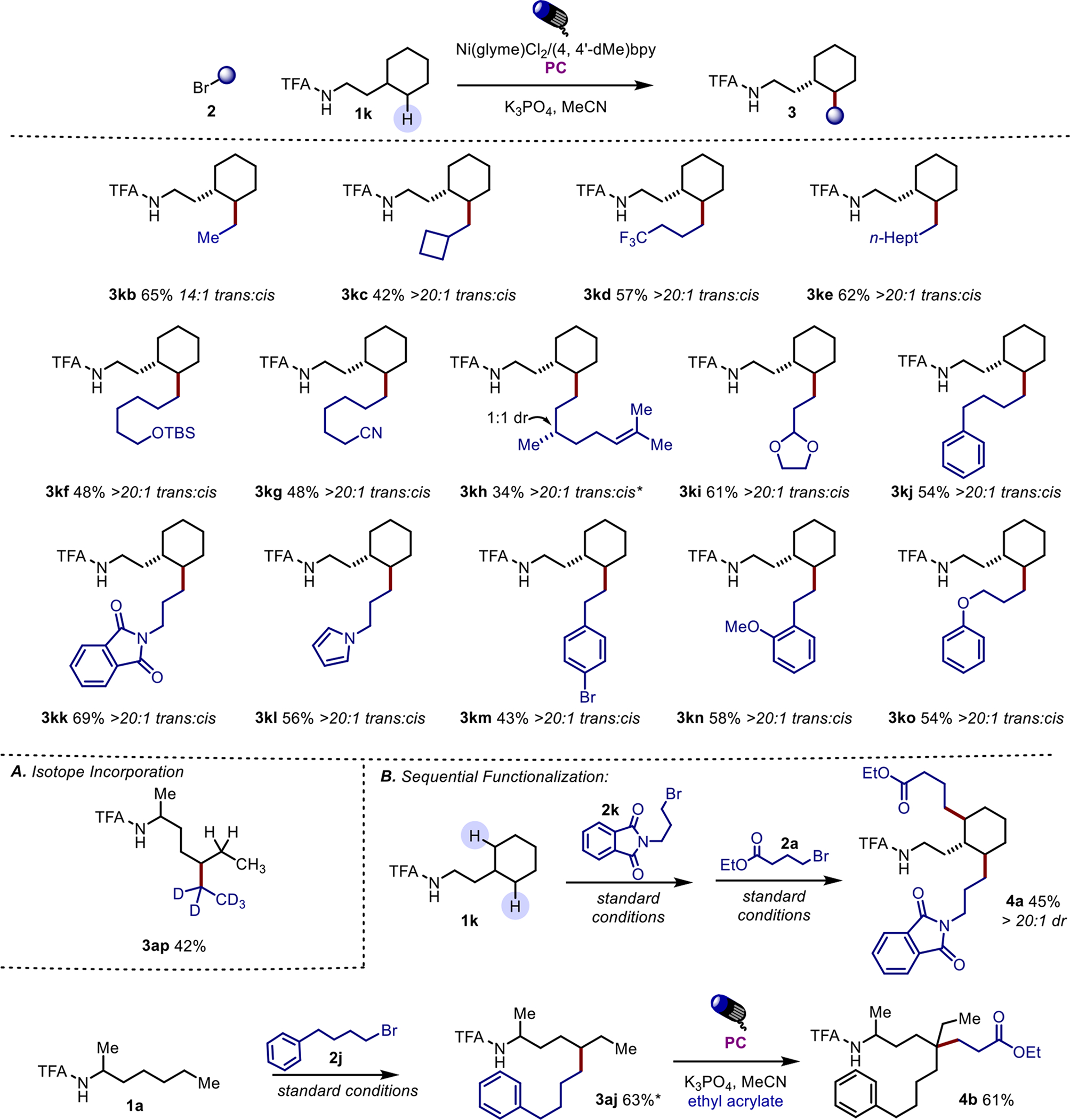
Reactions were run on 0.1 mmol scale with 2 equivalents of alkyl bromide, 10 mol % Ni(glyme)Cl2, 12 mol % 4,4’-dimethyl-2,2’-bipyridine, 1 mol% PC ([Ir(dF-CF3ppy)2dtbbpy]PF6) and 4 equivalents K3PO4 in 0.75 mL of MeCN for 36 hours. A. Deuterotopically labeled alkyl bromides can be utilized to incorporate deuterated fragments into substrates otherwise lacking reactive handles. B. Sequential functionalization is available through this reactivity as symmetrical reactive sites can be functionalized without degradation of diasteroselectivity. The reactivity can also be paired with prior disclosed methods to effectively furnish quaternary sites. *Product assay purity 95–98% (contains 2–5% inseparable impurity of product-derived olefin). (See SI)
Isotopically labeled tags can be easily incorporated under these conditions allowing for the isotopic differentiation of otherwise symmetrical groups. The reactivity can also be performed sequentially when there are more potential abstraction sites, allowing for the difunctionalization of 2k with two distinct coupling partners in excellent diastereoselectivity. Alternatively, orthogonal reactivity can be performed to generate quaternary carbon centers through the direct radical capture of a suitable radicalophile.10a This allows for the selective, mild generation of a distal quaternary carbon from a previously unactivated or functionalized methylene site, 4b.
A potential mechanism that accounts for the observed transformation and regioselectivity is represented by the catalyst system depicted in Scheme 5 (A). The trifluoroacetamide directing group, which is sufficiently acidic to be deprotonated by the phosphate base, is oxidized by an excited-state photocatalyst to generate a neutral amidyl radical, V. This amidyl radical can then abstract a hydrogen atom in a 1,5-HAT fashion from the δ-methylene to afford a carbon-centered radical at the distal position, VI. Meanwhile, a low-valent nickel complex, I, can oxidatively insert into the alkyl bromide to generate a NiIII alkyl species, II. This NiIII can be reduced to a NiII species by the reduced-state photocatalyst which can then trap the distal carbon radical to afford a transient NiIII, VII, which undergoes reductive elimination to afford the newly formed C-C bond and regenerate a catalytically active NiI.
Scheme 5. Mechanistic insights into the distal C-H alkylation.
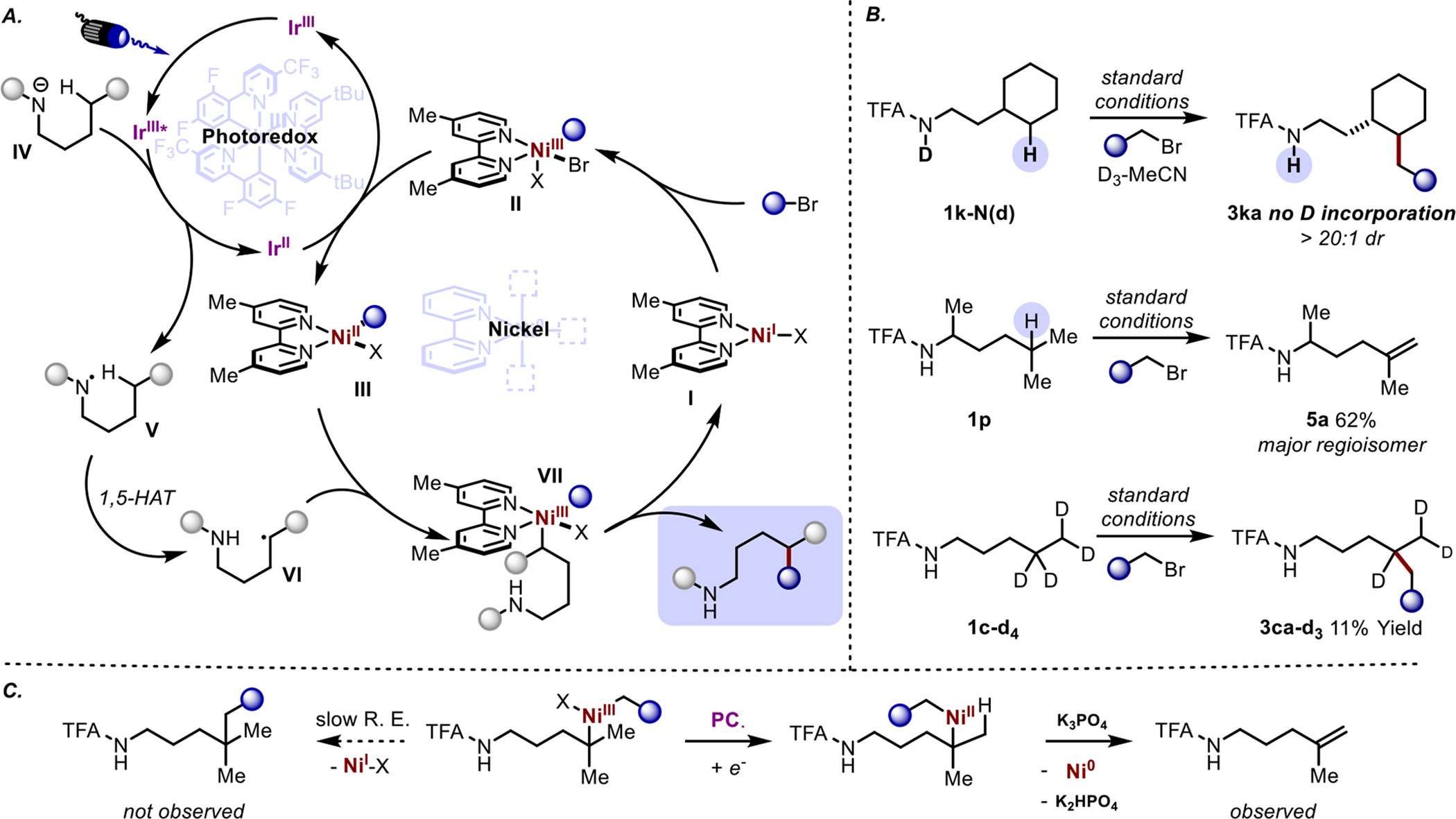
A. Potential mechanism. B. Deuteration studies and reactivity with tertiary substrates. C. A proposed reaction pathway describing the fate of tertiary nickel intermediates in which slow reductive elimination allows for competing substrate oxidation.
It should be noted that this reaction proceeds when using either Ni(cod)2 or Ni(glyme)Cl2 as the precatalyst. When using a NiII precatalyst, as in the optimized conditions, a reduction of the catalyst to a low valent Ni species is likely required before entering the catalytic cycle. In the cases where a Ni0 catalyst is used, increased amount of reductively homocoupled alkyl bromide is observed, which likely helps generate the necessary NiI.15
One perplexing caveat of the reactivity exhibited by the 1,5 HAT/Nickel cross-coupling is the lack of observed reactivity at tertiary positions, especially considering that these positions exhibit a multitude of functionalization methods through 1,5-HAT strategies.13 This lack of reactivity also mirrors that of the more sterically demanding alkyl bromides, as the presumably more sterically encumbered Ni active species demonstrates diminished catalytic efficiency. Despite screening a number of ligands, none produced cross-coupled product in either set of conditions.
In order to more thoroughly probe the fate of the tertiary radical, several mechanistic studies were performed (Scheme 5). We tested this hypothesis through deuterium labeling of the amidyl N-H and conducting the reaction in deuterated solvent to ensure that any alkyl radical abstraction or reversible 1,5-HAT would incorporate deuterium into the product. The absence of deuterium in the product demonstrates that C-C bond formation is diastereoselective rather than a result of epimerization to the more stable diastereomer after alkylation. Due to the low reactivity of tertiary reactive sites, we hypothesize that the reductive elimination of the tertiary alkyl group to generate a quaternary carbon is relatively slow (C). Instead, it is possible that the long-lived NiIII intermediate could in turn be reduced by the photocatalyst and undergo elimination to generate Ni0 and an alkene.16 Olefinic products are observed with more sluggish substrates which supports the likelihood of this pathway. This was further corroborated by the fact that a variety of olefinic products, primarily the δ-ε alkene, are observed when utilizing substrate 1p. This reactivity of the generated tertiary position enables the formation of unsaturated elimination products that remain as inseparable impurities in some isolated products. Deuteration of the activated position (1c-d4) results in the conservation of the second deuteron in the alkylated product. This indicates that if 1,5-DAT occurs on the generated tertiary center then the process is reversible through either a retro-1,5-DAT or a Ni mediated delivery of a hydrogen atom to the alkyl radical through metalation, reduction, and protonation. It seems likely, therefore, that the 1,5-abstraction and Ni capture may occur in the case of δ-methines, but then results in elimination upon reduction or other unproductive side reactions.
We have demonstrated functionalization of distal unactivated C(sp3)-H bonds through a photoredox mediated 1,5-HAT and nickel cross-coupling strategy. This allows for the broad diversification of possible synthetic building blocks that can be selectively incorporated at an unactivated distal methylene site. Studies indicate that this coupling is selective for mono-alkylation, resulting in distal tertiary carbon centers.
Supplementary Material
Scheme 1. Inspiration for the development for the regioselective alkylation of C(sp3)-H bonds.
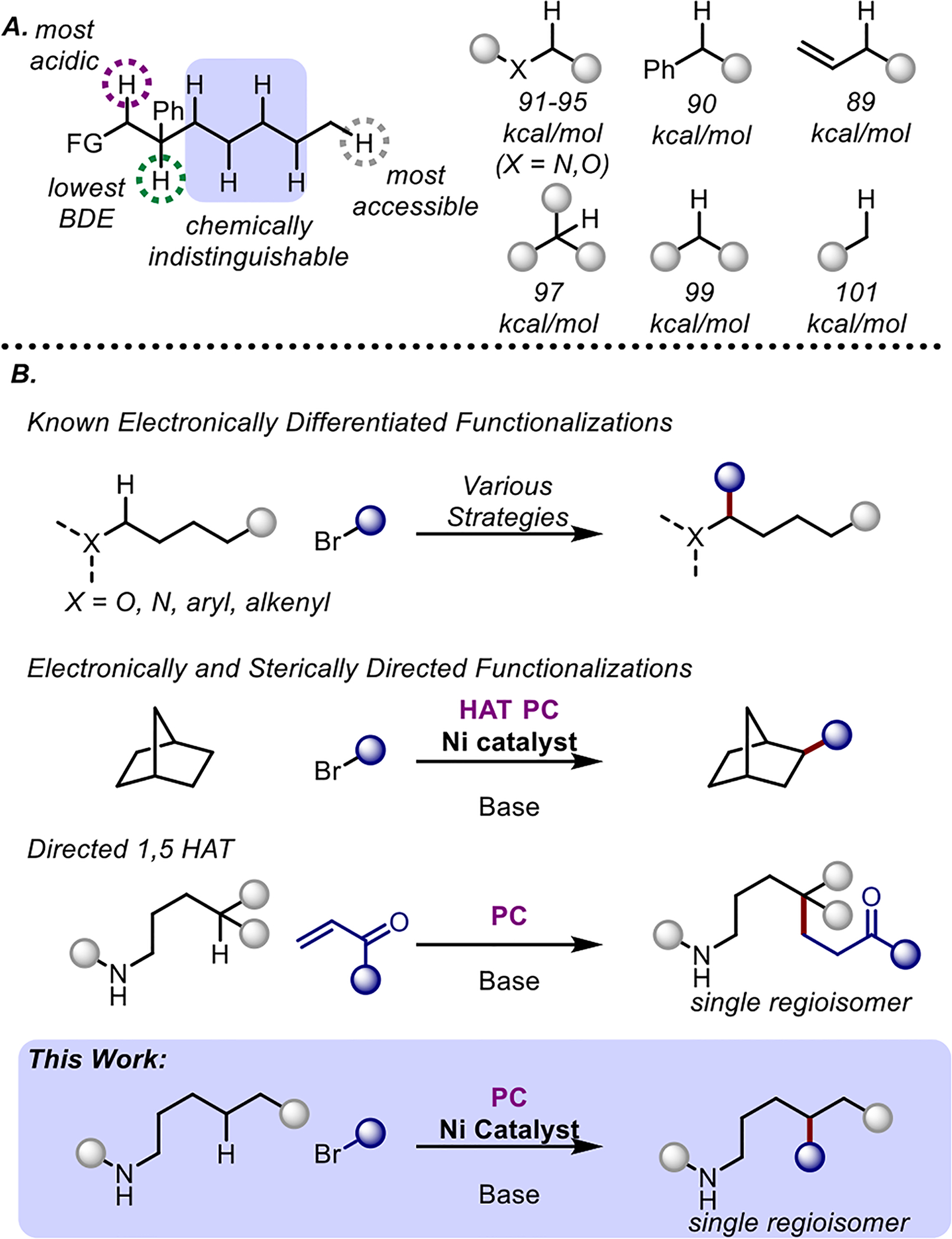
A. Selectivity challenges present in the functionalization of C-H bonds in organic molecules B. Prior art of photoredox-enabled HAT strategies for the functionalization of C(sp3)-H bonds in comparison to this work.
ACKNOWLEDGMENT
We thank NIGMS (GM125206) for support.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and compound characterization (PDF)
The authors declare no competing financial interests.
REFERENCES
- (1).Bergman RG Organometallic Chemistry: C-H Activation. Nature 2007, 446, 391. [DOI] [PubMed] [Google Scholar]
- (2).(a) Lyons TW; Sanford MS Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev 2010, 110, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McMurray L; O’Hara F; Gaunt MJ Recent Developments in Natural Product Synthesis Using Metal-Catalysed C–H Bond Functionalisation. Chem. Soc. Rev 2011, 40, 1885. [DOI] [PubMed] [Google Scholar]; (c) Hartwig JF Evolution of C–H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc 2016, 138, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dong Z; Ren Z; Thompson SJ; Xu Y; Dong G Transition-Metal-Catalyzed C–H Alkylation Using Alkenes. Chem. Rev 2017, 117, 9333. [DOI] [PubMed] [Google Scholar]; (e) Gensch T; Hopkinson MN; Glorius F; Wencel-Delord J Mild Metal-Catalyzed C–H Activation: Examples and Concepts. Chem. Soc. Rev 2016, 45, 2900. [DOI] [PubMed] [Google Scholar]; (f) Colby DA; Bergman RG; Ellman JA Rhodium-Catalyzed C–C Bond Formation via Heteroatom-Directed C–H Bond Activation. Chem. Rev 2010, 110, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Giri R; Shi B-F; Engle KM; Maugel N; Yu J-Q Transition Metal-Catalyzed C–H Activation Reactions: Diastereoselectivity and Enantioselectivity. Chem Soc Rev.2009, 38, 3242. [DOI] [PubMed] [Google Scholar]; (h) Karimov RR; Hartwig JF Transition-Metal-Catalyzed Selective Functionalization of C(Sp3)–H Bonds in Natural Products. Angew. Chem. Int. Ed 2018, 57, 4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chu JCK; Rovis T Complementary Strategies for Directed C(Sp3)–H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed 2018, 57, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Newhouse T; Baran PS If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem. Int. Ed 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Skubi KL; Blum TR; Yoon TP Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev 2016, 116, 10035. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Shaw MH; Twilton J; MacMillan DWC Photoredox Catalysis in Organic Chemistry. J. Org. Chem 2016, 81, 6898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Narayanam JMR; Stephenson CRJ Visible Light Photoredox Catalysis: Applications in Organic Synthesis. Chem. Soc. Rev 2010, 40, 102. [DOI] [PubMed] [Google Scholar]
- (6).(a) Mukherjee S; Maji B; Tlahuext-Aca A; Glorius F Visible-Light-Promoted Activation of Unactivated C(Sp3)–H Bonds and Their Selective Trifluoromethylthiolation. J. Am. Chem. Soc 2016, 138, 16200. [DOI] [PubMed] [Google Scholar]; (b) Margrey KA; Czaplyski WL; Nicewicz DA; Alexanian EJ A General Strategy for Aliphatic C-H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 4213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schultz DM; Levesque F; DiRocco DA; Reibarkh M; Ji Y; Joyce LA; Dropinski JF; Sheng H; Sherry BD; Davies IW Oxyfunctionalization of the Remote C–H Bonds of Aliphatic Amines by Decatungstate Photocatalysis. Angew. Chem. Int. Ed 2017, 56, 15274. [DOI] [PubMed] [Google Scholar]; (d) Ravelli D; Fagnoni M; Fukuyama T; Nishikawa T; Ryu I Site-Selective C–H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope. ACS Catal. 2018, 8, 701. [Google Scholar]; (e) Salamone M; Bietti M Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C–H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects. Acc. Chem. Res 2015, 48, 2895. [DOI] [PubMed] [Google Scholar]
- (7).(a) Tellis JC; Primer DN; Molander GA Single-Electron Transmetalation in Organoboron Cross-Coupling by Photoredox/Nickel Dual Catalysis. Science 2014, 345, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuo Z; Ahneman DT; Chu L; Terrett JA; Doyle AG; MacMillan DWC Merging Photoredox with Nickel Catalysis: Coupling of α-Carboxyl Sp3-Carbons with Aryl Halides. Science 2014, 345, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Shaw MH; Shurtleff VW; Terrett JA; Cuthbertson JD; MacMillan DWC Native Functionality in Triple Catalytic CrossCoupling: Sp3 C–H Bonds as Latent Nucleophiles. Science 2016, 352, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shields BJ; Doyle AG Direct C(Sp3)-H Cross Coupling Enabled by Catalytic Generation of Chlorine Radicals. J. Am. Chem. Soc 2016, 138, 12719. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C-H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12715. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shen Y; Gu Y; Martin R Sp3 C–H Arylation and Alkylation Enabled by the Synergy of Triplet Excited Ketones and Nickel Catalysts. J. Am. Chem. Soc 2018, 140, 12200. [DOI] [PubMed] [Google Scholar]; (e) Dauncey E; Dighe SU; Douglas JJ; Leonori D A Dual Photoredox-Nickel Strategy for Remote Functionalization via Iminyl Radicals: Radical Ring-Opening–Arylation, –Vinylation and –Alkylation Cascades. Chem. Sci 2019. DOI: 10.1039/c9sc02616a [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct Arylation of Strong Aliphatic C–H Bonds. Nature 2018, 560, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ackerman LKG; Martinez Alvarado JI; Doyle AG Direct C–C Bond Formation from Alkanes Using Ni-Photoredox Catalysis. J. Am. Chem. Soc 2018, 140, 14059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Chu JCK; Rovis T Amide-Directed Photoredox-Catalysed C–C Bond Formation at Unactivated Sp3 C–H Bonds. Nature 2016, 539, 272. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi GJ; Zhu Q; Miller DC; Gu CJ; Knowles RR Catalytic Alkylation of Remote C–H Bonds Enabled by Proton-Coupled Electron Transfer. Nature 2016, 539, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Twilton J; Le C. (Chip); Zhang P; Shaw MH; Evans RW; MacMillan DWC The Merger of Transition Metal and Photocatalysis. Nature Reviews Chemistry 2017, 1, 52. [Google Scholar]
- (12).(a) Similar strategies have been implemented for remote arylation from a homolytically cleaved N-F bond; see:Li Z; Wang Q; Zhu J Copper-Catalyzed Arylation of Remote C(Sp3)–H Bonds in Carboxamides and Sulfonamides. Angew. Chem. Int. Ed 2018, 130, 13472. [DOI] [PubMed] [Google Scholar]; (b) Zhang Z; Stateman LM; Nagib DA δ C–H (Hetero)Arylation via Cu-Catalyzed Radical Relay. Chem. Sci 2019, 10, 1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).(a) Morcillo SP; Dauncey EM; Kim JH; Douglas JJ; Sheikh NS; Leonori D Photoinduced Remote Functionalization of Amides and Amines Using Electrophilic Nitrogen Radicals. Angew. Chem. Int. Ed 2018, 57, 12945. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xia Y; Wang L; Studer A Site-Selective Remote Radical C–H Functionalization of Unactivated C–H Bonds in Amides Using Sulfone Reagents. Angew. Chem. Int. Ed 2018, 57, 12940. [DOI] [PubMed] [Google Scholar]; (c) Chen D-F; Chu JCK; Rovis T Directed γ-C(Sp3)–H Alkylation of Carboxylic Acid Derivatives through Visible Light Photoredox Catalysis. J. Am. Chem. Soc 2017, 139, 14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Secondary alkyl bromides have also been explored. The model secondary alkyl bromide, cyclohexyl bromide, usually generates desired product in 5–15% yield. Efforts to mitigate this through ligand substitution and excess alkyl bromide has thus far proven unproductive.
- (15).Everson DA; Jones BA; Weix DJ Replacing conventional carbon nucleophiles with electrophiles: Nickel-catalyzed reductive alkylation of aryl bromides and chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sommer H; Juliá-Hernández F; Martin R; Marek I Walking metals for remote functionalizations. ACS Cent. Sci 2018, 4, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.