

Staphylococcus aureus remains a global health concern and exemplifies the ability of an opportunistic pathogen to adapt and persist within multiple environments, including host tissue. Not only does biofilm contribute to persistence and immune evasion in the host environment, it also may aid in the transition to invasive disease. Thus, understanding how biofilms form is critical for developing strategies for dispersing biofilms and improving biofilm disease-related outcomes. Using biochemical, genetic, and cell biology approaches, we reveal a synergistic interaction between PIA and eDNA that promotes cell aggregation and biofilm formation in a CodY-dependent manner in S. aureus. We also reveal that envelope-associated lipoproteins mediate attachment of the biofilm matrix to the cell surface.
KEYWORDS: CodY, PIA, Staphylococcus aureus, biofilm, eDNA, exopolysaccharide, lipoproteins
ABSTRACT
The global regulator CodY links nutrient availability to the regulation of virulence factor gene expression in Staphylococcus aureus, including many genes whose products affect biofilm formation. Antithetical phenotypes of both biofilm deficiency and accumulation have been reported for codY-null mutants; thus, the role of CodY in biofilm development remains unclear. codY mutant cells of a strain producing a robust biofilm elaborate proaggregation surface-associated features not present on codY mutant cells that do not produce a robust biofilm. Biochemical analysis of the clinical isolate SA564, which aggregates when deficient for CodY, revealed that these features are sensitive to nuclease treatment and are resistant to protease exposure. Genetic analyses revealed that disrupting lgt (the diacylglycerol transferase gene) in codY mutant cells severely weakened aggregation, indicating a role for lipoproteins in the attachment of the biofilm matrix to the cell surface. An additional and critical role of IcaB in producing functional poly-N-acetylglucosamine (PIA) polysaccharide in extracellular DNA (eDNA)-dependent biofilm formation was shown. Moreover, overproducing PIA is sufficient to promote aggregation in a DNA-dependent manner regardless of source of nucleic acids. Taken together, our results point to PIA synthesis as the primary determinant of biofilm formation when CodY activity is reduced and suggest a modified electrostatic net model for matrix attachment whereby PIA associates with eDNA, which interacts with the cell surface via covalently attached membrane lipoproteins. This work counters the prevailing view that polysaccharide- and eDNA/protein-based biofilms are mutually exclusive. Rather, we demonstrate that eDNA and PIA can work synergistically to form a biofilm.
IMPORTANCE Staphylococcus aureus remains a global health concern and exemplifies the ability of an opportunistic pathogen to adapt and persist within multiple environments, including host tissue. Not only does biofilm contribute to persistence and immune evasion in the host environment, it also may aid in the transition to invasive disease. Thus, understanding how biofilms form is critical for developing strategies for dispersing biofilms and improving biofilm disease-related outcomes. Using biochemical, genetic, and cell biology approaches, we reveal a synergistic interaction between PIA and eDNA that promotes cell aggregation and biofilm formation in a CodY-dependent manner in S. aureus. We also reveal that envelope-associated lipoproteins mediate attachment of the biofilm matrix to the cell surface.
INTRODUCTION
Microorganisms are adept at surviving and sometimes thriving in hostile environments. They compete with other microbes for limited nutrients, face possible desiccation, and experience fluctuations in temperature, osmolarity, and pH. During infection, pathogens experience these same chemical and environmental insults and must also contend with host immune defenses. Adopting a sessile biofilm lifestyle insulates microbes from these stresses, prevents phagocytosis and the penetration of toxic compounds, and promotes a pseudomulticellular existence with division of labor (1–5). Nutrient diffusion into biofilms is retarded, and this is thought to contribute to low growth rates, persister cell formation, and antibiotic tolerance (6–8). Thus, understanding mechanisms underlying biofilm development and dispersal can help to identify new strategies to combat microbial infections.
Staphylococcus aureus is a Gram-positive commensal bacterium that colonizes the nares of up to 30% of individuals (9, 10). As an opportunistic pathogen, S. aureus is the leading cause of devastating skin and soft tissue infections, endocarditis, and osteomyelitis, resulting in 20,000 deaths annually (11, 12). In recent years, the prevalence of antibiotic-resistant isolates has increased, as well as the frequency at which seemingly healthy individuals contract infections, exacerbating the problem and thwarting treatment (9, 13, 14). Further, S. aureus is one of the most commonly identified bacterial species that are able to form biofilms on indwelling medical devices such as surgical implants and catheters (15). S. aureus biofilms facilitate prolonged infections by promoting attachment to host surfaces (16–19) and are comprised of one to many species that assemble a self-produced matrix constructed primarily of polysaccharides, proteins, and extracellular DNA (eDNA) (20, 21). In S. aureus, biofilm development is thought to occur via two independent pathways that result in either a polysaccharide-based biofilm or an eDNA/protein-based biofilm (22–26).
S. aureus secretes a polysaccharide composed of repeating oligomers of poly-β-(1,6)-N-acetylglucosamine (PNAG), which is also called polysaccharide intercellular adhesin (PIA) (27, 28). The ability to produce PIA is encoded by the icaADBC locus, which was first revealed in Staphylococcus epidermidis during transposon mutagenesis to identify factors important for biofilm formation (29, 30). Later, orthologs of the ica genes were identified in S. aureus (31). Subsequent work revealed that the ica genes are positively regulated by factors including the alternative sigma factor B (SigB), SarA, SrrAB, and CcpA (32–35). In contrast, TcaR, Spx, CodY, and IcaR negatively regulate the ica operon (36–39). The synthesis of PIA is catalyzed primarily by IcaA, an N-acetylglucosamine transferase; IcaD increases the specificity of IcaA for polymers of ∼20 residues in length (30, 40). IcaC is a membrane-spanning protein that mediates the translocation of newly synthesized PIA to the cell surface, where approximately 43% of the glucosamine residues are deacetylated by the secreted enzyme IcaB (41, 42). This deacetylation imparts a net positive charge to the polymer and is essential for attachment to the cell surface and for intercellular adhesion (i.e., biofilm formation). It was previously thought that the positively charged PIA polymer interacted with negatively charged teichoic acids (TAs) for attachment to the cell surface, as they are the most abundant anions in the cell envelope (43, 44). However, PIA levels and function were not affected in a mutant lacking wall-associated TAs, indicating that they are dispensable (45). Precisely how PIA attaches to the staphylococcal cell surface remains unclear.
In PIA-independent biofilm formation, eDNA and proteins form the biofilm matrix. Genomic DNA (gDNA) is released from bacterial cells into the environment by active secretion, by cell lysis via autolysis, or by phage induction (46). The murein hydrolase AtlA is the major autolysin in S. aureus and was shown to be important for PIA-independent biofilm formation, as mutants fail to release DNA and proteins into the environment (47). In S. aureus, microbial surface component recognizing adhesive matrix molecule (MSCRAMM) proteins mediate the initial attachment to surfaces (17). Following attachment, biofilm development occurs via stages of multiplication, exodus, and maturation, each of which is associated with changes in matrix composition (4). For instance, during early development under biologically relevant flow conditions, biofilms are exquisitely sensitive to proteinase K treatment, while later the biofilm matrix becomes sensitive to DNase I (5, 48). Recent work by Losick and colleagues suggests that upon lysis an electrostatic interaction occurs between cytoplasmic “moonlighting” proteins and eDNA to form a net around bacterial cells in a pH-dependent manner (49, 50).
CodY is a global transcriptional regulator found in Firmicutes that, in response to the availability of the branched-chain amino acids (BCAAs) (isoleucine, leucine, and valine [ILV]) and GTP, adjusts the expression of hundreds of genes whose products broadly mediate the search for, uptake, and processing of alternative nutrient sources through multiple metabolic pathways (51, 52). When intracellular levels of ILV and GTP are high, CodY is activated as a DNA-binding protein and typically represses gene expression (53, 54). In response to diminishing levels of ILV and GTP, the active fraction of CodY protein in the cell decreases, resulting in the remodeling of the transcriptome (55, 56). In pathogenic species such as S. aureus, CodY also controls the production of important virulence factors, including secreted enzymes and toxins that likely enable the bacterium to liberate nutrients from the host tissue (38, 56–59). Further, CodY regulates the expression of genes whose products either build or modulate the biofilm matrix (38, 56–58). For instance, CodY positively regulates the MSCRAMM proteins FnbAB and SasG, which help facilitate the initial attachment of the bacterial cell to host tissue, and has been shown to be required for biofilm formation (22, 56, 60). In contrast, CodY represses the expression of genes coding for secreted proteases that negatively impact biofilm formation and represses the expression of nuclease (Nuc), which is necessary for the exodus stage of biofilm development (57, 61). Additionally, it was reported previously that the ica locus is overexpressed up to 225-fold in a codY-null mutant of the methicillin-susceptible USA200 osteomyelitis isolate UAMS-1 (62), suggesting that CodY is a key regulator of both PIA-dependent and PIA-independent biofilm formation (38, 56).
The exact role of CodY in biofilm formation has remained unclear based on phenotypes reported in a limited number of clinical isolates. We sought to understand the mechanistic underpinnings through which CodY controls biofilm formation among an array of isolates. Herein, we show that CodY-dependent biofilm phenotypes correlate with the extent of ica expression and PIA production. Importantly, in contrast to the prevailing view that PIA-based biofilms and eDNA-based biofilms are mutually exclusive, we reveal a previously unidentified mixed PIA and eDNA matrix that works synergistically to promote cell aggregation and biofilm formation. Additionally, we demonstrate that lipidation of one or more prelipoproteins contributes to the interaction of the PIA/eDNA complex with the cell envelope.
RESULTS
CodY suppresses cell aggregation in planktonic cultures.
Previous reports have implicated CodY in controlling biofilm formation (38, 63). Supporting these findings, analysis of CodY-regulated genes by transcriptome sequencing (RNA-seq) and by in vitro pulldown assays revealed that many genes known to be involved in biofilm formation are under CodY control (i.e., fnbA, ica, sspA, nuc, and hlb) (38, 56–58). As had been noted previously (38), during routine culturing we observed that codY-null mutant (ΔcodY) colonies of methicillin-susceptible osteomyelitis isolate UAMS-1 (62) and sepsis isolate SA564 (64) were exceptionally viscous on solid medium and formed both prominent aggregates and a thick ring of biomass on glass culture vessels during exponential growth in tryptic soy broth (a rich, complex medium). This does not occur during cultivation of wild-type (WT) staphylococci even though they cluster due to incomplete separation of daughter cells following division in alternating planes (Fig. 1A) (65, 66). In contrast, community-associated, methicillin-resistant USA300 LAC* (here referred to as LAC) ΔcodY mutant cells did not exhibit these phenotypes (see Fig. S1A in the supplemental material). It has previously been reported that ΔcodY mutant cells of USA300 LAC fail to form biofilm during static culturing (63), suggesting that the lack of cell aggregation we observed may be due to a defect in biofilm formation. Given the particularly strong aggregation phenotype of SA564 ΔcodY mutant cells during aerobic growth, we focused our attention on this clinical isolate and used scanning electron microscopy (SEM) to determine whether aggregation occurs by cell-to-cell interaction or via extracellular matrix production. SEM revealed large aggregates of SA564 ΔcodY mutant cells compared to wild-type cells. Upon closer inspection, the ΔcodY mutant cell aggregates appeared to consist of cells connected to one another by a stringlike extracellular matrix consisting of filaments ranging from 20 to 50 nm in width and up to 2 μm in length, with most being between 0.5 and 1 μm in length (Fig. 1A). We note that this is the defining feature of these aggregates.
FIG 1.

ΔcodY mutant cells of diverse S. aureus clinical isolates form large cell aggregates tethered by a stringlike matrix. (A) Scanning electron microscopy was performed on SA564 and ΔcodY mutant cells during exponential growth in tryptic soy broth. Images are representative of multiple experiments. Images are at the same magnification. Representative images of biofilm observed in overnight culture growth are shown to the left of each micrograph. (B and C) Percent aggregation of S. aureus clinical isolates and their ΔcodY mutant derivatives (B) and the complemented SA564 codY-null mutant (C) using the settling assay from samples obtained during exponential growth in TSB as described in Materials and Methods. Data indicate the mean ± standard error of the mean (SEM) values from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (relative to WT using Student’s t test [B] and analysis of variance [ANOVA] with Dunnett’s postanalysis relative to SA564 WT [C]). VOC, vector-only control.
To determine the extent to which CodY’s role in suppressing aggregation is dependent on the strain background, we used a settling assay (67) to survey S. aureus isolates across multiple clonal complexes, sequence types, and methicillin susceptibilities. Strains were grown aerobically in tryptic soy broth for 3 h (optical density at 600 nm [OD600] of ∼2 to 4), at which time the cells were allowed to settle for 45 min. After the settling period, wild-type cells remained suspended in broth (Fig. 1B, black bars). In contrast, ΔcodY mutant cells of SA564, UAMS-1, MW2, and COL settled at a significantly higher rate during the settling period, as indicated by the drop in the optical density of the mutant sample, consistent with larger cell aggregates (Fig. 1B, gray bars). The aggregation phenotype in SA564 was complementable by introducing a plasmid containing a wild-type copy of codY under the control of its native promoter (Fig. 1C). The LAC and Newman ΔcodY mutants did not settle during the course of the assay, revealing that CodY suppresses cell aggregation in many (but not all) isolates. Notably, ΔcodY mutant cells of strains that aggregated also formed static biofilms (Fig. S1B).
Multicellular aggregates of ΔcodY mutant cells contain DNA in the extracellular matrix.
We next sought to further characterize the composition of the extracellular matrix binding the SA564 ΔcodY mutant cells together. Based on the SEM images and the viscous texture on solid medium, we hypothesized that ΔcodY mutant cells were tethered by eDNA. To test this hypothesis, we sampled cells during exponential growth and stained them with Syto 40 (a membrane-permeable double-stranded DNA [dsDNA] dye [68]; blue signal) and TOTO-1 (a nonpermeable dsDNA dye [69]; green signal) and then imaged the samples using confocal scanning laser microcopy (CSLM). Similar to the results obtained by SEM, CSLM revealed dense aggregates of ΔcodY mutant cells, which were absent when wild-type cells were examined. Further, cell aggregates colocalized with an abundance of TOTO-1 signal, indicating the presence of copious amounts of eDNA (Fig. 2A). Moreover, the addition of 100 U ml−1 of DNase I to cultures during planktonic shake flask growth eliminated ΔcodY mutant cell aggregation and the filamentous matrix material observed by SEM (Fig. 2B and C). Similar results were observed when these cells were seeded into microtiter plates and static biofilm development was assessed (Fig. 2D). Taken together, these results indicate that eDNA is critical for cell aggregation of and biofilm formation by ΔcodY mutant cells.
FIG 2.

ΔcodY mutant cell aggregates are associated with extracellular DNA (eDNA) and are sensitive to DNase I treatment. (A) Cells were grown to exponential phase in TSB, stained with Syto 40 and TOTO-1, and then visualized using confocal scanning laser microscopy (CSLM). Live cells are blue (Syto 40), while eDNA and dead cells are green (TOTO-1). Each panel is viewed at the same magnification. Insets are at ×10 magnification. Asterisks indicate the areas used for insets. (B) SEM micrographs of SA564 cells grown aerobically to exponential phase in TSB containing DNase I. All images are shown at the same magnification. (C) Settling assay of cells grown in TSB aerobically to exponential phase in shake flask culture. (D) Static biofilm development in the presence of increasing amounts of DNase I was measured at 20 h after inoculation in TSB as described in Materials and Methods. Data are the mean ± SEM values from at least three independent experiments. **, P < 0.01; ****, P < 0.0001 (relative to wild type using Student’s t test [C] or ANOVA with Dunnett’s postanalysis [relative to wild type, 0 U/ml] [D]). NS, not significant. All images are representative of multiple experiments.
eDNA is strongly associated with the cell envelope of ΔcodY mutant cells.
Secreted nuclease (Nuc) plays a role in shaping the biofilm and is required for the exodus phase during biofilm development (5, 70). Additionally, we and others showed that the expression of nuc is increased in ΔcodY mutant cells and results in higher nuclease activity in culture supernatants (56, 58). Although secreted products and their abundances can be lineage dependent, we reasoned that ΔcodY mutant cells that fail to aggregate may lack the necessary factor(s) for eDNA tethering or may overproduce nuclease and degrade the existing eDNA. As seen in Fig. S2A in the supplemental material, we measured relatively low nuc transcript abundance in wild-type cells. As expected, nuc transcript abundance increased approximately 15- to 60-fold in SA564 ΔcodY and LAC ΔcodY mutant cells relative to their wild-type parent strains. In parallel, we assayed secreted nuclease activity in culture supernatants as previously described (70). Consistent with quantitative real-time reverse transcription-PCR (qRT-PCR) analysis, an increase in secreted nuclease activity was observed in ΔcodY mutant culture supernatants compared to wild-type culture supernatants (P < 0.0001). However, when comparing aggregating (SA564) and nonaggregating (LAC) backgrounds, secreted nuclease activity in ΔcodY mutant supernatants was essentially identical (Fig. S2B). Collectively, these data suggest that nuclease activity does not account for the lack of cell aggregation in the LAC ΔcodY mutant and that aggregation is likely mediated by a cell-associated factor.
Given that eDNA is necessary for ΔcodY mutant cell aggregation and biofilm formation in SA564, we next sought to understand how the eDNA interacts with the cell envelope. Past studies revealed that the S. aureus biofilm extracellular matrix contains cytosolic and secreted proteins, many of which have an average pI of >8 (49, 50, 71). These proteins are thought to mediate cell association of eDNA via electrostatic interactions. That is, under the mildly acidic conditions that naturally occur in biofilms, these matrix proteins would carry a net positive charge and interact with negatively charged DNA, resulting in DNA incorporation (72). Under conditions where the pH approaches or exceeds the pI (i.e., more alkaline conditions), these proteins would be negatively charged and would not be expected to interact electrostatically with DNA. We found that the addition of proteinase K to culture medium had no effect on ΔcodY mutant cell aggregation during planktonic growth, suggesting either that the eDNA is interacting with the cell envelope independent of released cytoplasmic proteins, that the eDNA somehow protects surface-associated proteins from digestion with proteinase K, or that the proteinase K itself is degraded (Fig. 3A). With respect to DNA-binding proteins, uninoculated tryptic soy broth (TSB) has a pH of 7.3, which drops during exponential growth due to secretion of acidic by-products of metabolism (73). When we buffered the TSB medium to pH 7.5 and performed a settling assay with exponentially grown cells, ΔcodY mutant cells failed to form aggregates (Fig. 3B, gray bars). In parallel, we tested whether the eDNA can be released from the cell surface by resuspending a sample of TSB-grown cells in phosphate-buffered saline (PBS) buffered at pH 5.0 or pH 7.5. At pH 7.5, we would expect electrostatic interactions to be diminished. Consistent with previous results (74), when we suspended SA564 cells in PBS at pH 7.5, we measured an ∼35-fold increase in eDNA released from cells compared to the level of eDNA recovered from cells suspended in PBS at pH 5.0 (Fig. 3C, compare wild-type gray versus black bars). However, to our surprise, we did not observe DNA release when cells were suspended in PBS at pH 7.5. Notably, the suspension of ΔcodY mutant cells in PBS at pH 7.5 did not disperse aggregates as previously demonstrated when biofilm formation occurred via the electrostatic net model (Fig. 3C, compare ΔcodY gray to black bars and CSLM micrographs). Taken together, these data indicate that aggregate formation is protease tolerant and pH sensitive initially but is resistant to changes in pH once the aggregates have formed, suggesting that surface charge is an important factor for initial cell aggregation. However, how the matrix is assembled at the surface remains unclear.
FIG 3.

eDNA-based ΔcodY mutant cell aggregation is protease tolerant and initially dependent on electrostatic interactions. (A) The effect of proteinase K (0.1 mg ml−1) on wild-type and ΔcodY mutant cells of SA564 was examined during exponential growth in TSB using a settling assay. (B) A settling assay was performed on exponentially growing cells cultured in TSB buffered to pH 7.5 or unbuffered TSB, which acidified to ∼pH 6.5 during growth. (C) Cells cultured in TSB were resuspended in phosphate-buffered saline at either pH 5.0 or 7.5 for 15 min, and quantitative PCR was used to measure the amount of eDNA released from the samples, taken at the same OD600 value. Representative micrographs are shown for ΔcodY mutant cells from the same experiment. Syto 40 (blue signal) was used to stain all cells, while eDNA was visualized using TOTO-1 (green signal). Data are the mean ± SEM values from at least three independent experiments. *, P < 0.05; ***, P < 0.001 (by Student’s t test comparing ΔcodY to wild type [A and B] or Friedman’s test with Dunn’s postanalysis [C]). Here, results for the wild-type cell sample suspended in PBS pH 7.5 are significantly different from those for the ΔcodY mutant cell sample at pH 5.0 and 7.5 and trended higher than those for the wild type at pH 5.0 (but were not significantly different).
Cell aggregation depends on an elaborated factor in CodY-deficient cells.
Since the eDNA tethered to ΔcodY mutant cells could not be removed in the same manner as for wild-type cells, we reasoned that eDNA tethering depends on a more extensive network of interactions and/or additional factors specifically produced when CodY activity is reduced or eliminated. To address this, we cocultured SA564 wild-type cells with ΔcodY mutant cells. Cell genotypes were differentiated by the presence of the plasmid pKM16, which contains the dsRed fluorescent protein under the control of the constitutive sarA P1 promoter. Cells were mixed at a starting ratio of 1:1 and grown to exponential phase in TSB medium. Confocal imaging of cocultures revealed that cell aggregates are composed almost exclusively of ΔcodY mutant cells (Fig. 4A, red signal), whereas wild-type cells (Fig. 4B, red signal), though present, do not appear to be the major constituent of the aggregate. Indeed, image analysis indicates ∼7-fold more ΔcodY mutant cells in the aggregate. There is less than a 2-fold difference in the numbers of wild-type and ΔcodY mutant cells in a randomly selected area of dispersed cells (see Table S1 in the supplemental material).
FIG 4.

Coculture experiments reveal that eDNA-based aggregates consist of predominantly ΔcodY mutant cells. Cultures were inoculated with the indicated genotypes at approximately a 1:1 ratio, grown to exponential phase (OD600 of ∼0.5) in TSB, and then visualized by CSLM. All cells are labeled with Syto 40 (blue); eDNA and dead cells are labeled with TOTO-1 (green). In each panel a particular strain harbors a constitutive dsRed plasmid (pKM16) to discern genotypes. (A) SA564 wild-type cells mixed with isogenic ΔcodY mutant cells harboring pKM16. (B) SA564 wild-type cells harboring pKM16 mixed with isogenic ΔcodY mutant cells. (C) Nonaggregating LAC ΔcodY mutant cells mixed with SA564 ΔcodY mutant cells harboring pKM16. (D) SA564 ΔcodY mutant cells mixed with aggregating COL ΔcodY mutant cells harboring pKM16. All images are representative of multiple experiments. All panels are viewed at the same magnification.
To examine if ΔcodY mutants of nonaggregating lineages can adhere to a preexisting matrix, we next cocultured LAC ΔcodY mutant cells (nonaggregating) with SA564 ΔcodY mutant cells (aggregating). As seen in Fig. 4C, cell aggregates consist of mostly SA564 ΔcodY mutant cells (Fig. 4C, red signal). These results, together with the observation that secreted nuclease activities are similar in both lineages, might suggest that the low numbers of LAC ΔcodY mutant cells in the aggregate under these conditions are not due to a secreted factor that disrupts aggregation but, rather, may be due to a reduction in the abundance of a surface factor on which the aggregates form. This would likely weaken the affinity of the cells for the aggregate. To control for the possibility that segregation occurs when different lineages of S. aureus cells are mixed, we cocultured ΔcodY mutant cells of COL and SA564 (both aggregating lineages). Confocal images show cells of each lineage in the aggregate (Fig. 4D), indicating that the cell aggregates are not clonal. Taken together, these experiments are consistent with the notion that CodY regulates the production of a surface factor that promotes eDNA tethering to the cell envelope.
Isolation of a suppressor mutant reveals that PIA contributes to cell aggregation in a ΔcodY mutant.
While conducting this study, we serendipitously discovered a spontaneous suppressor mutant in the SA564 ΔcodY mutant background that failed to aggregate and form biofilm. We designated the mutant with the allele soa-1 (suppressor of aggregation) (Fig. 5A). Further characterization of this suppressor mutant using SEM confirmed the absence of cell aggregates and revealed that the cells were largely devoid of matrix material (Fig. 5B). To map the suppressor mutation(s), we performed whole-genome sequencing of the soa-1 suppressor mutant. Analysis of the sequence data revealed a missense mutation coding for a variant cell wall biosynthesis enzyme, MraYL113M, and a nonsense mutation coding for a truncated PIA biosynthetic enzyme (IcaBQ223*) (Table 1). Using allelic exchange, we reconstituted the mraY mutation in the ΔcodY mutant background and analyzed aggregation in this strain. Routine overnight cultures exhibited amounts of ring biomass material around the incubation tube that were similar to those for the ΔcodY mutant (see Fig. S3A in the supplemental material). Further, a settling assay revealed aggregation essentially equivalent to that of the isogenic ΔcodY mutant. This mraY* “hitchhiker” allele (75), though present in the isolated suppressor mutant, may confer some fitness advantage but does not suppress aggregation (Fig. 5C). In contrast, deleting the ica operon in the SA564 ΔcodY mutant was sufficient to alleviate cell aggregation and biofilm formation (Fig. 5C). This came as a surprise to us, as it has previously been reported that SA564 does not produce appreciable amounts of PIA (76). As an independent approach, we placed the coding sequence of icaB under the control of the constitutive sarA P1 promoter and introduced a plasmid containing this construct (pKM26) into the suppressor mutant. Expressing a wild-type copy of icaB in the suppressor mutant restored cell aggregation during exponential growth, and once again eDNA enshrouded the aggregates (Fig. 5D and E). Moreover, suppressor mutant cells form mixed aggregates with SA564 ΔcodY mutant cells, suggesting that IcaB production and secretion in the ΔcodY mutant trans-complements the lesion in the suppressor mutant (Fig. S3B). Thus, moving forward, we refer to the soa-1 mutant as an icaB mutant.
FIG 5.

Suppressor analysis reveals that ica is required for cell aggregation. (A) SRB1243 cells (SA564 ΔcodY soa-1) were grown to exponential phase in TSB and assayed for aggregation. ****, P < 0.0001 (by ANOVA with Dunnett’s postanalysis comparing samples to wild type). (B) SEM of suppressor mutant cells sampled during exponential growth in tryptic soy broth. The image is representative of multiple experiments. (C) Settling assay performed on isogenic strains during exponential growth in TSB. **, P < 0.01; ***, P < 0.001 (by ANOVA with Dunnett’s postanalysis). Here, the ΔcodY and mraY* ΔcodY mutants are significantly different from the wild type. (D) Complementation using the sarA P1 promoter to constitutively express icaB. ***, P < 0.001 (by two-tailed Student t test comparing +icaB [PsarAP1-icaB+] to +vector). (E) CSLM micrographs of ΔcodY soa-1 cells harboring pCN51 or pKM26 (PsarAP1-icaB+) during exponential growth in TSB. All cells were visualized using Syto 40 (blue signal), while eDNA and dead cells were stained by TOTO-1 (green signal). All images are representative of multiple experiments. All panels are viewed at the same magnification.
TABLE 1.
Summary of mutations found in ΔcodY soa-1 coding sequences
| Nucleotide | Mutation | Annotation | Product description | Gene |
|---|---|---|---|---|
| 1167134 | T→A | L113M | Phospho-N-acetylmuramoyl-pentapeptide transferase | mraY |
| 2741388 | C→T | Q223* (CAG→TAG) | Poly-beta-1,6-N-acetyl-d-glucosamine N-deacetylase | icaB |
It was previously shown that the ica operon is overexpressed in UAMS-1 ΔcodY mutant cells (38). Therefore, we hypothesized that nonaggregating cells are simply not producing PIA (or sufficient quantities of PIA) during exponential growth. To test this, we performed qRT-PCR and measured ica transcript abundance in the wild-type and ΔcodY mutant strains of SA564 and LAC. We measured an >600-fold increase in icaA transcript in the SA564 ΔcodY mutant compared to the wild-type parent. In contrast, we measured an ∼5-fold increase in the LAC ΔcodY mutant (Fig. 6A, compare ΔcodY with wild type). Immunoblot analysis of PIA production by exponentially growing SA564 and LAC cells mirrored the expression data, and the complemented suppressor mutant regained the ability to produce PIA (Fig. 6B). Furthermore, we note that aggregation in the strains surveyed in Fig. 1 is correlated with PIA production (Fig. 6C), and, at least for SA564, the aggregates can also be dispersed using sodium metaperiodate (which cleaves polysaccharide rings between vicinal diols) and dispersin B (which enzymatically degrades polymers of PIA) (Fig. 6D). Thus, the aggregates are PIA dependent. Finally, overexpressing the ica locus using the sarA P1 promoter was sufficient to induce aggregation in LAC and SA564 wild-type cells, and exposing the SA564 cultures to DNase blocked aggregation (Fig. 6E; see Fig. S4 in the supplemental material). Taken together, these results indicate that a critical level of PIA production is required for ΔcodY mutant cell aggregation and that deacetylated PIA resulting from IcaB activity is required for aggregation. These results also affirm the requirement for DNA along with PIA for aggregation.
FIG 6.

eDNA-based cell aggregation is dependent on the production of PIA in ΔcodY mutant cells. (A) SA564 and LAC cells were grown to exponential phase aerobically in TSB, and icaA transcript copy numbers in wild-type and ΔcodY mutant cells were determined by qRT-PCR. Data were normalized to rpoC transcript copy number. (B and C) Quantification of cell-associated PIA detected by immunoblot analysis using densitometry for SA564 or isogenic mutants (B) or the wild type and codY-null mutant of the indicated strains (C) obtained from cell pellets grown aerobically for 3 h in tryptic soy broth. When necessary, samples were diluted to avoid membrane saturation. (D) SA564 and ΔcodY mutant cells were grown aerobically in TSB containing sodium metaperiodate (40 μg ml−1) or dispersin B (5 μg ml−1), and aggregation was assessed using the settling assay. (E) Wild-type SA564 and LAC cells constitutively expressing icaADBC under the control of the sarA P1 promoter were grown planktonically in tryptic soy broth, and a settling assay was used to assess aggregation. SA564 was additionally cultured in the presence of DNase I (200 U ml−1). Data indicate the mean ± SEM values from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (using Student’s t test comparing the ΔcodY mutant to the wild type for each condition in panels A, D, and E). ns, not significantly different. Error bars are plotted for all data; in some cases, they are too small to see.
Surface-associated PIA may concentrate DNA from the environment to promote cell aggregation.
Our data thus far demonstrate that both PIA and eDNA contribute to cellular aggregation. However, the source of the eDNA remained unclear. A ΔguaA mutant of S. aureus is a guanine nucleotide auxotroph. TSB provides this required nutrient (77), suggesting that the DNA incorporated into the biofilm matrix might be derived from the medium. Consistent with this observation, ΔcodY mutant cells failed to aggregate during growth in chemically defined medium (CDM) lacking DNA (see Fig. S5A in the supplemental material). Because functional PIA is required for eDNA-dependent cell aggregation, we measured icaA transcript abundance as proxy for the ica locus. While we measured a marked increase in ica gene expression in ΔcodY mutant cells relative to wild-type cells grown in TSB, we saw relatively little icaA transcript in either strain during growth in CDM (Fig. S5B). Overexpression of the ica operon failed to restore cell aggregation in CDM. However, amending the medium with exogenous S. aureus chromosomal DNA purified from a commercial kit induced aggregation in an ica-dependent manner (Fig. 7A). The requirement for eDNA was nonspecific, as exogenous chromosomal DNA purified from Bacillus subtilis strain SMY and Pseudomonas aeruginosa strain PAO1 induced aggregation (Fig. 7B and C). To address the possibility that ΔcodY mutant cells exhibit increased cell lysis and DNA release, we measured nucleic acid levels in cultures grown in TSB and CDM as well as in uninoculated controls. As can be seen in Fig. S5C, we detected no strain-specific differences in nucleic acid concentration during growth in either medium. No differences in CFU counts were apparent, and we detected no obvious differences in staphylococcal chromosomal DNA released into culture supernatants (see Fig. S6 in the supplemental material). Compared to conditioned medium, uninoculated CDM contained no detectable eDNA, suggesting that wild-type and ΔcodY mutant cells experience mild lysis. eDNA levels were generally lower in inoculated TSB than in uninoculated TSB, suggesting that DNA was consumed during growth and/or incorporated into the biofilm matrix. We also examined the role of Atl in CodY-mediated cell aggregation given its well-defined role in eDNA release (47, 73). Notably, an Δatl ΔcodY double mutant formed dense cell aggregates; chemical inhibition of Atl using polyanethol sulfonate (PAS) failed to disrupt biofilm development (see Fig. S7A and B in the supplemental material). No differences were detected in acetate release or culture acidification, further suggesting that Atl is not required for eDNA-dependent cell aggregation in a ΔcodY mutant (Fig. S7C and D). Taken together, our data suggest that exogenous DNA, regardless of the source, can be incorporated into the biofilm matrix. We cannot exclude the possibility that nonspecific cell lysis in defined medium contributes eDNA in the matrix, but this DNA alone cannot promote aggregation even when PIA is present.
FIG 7.

PIA and bacterial chromosomal DNA promote cell aggregation in CDM. Wild-type SA564 cells containing pKM28 (PsarA-P1-icaADBC) or the vector-only control (pMRSI) were cultured in CDM lacking exogenous DNA. During exponential growth, a 1-ml sample of cells was mixed with purified genomic DNA from S. aureus LAC (A), Bacillus subtilis SMY (B), or Pseudomonas aeruginosa PAO1 (C), and a settling assay was performed. Data are plotted as the mean ± SEM values from at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (by one-way ANOVA with Dunnett’s posttest comparing samples to SA564 + pMRSI [0 ng/ml DNA]).
Lipoproteins contribute to PIA/eDNA interaction with the cell envelope.
Teichoic acids (TAs) were originally thought to facilitate the interaction of deacetylated PIA with the cell surface, given their abundance in the cell wall and overall negative charge (43, 44). However, the exact point of attachment of PIA to the cell surface remains unclear, as a tagO mutation, causing deficiency in wall TA synthesis, has little effect on PIA production or anchoring (45). Recently, a set of lipoproteins was shown to function as anchor points between eDNA in the biofilm matrix and the cell surface (72). The prelipoprotein diacylglycerol transferase Lgt catalyzes the first step in lipoprotein retention at the cell membrane by lipidating invariant cysteine residues, and lgt mutants eject lipoproteins into the extracellular milieu (78, 79). Consistent with the findings of Kavanaugh et al. (72), cell aggregates produced by Δlgt ΔcodY double mutant cells were easily disrupted by vigorous shaking during aerobic cultivation in shake flasks (no rings of biomass were present on culture vessels), and knocking out lgt in the ΔcodY mutant partially suppressed the aggregation phenotype in the settling assay (Fig. 8A, compare Δlgt ΔcodY strain to ΔcodY strain). Notably, immunoblot analysis revealed a >5-fold decrease in cell-associated PIA in the Δlgt ΔcodY double mutant compared to the ΔcodY mutant (compare Fig. 8B and 6C). We conclude that eDNA bound to membrane lipoproteins mediates in part the attachment of PIA to the cell surface to promote aggregation.
FIG 8.

Cell wall-anchored lipoproteins contribute to eDNA/PIA adherence to the cell surface. (A) An isogenic suite of SA564 mutant strains were grown in TSB, and a settling assay was performed as described in Materials and Methods. **, P < 0.01; ***, P < 0.001 (compared to the wild-type and Δlgt strains. #, P < 0.05 (comparing the ΔcodY mutant to the Δlgt ΔcodY double mutant). One-way ANOVA with Tukey postanalysis was used. SEMs are plotted for all data; in some cases the error bars are too small to see. (B) Immunoblot densitometry analysis of PIA production for the indicated strains is shown. When necessary, samples were diluted to avoid membrane saturation.
DISCUSSION
CodY plays a key role in linking nutrient availability to virulence gene expression and factor production. As a result, a drop in CodY activity may affect disease progression in a variety of Gram-positive pathogens when the amino acids are depleted in host tissues during infection (80–82). Notably, biofilm development promotes prolonged infection, and knocking out codY (which mimics severe nutrient depletion) either promotes or reduces biofilm formation in S. aureus clinical isolates. Herein, we provide an explanation for these divergent phenotypes and show that ica expression and PIA production (via complementation, suppressor analysis, and immunoblot analysis) are the primary determinants for CodY-mediated biofilm formation. Moreover, new evidence that eDNA and PIA function together to form biofilm matrix and promote cellular aggregation came unexpectedly from the analysis of ΔcodY mutant cells in planktonic culture. This is supported by genetic and biochemical evidence demonstrating (i) that the biofilm aggregates can be dispersed when either the PIA or the eDNA component is eliminated and (ii) that the presence of only one of these molecules is not sufficient to cause aggregation.
Historically, biofilm formation in S. aureus has largely been classified into two categories based on the matrix composition: PIA-dependent biofilms are often associated with methicillin-susceptible S. aureus (MSSA) isolates, and eDNA/protein-dependent biofilms are typically associated with methicillin-resistant S. aureus (MRSA) isolates (24). Our data demonstrate that these matrices are not mutually exclusive, and the conditions in which the biofilms or aggregates form as well as the regulatory circuitry governing the expression of factors (e.g., ica, nuc, proteases, etc.) dictate the relative proportions of matrix components and sensitivity to extracellular-matrix-degrading agents. For example, LAC strains, which normally do not form PIA-dependent biofilm aggregates due to low ica expression, are capable of doing so when icaADBC are overexpressed. However, our data reveal that aggregate formation is still dependent on the presence of eDNA despite icaADBC overexpression. In the absence of PIA, the biofilms formed by LAC are relatively weak and are dispersed when codY is knocked out, likely due to overexpression of proteases and reduction in the abundance of MSCRAAM proteins such as FnBPs, SpA, Map, EbpS, and CflA (61). Attenuation of the LAC ΔcodY mutant in an acute sepsis model of infection (83) is likely due to reduced attachment in vivo. On the other hand, UAMS-1 and SA564 codY mutant cells express ica to high levels, produce large amounts of PIA, and produce viscous biofilms. Given the apparent host niche-dependent effects on CodY-deficient strains (84), it would be beneficial to examine the role of S. aureus CodY in chronic, biofilm-associated infections such as infective endocarditis (85), osteomyelitis (62), polymicrobial infections (86, 87), or diabetic foot ulcers (88).
The cross-linking of eDNA to the surface via PIA and lipoproteins (shown in this study and by Kavanaugh et al. [72]) brings DNA in close proximity to the cell membrane. This eDNA may have a use beyond biofilm formation and aggregation. First, amino acid starvation triggers the induction of the stringent response as well as a reduction in CodY activity. Both responses elicit changes in secreted nuclease activity, nucleotide transport, and metabolism (77, 89, 90). Using PIA as a sponge for eDNA may allow for more efficient nucleotide scavenging. Second, fibrinogen was shown previously to promote S. aureus clumping, a condition that increases local concentrations of autoinducing peptide and activates the Agr quorum-sensing system and its RNA effector RNAIII (91). The primary mechanism by which CodY controls expression and activity of the Agr system remains unknown, as direct CodY binding to the agr P2/P3 promoter or a CodY binding motif in the coding sequence of agrC seems an unlikely explanation given the low affinity of CodY for these sites (38, 57, 92). It is conceivable that the PIA- and eDNA-induced aggregation may help explain the potent increases in agr (RNAII) and RNAIII transcripts seen in UAMS-1 (56, 57). Third, CodY-mediated cell aggregation using eDNA from other bacterial species may provide a prime opportunity for genetic exchange. Two lines of evidence support this notion: (i) CodY has been shown to control competence in B. subtilis in response to nutrient limitation (93) and (ii) the master regulator of competence in B. subtilis, ComK, is upregulated in ΔcodY mutant cells of S. aureus (56). Additional analysis of the CodY regulon reveals several target genes whose products share significant sequence homology with genes important for competence in B. subtilis and streptococci. These include the oppABCD oligopeptide ABC transporters and genes that direct the synthesis of a putative secretion apparatus (i.e., the comG operon) (94, 95).
Working model for PIA/eDNA-dependent cell aggregation.
Given the overall positive charge of PIA and the overall negative charge of DNA, we now extend the existing electrostatic model to explain aggregate formation and biofilm development when CodY activity is eliminated (Fig. 9). Under conditions that reduce CodY activity, PIA synthesis is upregulated. We propose that secreted PIA is capable of directly interacting with available eDNA and that this DNA binds one or more lipoproteins. These lipoproteins in the biofilm matrix strongly bind eDNA under slightly acidic conditions (72). Once these extensive interactions are established, the aggregates become recalcitrant to dispersal. This likely explains our observations that buffering TSB medium to pH 7.5 prevented eDNA-based ΔcodY mutant cell aggregates from forming during exponential growth and that eDNA could not be released from preformed aggregates after the cells were exposed to a more alkaline pH (Fig. 3B and C). It is noteworthy that strains overproducing PIA require a slightly higher concentration of DNase I to circumvent aggregation, suggesting that eDNA may be coated with PIA (Fig. 6E). This is reminiscent of a recent P. aeruginosa study that showed that the secreted polysaccharide Psl interacts with eDNA to stabilize and possibly even fuse strands of DNA together, providing a scaffold for cells to adhere (96). Depending on the density of these fibers, this might also block proteinase K from reaching the cell surface, explaining the proteinase K-tolerant phenotype of the aggregates. As mentioned above, our data indicate that one or more lipoproteins on the cell surface likely bind negatively charged eDNA, which interacts with positively charged PIA. It is worth noting that an lgt mutation does not completely suppress ΔcodY mutant cell aggregation (Fig. 8). This suggests that the noncovalent electrostatic interactions between the matrix and the cell surface are weakened but that additional surface factors, including teichoic acids, seem to contribute to aggregate formation. Although redundancy may thwart our efforts, a genetic screen is under way to identify factors required for aggregate formation in codY mutant cells.
FIG 9.
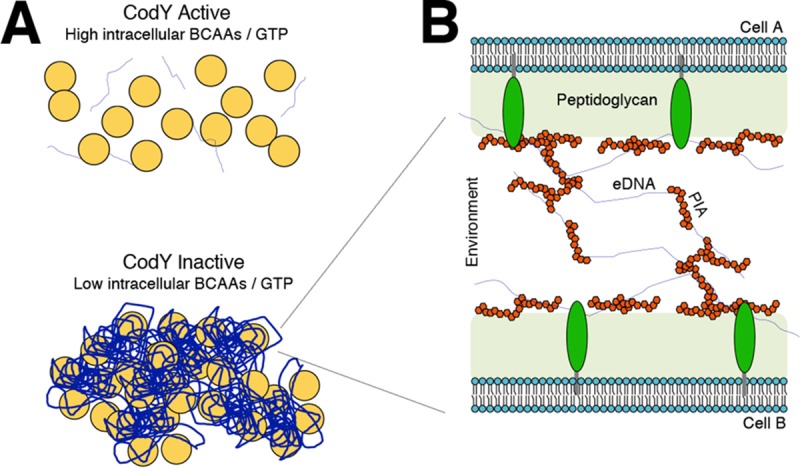
Working model of PIA/eDNA-dependent cell aggregation. (A) As the abundance of key nutrients (i.e., branched-chain amino acids and GTP) drops intracellularly, CodY activity decreases, promoting cell aggregation using available eDNA and PIA. (B) Cell-to-cell interaction occurs in a CodY-dependent manner whereby eDNA and PIA interact synergistically with the cell surface, mediated by one or more lipoproteins. eDNA, blue threads; PIA, red polygons; lipoproteins, green ovals.
MATERIALS AND METHODS
Bacterial strains and culturing.
All strains used in this study are listed in Table S2 in the supplemental material. Staphylococcus aureus strains were cultured in tryptic soy broth (TSB) (Becton Dickinson formulation containing 0.25% [wt/vol] dextrose) at 37°C with shaking at 280 rpm unless otherwise noted. Escherichia coli strains were cultured in modified Lennox (L) medium consisting of 10 g liter−1 tryptone, 5 g liter−1 yeast extract, and 5 g liter−1 NaCl (97). P. aeruginosa PAO1 was cultivated on Pseudomonas isolation agar and in liquid using TSB. When necessary, media were solidified with agar to 1.5% (wt/vol), and antibiotics were included in media at the following concentrations to maintain selection: ampicillin (Ap), 50 μg ml−1, chloramphenicol (Cm), 10 μg ml−1, tetracycline (Tc), 3 μg ml−1, trimethoprim (Tm) 10 μg ml−1; and erythromycin (Em), 5 μg ml−1. Unless otherwise noted, where indicated, cultures were supplemented with micrococcal nuclease (Worthington Biochemical) at 100 U ml−1 or buffered using 50 mM Tris-HCl to maintain a pH of 7.5. Planktonic growth was performed as previously described (56), with the exception that 250-ml DeLong flasks were used at with a 10:1 flask-to-medium ratio in TSB or chemically defined medium (CDM) (98). Briefly, overnight cultures were diluted to an initial optical density at 600 nm (OD600) of 0.05, grown to an OD600 of ∼1, and rediluted to an OD600 of 0.05 to ensure that cells were in exponential phase. For coculture experiments using multiple genotypes, cells were mixed at approximately 1:1 to obtain an OD600 of 0.05. CFU counts were verified by dilution plating on tryptic soy agar (TSA) plates when samples were withdrawn for microscopy.
Recombinant DNA and genetic techniques.
Oligonucleotides for this study were synthesized by Integrated DNA Technologies (Coralville, IA) and are listed in Table S3 in the supplemental material. Restriction enzymes, T4 DNA ligase, and Q5 DNA polymerase were purchased from New England Biolabs. Plasmid minipreparation kits were purchased from Promega, and PCR purification and gel extraction kits were purchased from Qiagen. Plasmids used in this study are listed in Table S4 in the supplemental material. E. coli NEB 5α (New England Biolabs) was used as the host for plasmid constructions, and all plasmids were confirmed by restriction digestion and nonradioactive Sanger sequencing (Genewiz). Plasmids were introduced into S. aureus strain RN4220 by electroporation as previously described (99). As needed, plasmids and marked chromosomal mutations were transferred into select strain backgrounds using ϕ11-mediated transduction (99).
Construction of plasmids.
(i) Complementation plasmids. (a) codY. The native promoter of the operon containing the codY gene (RT87_06190 to RT87_06205) was amplified from wild-type SA564 genomic DNA using oKM120 and oKM111 to generate a 392-bp fragment containing 26 bp of homology to the codY coding sequence. The open reading frame of codY was amplified using oKM112 and oKM113. Fusion PCR (100) was performed using oKM120 and oKM113 with a 1:1 (mol/mol) mixture of both fragments as the template, creating a 1,140-bp product, which was subsequently cloned into pKK30 using NotI/BamHI, resulting in pKM25.
(b) icaB. A 270-bp DNA fragment containing the strong constitutive sarA P1 promoter (101) and translational initiation region (TIR) ribosome-binding site previously shown to enhance protein synthesis (102) was amplified from SA564 genomic DNA using oKM121 and oKM122. In addition, an 873-bp DNA fragment containing the icaB coding sequence was amplified using oKM123 and oKM124. The two fragments were purified, mixed in equal amounts, and used as the template in a fusion PCR using oKM121 and oKM124. The 1,143-bp PCR product was digested with SphI and EcoRI and ligated to the same sites of pCN51 (103), resulting in pKM26.
(ii) Constitutive fluorescent reporter plasmids. A 232-bp DNA fragment containing the sarA P1 promoter region was amplified from SA564 using oligonucleotides oKM074 and oKM075 for superfolder green fluorescent protein (sGFP) labeling and oKM076 and oKM077 for sDsRed labeling. The resulting fragments were cloned into pMRSI upstream of the TIR site using SphI and EcoRI or EcoRI and SalI to drive sGFP (pKM15) or sDsRed (pKM16) expression, respectively. We note that sGFP was below the limit of detection when cells were counterstained with TOTO-1 to reveal eDNA, allowing pKM15 to provide drug resistance in coculture experiments. For rigor, all coculture experiments were repeated with each genotype harboring pKM16.
(iii) Construction of mraY* (encoding MraYL113M). A 2,001-bp DNA fragment containing 1,000 bp of homology flanking each side of the T-to-A transversion at nucleotide 1167134 in the SA564 mraY gene (RT87_05830) was amplified from SRB1243 using oKM102 and oKM103. The purified PCR fragment was digested with EcoRI and SalI and ligated to the same sites of pJB38, resulting in pKM22. Allelic exchange was performed as previously described (102). The presence of the mutant allele was confirmed by nonradioactive Sanger sequencing.
(iv) Overexpression of icaADBC. The SA564 icaADBC genes (RT87_13865 through RT87_13880) were amplified using oKM125 and oKM126. The 3,446-bp PCR product was digested using SphI and BglII and ligated to pKM16 digested with SphI and BamHI, replacing sDsRed. The resulting plasmid, pKM28, places the ica operon under the control of the strong constitutive sarA P1 promoter.
Settling assay.
A previously described settling assay was performed with the following modifications (67). Briefly, cells were cultured in TSB as described above. Where indicated, treatment (e.g., DNase I, proteinase K, or dispersin B) was applied at the indicated concentration after subculturing from overnight cultures and maintained through the duration of the experiment. After the second dilution to an OD600 of 0.05, cells were grown for 3 h (OD600 of ∼2 to 4), at which time a 5-ml sample was obtained and the initial OD600 (IOD) was measured in duplicate using 200 μl from the upper 5 mm of the sample. The sample was allowed to settle without disturbance for 45 min, and then a second OD600 measurement was obtained (FOD). Where indicated, sodium metaperiodate was added to the 5-ml sample. The background (medium only) was subtracted from each sample, and the percentage of the initial OD600 was calculated using the formula (FOD/IOD) × 100. In experiments requiring the addition of exogenous DNA, cells were grown to an OD600 of ∼1 in CDM, after which a 1-ml sample was transferred to a 1.75-ml microcentrifuge tube containing gDNA at the concentrations indicated in Fig. 7. The percent settling was obtained by sampling 20 μl at the air-liquid interface at time zero and at 1 h in duplicate.
Static biofilm assay.
Biofilm formation during static culture was measured as previously described (56), with slight modification. In short, strains were cultured in 4 ml TSB for 7 h and then diluted to an optical density of 0.01. Next, 200 μl of each dilution was transferred in triplicate to an untreated flat-bottom 96-well polystyrene plate (Greiner Bio-One). Plates were then incubated statically for 18 h at 37°C. After incubation, plates were gently inverted to remove culture medium and then washed four times with 200 μl PBS to remove nonadherent cells. After each wash, plates were gently blotted on absorbent paper to remove excess liquid. Plates were then dried at room temperature for 10 min. Biofilms were fixed with 200 μl of 100% ethanol for 20 min, after which each well was stained with 0.41% (wt/vol) crystal violet (in 12% [vol/vol] ethanol) for 8 min. The crystal violet was decanted, and each well was washed four times with 200 μl PBS. Plates were blotted on absorbent paper and dried for 10 min, and then the crystal violet was eluted with 200 μl of 95% ethanol for 10 min. All 200 μl from each well was transferred to a clear flat-bottom microtiter plate (Corning), and the absorbance at 600 nm was measured for each well in a Synergy H1 microplate reader (BioTek). Samples were diluted 2-fold in water when necessary to remain within the linear range of the plate reader.
Confocal microscopy.
A 1-ml sample was obtained from exponentially growing cultures, pelleted by centrifugation for 3 min at 21,000 × g, and resuspended in an equal volume of phosphate-buffered saline (PBS) (pH 7.5). Cells were stained with Syto 9 and propidium iodide for 15 min at final concentrations of 3.5 μM and 20 μM, respectively. When using the sDsRed reporter fusion to label cells (pKM16), cells were counterstained with Syto 40 (68) and TOTO-1 (69) at final concentrations of 5 μM and 2 μM, respectively. Cells were allowed to adhere to a poly-l-lysine-treated coverslip for 15 min, after which time nonadherent cells were removed using a brief PBS wash as previously described (59). Images were obtained using a Zeiss LSM 880 confocal scanning laser microscope and processed using ImageJ (104). For the image analysis shown in Fig. 4B, the fluorescence signal attributed to wild-type cells was subtracted from the total fluorescence in either the aggregate or an area of dispersed cells chosen at random. These normalized fluorescence values for wild-type and ΔcodY mutant cells were then compared.
SEM.
A 12-mm round coverslip was treated with 20 μl of poly-l-lysine and allowed to dry for 30 min. Next, a 1-ml sample was obtained from exponentially growing cells at an OD600 of 0.5 and washed once with PBS (pH 5.0). Twenty microliters of the washed cells was applied to the treated coverslip and incubated at room temperature for 30 min. Nonadherent cells were removed from the coverslip by washing twice with PBS (pH 7.5). Samples were fixed overnight at 4°C in PBS (pH 5.0) containing 2% (vol/vol) glutaraldehyde. Subsequently, a secondary fixing step was performed using 1% (vol/vol) osmium tetroxide for 1 h. Samples were then dehydrated using three incremental ethanol washes (70% for 10 min followed by 95% for 10 min and finally 100% for 10 min) and dried by immersion in hexamethyldisilazane (HMDS) overnight. Samples were then sputter coated with a gold-palladium alloy (60:40) and mounted to a stub. Images were obtained on a Hitachi S-4700 field emission scanning electron microscope equipped with a transmitted electron detector and backscattered electron detector at the Laboratory of Biological Ultrastructure at the University of Maryland, College Park, MD.
RNA extraction and qRT-PCR.
RNA was extracted as previously described (59). Briefly, a 4-ml sample of exponentially growing cells (OD600 of ∼0.5) was quenched by mixing with an equal volume of 1:1 (vol/vol) ethanol-acetone prechilled to −20°C and immediately frozen on dry ice or in liquid nitrogen. To process, thawed samples were washed twice with TE buffer (10 mM Tris-Cl [pH 8], 1 mM EDTA) and mechanically disrupted in TRIzol using a Precellys 24 homogenizer with three 30-s pulses at 6,800 rpm, with incubation on wet ice for 1 min between pulses. Nucleic acids were extracted using the Direct-Zol kit (Zymo Research Corporation) following the manufacturer’s instructions, and genomic DNA was depleted in each sample using the Turbo DNA-free DNase removal kit (Ambion) according to the manufacturer’s instructions. Transcript abundance was determined using standard curves as previously described (56) and normalized to rpoC because the abundance was constant across all samples analyzed.
FRET to quantify nuclease activity.
Secreted nuclease activity was quantified as described previously using fluorescent resonance energy transfer (FRET) (70). Briefly, supernatants from exponentially growing cultures were sterilized using a 0.22-μm spin filter (Corning), diluted in buffer A (10 mM Tris [pH 8.0], 5 mM CaCl2) such that ∼10% of substrate is cleaved during the assay, mixed with 1 μM FRET substrate, and incubated at 30°C. Fluorescence (535-nm and 590-nm excitation and emission filters, respectively) was monitored using a computer-controlled Tecan Infinite F200 Pro instrument. Nuclease activity was determined with a standard curve generated using purified micrococcal nuclease enzyme (Worthington Biochemicals).
Quantification of eDNA released by pH shift.
A 2-ml sample was obtained from cells cultured in TSB as described above at an OD600 of ∼0.5, washed once with PBS (pH 5.0 or 7.5), and then resuspended in 1 ml of fresh PBS at either pH 5 or 7.5 and incubated for 15 min at room temperature. CFU were measured by dilution plating on tryptic soy agar (TSA) to ensure that cells were not dying during treatment. Next, the cells were pelleted by centrifugation at 18,500 × g for 3 min, and 500 μl of each supernatant was subjected to ethanol precipitation to obtain released eDNA. As done similarly elsewhere (49), quantitative PCR (qPCR) was used to determine the copy number of rpoC for each sample relative to a standard curve produced using SA564 genomic DNA and the primer set oSRB239 and oSRB240. The remaining sample containing cells was immediately resuspended, stained with Syto 40 and TOTO-1, and analyzed by CSLM as described above.
Nucleic acid measurements in TSB and CDM.
The concentration of nucleic acid was measured in inoculated and uninoculated TSB and CDM after overnight incubation. Strains were grown in 4 ml of TSB or CDM overnight. All 4 ml of each sample was sonicated at 10% amplitude, alternating 10 s on and 5 s off, for 3 min to release nucleic acids and cells from the aggregates. CFU were measured for each sample before and after sonication to ensure that there was no cell death. After sonication, cells were pelleted by centrifugation for 10 min at 21,000 × g, and 500 μl of supernatant was collected from each sample. Nucleic acids were then extracted by ethanol precipitation. The concentration of nucleic acid in each sample was measured by absorbance at 260 nm in a NanoDrop 2000 spectrophotometer (Thermo Scientific).
Illumina MiSeq genome resequencing.
Genomic DNA was purified from overnight cultures of SA564 wild-type (SRB1211), isogenic ΔcodY mutant (SRB1218), and spontaneous suppressor mutant (SRB1243) cells using the Wizard genomic DNA purification kit (Promega) following the manufacturer’s instructions. Whole-genome DNA libraries were constructed using the Nextera DNA XT Library Prep kit (Illumina) following the manufacturer’s instructions and sequenced at the Tufts University Genomics Core Facility with MiSeq V2 chemistry in paired-end 150-base format. The resulting reads were mapped to a previously published genome of SA564 (GenBank accession number CP010890.01), and single nucleotide polymorphisms (SNPs) were identified using the BreSeq pipeline (105).
Quantification of PIA by immunoblot analysis.
PIA accumulation was determined as previously described (106). Briefly, TSB medium containing 0.25% glucose was inoculated with equal numbers of bacteria from overnight cultures. The cultures were grown for 3 h at 37°C with a flask-to-medium ratio of 10:1 and aerated at 250 rpm. Equal numbers of bacteria were harvested by centrifugation (2.0 OD600 units), and the PIA was extracted in 0.5 M EDTA by boiling for 10 min and freezing overnight. Samples were incubated with proteinase K for 1 h at 37°C, followed by boiling for 5 min to inactivate proteinase K. Aliquots of PIA were applied to a neutral nylon membrane (GVS North America) and blocked with 5% skim milk for 6 h. The nylon membrane was incubated overnight with PIA-specific antibodies, followed by a 4-h incubation with an anti-rabbit immunoglobulin G–peroxidase conjugate. The presence of PIA was detected using SuperSignal West Pico chemiluminescent substrate (Pierce). The integrated density values of bands on autoradiographs were determined with the TotalLab software (Nonlinear Dynamics Ltd.).
Statistical analysis.
Data shown are the results of at least three independent experiments. Statistical significance was determined using PRISM 7 (GraphPad Software) with the indicated tests. The normality of each data set was assessed with a Shapiro-Wilk test.
Supplementary Material
ACKNOWLEDGMENTS
We thank Friedrich Götz (University of Tübingen) for the gift of the Δlgt::erm mutant, James O’Gara (National University of Ireland, Galway, Ireland) for the gift of PIA-specific antibodies, Amanda Oglesby-Sherrouse (University of Maryland School of Medicine) for the gift of Pseudomonas aeruginosa PAO1, Tim Maugel (University of Maryland, College Park) for help and expertise with electron microscopy, Albert Tai (Tufts University School of Medicine) for help with the analysis of whole-genome sequencing data, Jeffrey Kaplan (American University) for the generous gift of dispersin B, and Mark Rose for helpful discussions. We also thank Sarah Reuter and Steven Min for technical assistance.
This work was supported by National Institutes of Health grants P01-AI83211 and R01-AI125589 to K.W.B. and R21 AI123708 and R01 AI137403 to S.R.B.
Footnotes
Supplemental material is available online only.
Luke J. Curran met authorship criteria but was unreachable for final approval of the byline and article.
REFERENCES
- 1.Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. 2016. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol 14:563–575. doi: 10.1038/nrmicro.2016.94. [DOI] [PubMed] [Google Scholar]
- 2.Otto M. 2006. Bacterial evasion of antimicrobial peptides by biofilm formation. Curr Top Microbiol Immunol 306:251–258. doi: 10.1007/3-540-29916-5_10. [DOI] [PubMed] [Google Scholar]
- 3.Mah TF, O'Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 4.Moormeier DE, Bayles KW. 2017. Staphylococcus aureus biofilm: a complex developmental organism. Mol Microbiol 104:365–376. doi: 10.1111/mmi.13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moormeier DE, Bose JL, Horswill AR, Bayles KW. 2014. Temporal and stochastic control of Staphylococcus aureus biofilm development. mBio 5:e01341-14. doi: 10.1128/mBio.01341-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lewis K. 2001. Riddle of biofilm resistance. Antimicrob Agents Chemother 45:999–1007. doi: 10.1128/AAC.45.4.999-1007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsek MR, Singh PK. 2003. Bacterial biofilms: an emerging link to disease pathogenesis. Annu Rev Microbiol 57:677–701. doi: 10.1146/annurev.micro.57.030502.090720. [DOI] [PubMed] [Google Scholar]
- 8.Waters EM, Rowe SE, O'Gara JP, Conlon BP. 2016. Convergence of Staphylococcus aureus persister and biofilm research: can biofilms be defined as communities of adherent persister cells? PLoS Pathog 12:e1006012. doi: 10.1371/journal.ppat.1006012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core Surveillance MRSA Investigators. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 10.Wertheim HF, Vos MC, Ott A, van Belkum A, Voss A, Kluytmans JA, van Keulen PH, Vandenbroucke-Grauls CM, Meester MH, Verbrugh HA. 2004. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet 364:703–705. doi: 10.1016/S0140-6736(04)16897-9. [DOI] [PubMed] [Google Scholar]
- 11.Kourtis AP, Hatfield K, Baggs J, Mu Y, See I, Epson E, Nadle J, Kainer MA, Dumyati G, Petit S, Ray SM, Emerging Infections Program MRSA Author Group, Ham D, Capers C, Ewing H, Coffin N, McDonald LC, Jernigan J, Cardo D. 2019. Vital signs: epidemiology and recent trends in methicillin-resistant and in methicillin-susceptible Staphylococcus aureus bloodstream infections—United States. MMWR Morb Mortal Wkly Rep 68:214–219. doi: 10.15585/mmwr.mm6809e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowy FD. 1998. Staphylococcus aureus infections. N Engl J Med 339:520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 13.Prestinaci F, Pezzotti P, Pantosti A. 2015. Antimicrobial resistance: a global multifaceted phenomenon. Pathog Glob Health 109:309–318. doi: 10.1179/2047773215Y.0000000030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chambers HF, Deleo FR. 2009. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7:629–641. doi: 10.1038/nrmicro2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Percival SL, Suleman L, Vuotto C, Donelli G. 2015. Healthcare-associated infections, medical devices and biofilms: risk, tolerance and control. J Med Microbiol 64:323–334. doi: 10.1099/jmm.0.000032. [DOI] [PubMed] [Google Scholar]
- 16.Patti JM, Allen BL, McGavin MJ, Hook M. 1994. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu Rev Microbiol 48:585–617. doi: 10.1146/annurev.mi.48.100194.003101. [DOI] [PubMed] [Google Scholar]
- 17.Foster TJ, Geoghegan JA, Ganesh VK, Hook M. 2014. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 12:49–62. doi: 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Josse J, Laurent F, Diot A. 2017. Staphylococcal adhesion and host cell invasion: fibronectin-binding and other mechanisms. Front Microbiol 8:2433. doi: 10.3389/fmicb.2017.02433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Otto M. 2013. How colonization factors are linked to outbreaks of methicillin-resistant Staphylococcus aureus: the roles of SasX and ACME. Biomol Concepts 4:533–537. doi: 10.1515/bmc-2013-0025. [DOI] [PubMed] [Google Scholar]
- 20.Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M, Marrie TJ. 1987. Bacterial biofilms in nature and disease. Annu Rev Microbiol 41:435–464. doi: 10.1146/annurev.mi.41.100187.002251. [DOI] [PubMed] [Google Scholar]
- 21.Flemming HC, Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 22.O'Neill E, Pozzi C, Houston P, Humphreys H, Robinson DA, Loughman A, Foster TJ, O'Gara JP. 2008. A novel Staphylococcus aureus biofilm phenotype mediated by the fibronectin-binding proteins, FnBPA and FnBPB. J Bacteriol 190:3835–3850. doi: 10.1128/JB.00167-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Speziale P, Pietrocola G, Foster TJ, Geoghegan JA. 2014. Protein-based biofilm matrices in staphylococci. Front Cell Infect Microbiol 4:171. doi: 10.3389/fcimb.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O’Gara JP. 2007. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett 270:179–188. doi: 10.1111/j.1574-6968.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 25.McCarthy H, Rudkin JK, Black NS, Gallagher L, O'Neill E, O'Gara JP. 2015. Methicillin resistance and the biofilm phenotype in Staphylococcus aureus. Front Cell Infect Microbiol 5:1. doi: 10.3389/fcimb.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto M. 2013. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med 64:175–188. doi: 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- 27.Maira-Litran T, Kropec A, Abeygunawardana C, Joyce J, Mark G III, Goldmann DA, Pier GB. 2002. Immunochemical properties of the staphylococcal poly-N-acetylglucosamine surface polysaccharide. Infect Immun 70:4433–4440. doi: 10.1128/iai.70.8.4433-4440.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mack D, Fischer W, Krokotsch A, Leopold K, Hartmann R, Egge H, Laufs R. 1996. The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear beta-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol 178:175–183. doi: 10.1128/jb.178.1.175-183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heilmann C, Schweitzer O, Gerke C, Vanittanakom N, Mack D, Gotz F. 1996. Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol Microbiol 20:1083–1091. doi: 10.1111/j.1365-2958.1996.tb02548.x. [DOI] [PubMed] [Google Scholar]
- 30.Gerke C, Kraft A, Sussmuth R, Schweitzer O, Gotz F. 1998. Characterization of the N-acetylglucosaminyltransferase activity involved in the biosynthesis of the Staphylococcus epidermidis polysaccharide intercellular adhesin. J Biol Chem 273:18586–18593. doi: 10.1074/jbc.273.29.18586. [DOI] [PubMed] [Google Scholar]
- 31.Cramton SE, Gerke C, Schnell NF, Nichols WW, Gotz F. 1999. The intercellular adhesion (ica) locus is present in Staphylococcus aureus and is required for biofilm formation. Infect Immun 67:5427–5433. doi: 10.1128/IAI.67.10.5427-5433.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rachid S, Ohlsen K, Wallner U, Hacker J, Hecker M, Ziebuhr W. 2000. Alternative transcription factor sigma(B) is involved in regulation of biofilm expression in a Staphylococcus aureus mucosal isolate. J Bacteriol 182:6824–6826. doi: 10.1128/jb.182.23.6824-6826.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seidl K, Goerke C, Wolz C, Mack D, Berger-Bächi B, Bischoff M. 2008. Staphylococcus aureus CcpA affects biofilm formation. Infect Immun 76:2044–2050. doi: 10.1128/IAI.00035-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulrich M, Bastian M, Cramton SE, Ziegler K, Pragman AA, Bragonzi A, Memmi G, Wolz C, Schlievert PM, Cheung A, Doring G. 2007. The staphylococcal respiratory response regulator SrrAB induces ica gene transcription and polysaccharide intercellular adhesin expression, protecting Staphylococcus aureus from neutrophil killing under anaerobic growth conditions. Mol Microbiol 65:1276–1287. doi: 10.1111/j.1365-2958.2007.05863.x. [DOI] [PubMed] [Google Scholar]
- 35.Valle J, Toledo-Arana A, Berasain C, Ghigo J-M, Amorena B, Penadés JR, Lasa I. 2003. SarA and not sigmaB is essential for biofilm development by Staphylococcus aureus. Mol Microbiol 48:1075–1087. doi: 10.1046/j.1365-2958.2003.03493.x. [DOI] [PubMed] [Google Scholar]
- 36.Conlon KM, Humphreys H, O'Gara JP. 2002. Regulation of icaR gene expression in Staphylococcus epidermidis. FEMS Microbiol Lett 216:171–177. doi: 10.1111/j.1574-6968.2002.tb11432.x. [DOI] [PubMed] [Google Scholar]
- 37.Jefferson KK, Pier DB, Goldmann DA, Pier GB. 2004. The teicoplanin-associated locus regulator (TcaR) and the intercellular adhesin locus regulator (IcaR) are transcriptional inhibitors of the ica locus in Staphylococcus aureus. J Bacteriol 186:2449–2456. doi: 10.1128/jb.186.8.2449-2456.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Majerczyk CD, Sadykov MR, Luong TT, Lee C, Somerville GA, Sonenshein AL. 2008. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol 190:2257–2265. doi: 10.1128/JB.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pamp SJ, Frees D, Engelmann S, Hecker M, Ingmer H. 2006. Spx is a global effector impacting stress tolerance and biofilm formation in Staphylococcus aureus. J Bacteriol 188:4861–4870. doi: 10.1128/JB.00194-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cue D, Lei MG, Lee CY. 2012. Genetic regulation of the intercellular adhesion locus in staphylococci. Front Cell Infect Microbiol 2:38. doi: 10.3389/fcimb.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joyce JG, Abeygunawardana C, Xu Q, Cook JC, Hepler R, Przysiecki CT, Grimm KM, Roper K, Ip CC, Cope L, Montgomery D, Chang M, Campie S, Brown M, McNeely TB, Zorman J, Maira-Litran T, Pier GB, Keller PM, Jansen KU, Mark GE. 2003. Isolation, structural characterization, and immunological evaluation of a high-molecular-weight exopolysaccharide from Staphylococcus aureus. Carbohydr Res 338:903–922. doi: 10.1016/s0008-6215(03)00045-4. [DOI] [PubMed] [Google Scholar]
- 42.Vuong C, Kocianova S, Voyich JM, Yao Y, Fischer ER, DeLeo FR, Otto M. 2004. A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem 279:54881–54886. doi: 10.1074/jbc.M411374200. [DOI] [PubMed] [Google Scholar]
- 43.Neuhaus FC, Baddiley J. 2003. A continuum of anionic charge: structures and functions of d-alanyl-teichoic acids in Gram-positive bacteria. Microbiol Mol Biol Rev 67:686–723. doi: 10.1128/mmbr.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naumova IB, Shashkov AS, Tul'skaya EM, Streshinskaya GM, Kozlova YI, Potekhina NV, Evtushenko LI, Stackebrandt E. 2001. Cell wall teichoic acids: structural diversity, species specificity in the genus Nocardiopsis, and chemotaxonomic perspective. FEMS Microbiol Rev 25:269–284. doi: 10.1111/j.1574-6976.2001.tb00578.x. [DOI] [PubMed] [Google Scholar]
- 45.Vergara-Irigaray M, Maira-Litrán T, Merino N, Pier GB, Penadés JR, Lasa I. 2008. Wall teichoic acids are dispensable for anchoring the PNAG exopolysaccharide to the Staphylococcus aureus cell surface. Microbiology 154:865–877. doi: 10.1099/mic.0.2007/013292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ibáñez de Aldecoa AL, Zafra O, González-Pastor JE. 2017. Mechanisms and regulation of extracellular DNA release and its biological roles in microbial communities. Front Microbiol 8:1390. doi: 10.3389/fmicb.2017.01390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bose JL, Lehman MK, Fey PD, Bayles KW. 2012. Contribution of the Staphylococcus aureus Atl AM and GL murein hydrolase activities in cell division, autolysis, and biofilm formation. PLoS One 7:e42244. doi: 10.1371/journal.pone.0042244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moormeier DE, Endres JL, Mann EE, Sadykov MR, Horswill AR, Rice KC, Fey PD, Bayles KW. 2013. Use of microfluidic technology to analyze gene expression during Staphylococcus aureus biofilm formation reveals distinct physiological niches. Appl Environ Microbiol 79:3413–3424. doi: 10.1128/AEM.00395-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dengler V, Foulston L, DeFrancesco AS, Losick R. 2015. An electrostatic net model for the role of extracellular DNA in biofilm formation by Staphylococcus aureus. J Bacteriol 197:3779–3787. doi: 10.1128/JB.00726-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeFrancesco AS, Masloboeva N, Syed AK, DeLoughery A, Bradshaw N, Li GW, Gilmore MS, Walker S, Losick R. 2017. Genome-wide screen for genes involved in eDNA release during biofilm formation by Staphylococcus aureus. Proc Natl Acad Sci U S A 114:E5969–E5978. doi: 10.1073/pnas.1704544114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stenz L, Francois P, Whiteson K, Wolz C, Linder P, Schrenzel J. 2011. The CodY pleiotropic repressor controls virulence in Gram-positive pathogens. FEMS Immunol Med Microbiol 62:123–139. doi: 10.1111/j.1574-695X.2011.00812.x. [DOI] [PubMed] [Google Scholar]
- 52.Brinsmade SR. 2017. CodY, a master integrator of metabolism and virulence in Gram-positive bacteria. Curr Genet 63:417–425. doi: 10.1007/s00294-016-0656-5. [DOI] [PubMed] [Google Scholar]
- 53.den Hengst CD, van Hijum SA, Geurts JM, Nauta A, Kok J, Kuipers OP. 2005. The Lactococcus lactis CodY regulon: identification of a conserved cis-regulatory element. J Biol Chem 280:34332–34342. doi: 10.1074/jbc.M502349200. [DOI] [PubMed] [Google Scholar]
- 54.Belitsky BR, Sonenshein AL. 2008. Genetic and biochemical analysis of CodY-binding sites in Bacillus subtilis. J Bacteriol 190:1224–1236. doi: 10.1128/JB.01780-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brinsmade SR, Alexander EL, Livny J, Stettner AI, Segre D, Rhee KY, Sonenshein AL. 2014. Hierarchical expression of genes controlled by the Bacillus subtilis global regulatory protein CodY. Proc Natl Acad Sci U S A 111:8227–8232. doi: 10.1073/pnas.1321308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waters NR, Samuels DJ, Behera RK, Livny J, Rhee KY, Sadykov MR, Brinsmade SR. 2016. A spectrum of CodY activities drives metabolic reorganization and virulence gene expression in Staphylococcus aureus. Mol Microbiol 101:495–514. doi: 10.1111/mmi.13404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Majerczyk CD, Dunman PM, Luong TT, Lee CY, Sadykov MR, Somerville GA, Bodi K, Sonenshein AL. 2010. Direct targets of CodY in Staphylococcus aureus. J Bacteriol 192:2861–2877. doi: 10.1128/JB.00220-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pohl K, Francois P, Stenz L, Schlink F, Geiger T, Herbert S, Goerke C, Schrenzel J, Wolz C. 2009. CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J Bacteriol 191:2953–2963. doi: 10.1128/JB.01492-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mlynek KD, Sause WE, Moormeier DE, Sadykov MR, Hill KR, Torres VJ, Bayles KW, Brinsmade SR. 2018. Nutritional regulation of the Sae two-component system by CodY in Staphylococcus aureus. J Bacteriol 200:e00012-18. doi: 10.1128/JB.00012-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McCourt J, O'Halloran DP, McCarthy H, O'Gara JP, Geoghegan JA. 2014. Fibronectin-binding proteins are required for biofilm formation by community-associated methicillin-resistant Staphylococcus aureus strain LAC. FEMS Microbiol Lett 353:157–164. doi: 10.1111/1574-6968.12424. [DOI] [PubMed] [Google Scholar]
- 61.Rivera FE, Miller HK, Kolar SL, Stevens SM Jr, Shaw LN. 2012. The impact of CodY on virulence determinant production in community-associated methicillin-resistant Staphylococcus aureus. Proteomics 12:263–268. doi: 10.1002/pmic.201100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. doi: 10.1128/IAI.63.9.3373-3380.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atwood DN, Loughran AJ, Courtney AP, Anthony AC, Meeker DG, Spencer HJ, Gupta RK, Lee CY, Beenken KE, Smeltzer MS. 2015. Comparative impact of diverse regulatory loci on Staphylococcus aureus biofilm formation. Microbiologyopen 4:436–451. doi: 10.1002/mbo3.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Musser JM, Schlievert PM, Chow AW, Ewan P, Kreiswirth BN, Rosdahl VT, Naidu AS, Witte W, Selander RK. 1990. A single clone of Staphylococcus aureus causes the majority of cases of toxic shock syndrome. Proc Natl Acad Sci U S A 87:225–229. doi: 10.1073/pnas.87.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tzagoloff H, Novick R. 1977. Geometry of cell division in Staphylococcus aureus. J Bacteriol 129:343–350. doi: 10.1128/JB.129.1.343-350.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giesbrecht P, Kersten T, Maidhof H, Wecke J. 1998. Staphylococcal cell wall: morphogenesis and fatal variations in the presence of penicillin. Microbiol Mol Biol Rev 62:1371–1414. doi: 10.1128/MMBR.62.4.1371-1414.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crosby HA, Schlievert PM, Merriman JA, King JM, Salgado-Pabón W, Horswill AR. 2016. The Staphylococcus aureus global regulator MgrA modulates clumping and virulence by controlling surface protein expression. PLoS Pathog 12:e1005604. doi: 10.1371/journal.ppat.1005604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frey T. 1995. Nucleic acid dyes for detection of apoptosis in live cells. Cytometry 21:265–274. doi: 10.1002/cyto.990210307. [DOI] [PubMed] [Google Scholar]
- 69.Okshevsky M, Meyer RL. 2014. Evaluation of fluorescent stains for visualizing extracellular DNA in biofilms. J Microbiol Methods 105:102–104. doi: 10.1016/j.mimet.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 70.Kiedrowski MR, Kavanaugh JS, Malone CL, Mootz JM, Voyich JM, Smeltzer MS, Bayles KW, Horswill AR. 2011. Nuclease modulates biofilm formation in community-associated methicillin-resistant Staphylococcus aureus. PLoS One 6:e26714. doi: 10.1371/journal.pone.0026714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Graf AC, Leonard A, Schäuble M, Rieckmann LM, Hoyer J, Maass S, Lalk M, Becher D, Pané-Farré J, Riedel K. 2019. Virulence factors produced by Staphylococcus aureus biofilms have a moonlighting function contributing to biofilm integrity. Mol Cell Proteomics 18:1036–1053. doi: 10.1074/mcp.RA118.001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kavanaugh JS, Flack CE, Lister J, Ricker EB, Ibberson CB, Jenul C, Moormeier DE, Delmain EA, Bayles KW, Horswill AR. 2019. Identification of extracellular DNA-binding proteins in the biofilm matrix. mBio 10:e01137-19. doi: 10.1128/mBio.01137-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomas VC, Sadykov MR, Chaudhari SS, Jones J, Endres JL, Widhelm TJ, Ahn JS, Jawa RS, Zimmerman MC, Bayles KW. 2014. A central role for carbon-overflow pathways in the modulation of bacterial cell death. PLoS Pathog 10:e1004205. doi: 10.1371/journal.ppat.1004205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Foulston L, Elsholz AK, DeFrancesco AS, Losick R. 2014. The extracellular matrix of Staphylococcus aureus biofilms comprises cytoplasmic proteins that associate with the cell surface in response to decreasing pH. mBio 5:e01667-14. doi: 10.1128/mBio.01667-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lang GI, Rice DP, Hickman MJ, Sodergren E, Weinstock GM, Botstein D, Desai MM. 2013. Pervasive genetic hitchhiking and clonal interference in forty evolving yeast populations. Nature 500:571–574. doi: 10.1038/nature12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sadykov MR, Mattes TA, Luong TT, Zhu Y, Day SR, Sifri CD, Lee CY, Somerville GA. 2010. Tricarboxylic acid cycle-dependent synthesis of Staphylococcus aureus type 5 and 8 capsular polysaccharides. J Bacteriol 192:1459–1462. doi: 10.1128/JB.01377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.King AN, Borkar SA, Samuels DJ, Batz Z, Bulock LL, Sadykov MR, Bayles KW, Brinsmade SR. 2018. Guanine limitation results in CodY-dependent and -independent alteration of Staphylococcus aureus physiology and gene expression. J Bacteriol 200:00136-18. doi: 10.1128/JB.00136-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hayashi S, Wu HC. 1990. Lipoproteins in bacteria. J Bioenerg Biomembr 22:451–471. doi: 10.1007/bf00763177. [DOI] [PubMed] [Google Scholar]
- 79.Stoll H, Dengjel J, Nerz C, Götz F. 2005. Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73:2411–2423. doi: 10.1128/IAI.73.4.2411-2423.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hendriksen WT, Bootsma HJ, Estevao S, Hoogenboezem T, de Jong A, de Groot R, Kuipers OP, Hermans PW. 2008. CodY of Streptococcus pneumoniae: link between nutritional gene regulation and colonization. J Bacteriol 190:590–601. doi: 10.1128/JB.00917-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dineen SS, McBride SM, Sonenshein AL. 2010. Integration of metabolism and virulence by Clostridium difficile CodY. J Bacteriol 192:5350–5362. doi: 10.1128/JB.00341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Richardson AR, Somerville GA, Sonenshein AL. 2015. Regulating the intersection of metabolism and pathogenesis in Gram-positive bacteria. Microbiol Spectr 3. doi: 10.1128/microbiolspec.MBP-0004-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rom JS, Atwood DN, Beenken KE, Meeker DG, Loughran AJ, Spencer HJ, Lantz TL, Smeltzer MS. 2017. Impact of Staphylococcus aureus regulatory mutations that modulate biofilm formation in the USA300 strain LAC on virulence in a murine bacteremia model. Virulence 8:1776–1790. doi: 10.1080/21505594.2017.1373926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Montgomery CP, Boyle-Vavra S, Roux A, Ebine K, Sonenshein AL, Daum RS. 2012. CodY deletion enhances in vivo virulence of community-associated methicillin-resistant Staphylococcus aureus clone USA300. Infect Immun 80:2382–2389. doi: 10.1128/IAI.06172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abdelhady W, Bayer AS, Seidl K, Moormeier DE, Bayles KW, Cheung A, Yeaman MR, Xiong YQ. 2014. Impact of vancomycin on SarA-mediated biofilm formation: role in persistent endovascular infections due to methicillin-resistant Staphylococcus aureus. J Infect Dis 209:1231–1240. doi: 10.1093/infdis/jiu007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yadav MK, Chae SW, Go YY, Im GJ, Song JJ. 2017. In vitro multi-species biofilms of methicillin-resistant Staphylococcus aureus and Pseudomonas aeruginosa and their host interaction during in vivo colonization of an otitis media rat model. Front Cell Infect Microbiol 7:125. doi: 10.3389/fcimb.2017.00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee YJ, Jang HJ, Chung IY, Cho YH. 2018. Drosophila melanogaster as a polymicrobial infection model for Pseudomonas aeruginosa and Staphylococcus aureus. J Microbiol 56:534–541. doi: 10.1007/s12275-018-8331-9. [DOI] [PubMed] [Google Scholar]
- 88.Dunyach-Remy C, Ngba Essebe C, Sotto A, Lavigne JP. 2016. Staphylococcus aureus toxins and diabetic foot ulcers: role in pathogenesis and interest in diagnosis. Toxins (Basel) 8:209. doi: 10.3390/toxins8070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Geiger T, Francois P, Liebeke M, Fraunholz M, Goerke C, Krismer B, Schrenzel J, Lalk M, Wolz C. 2012. The stringent response of Staphylococcus aureus and its impact on survival after phagocytosis through the induction of intracellular PSMs expression. PLoS Pathog 8:e1003016. doi: 10.1371/journal.ppat.1003016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Geiger T, Wolz C. 2014. Intersection of the stringent response and the CodY regulon in low GC Gram-positive bacteria. Int J Med Microbiol 304:150–155. doi: 10.1016/j.ijmm.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 91.Rothfork JM, Dessus-Babus S, Van Wamel WJ, Cheung AL, Gresham HD. 2003. Fibrinogen depletion attenuates Staphyloccocus aureus infection by preventing density-dependent virulence gene up-regulation. J Immunol 171:5389–5395. doi: 10.4049/jimmunol.171.10.5389. [DOI] [PubMed] [Google Scholar]
- 92.Roux A, Todd DA, Velazquez JV, Cech NB, Sonenshein AL. 2014. CodY-mediated regulation of the Staphylococcus aureus Agr system integrates nutritional and population density signals. J Bacteriol 196:1184–1196. doi: 10.1128/JB.00128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fisher SH. 1999. Regulation of nitrogen metabolism in Bacillus subtilis: vive la différence! Mol Microbiol 32:223–232. doi: 10.1046/j.1365-2958.1999.01333.x. [DOI] [PubMed] [Google Scholar]
- 94.Claverys JP, Martin B, Polard P. 2009. The genetic transformation machinery: composition, localization, and mechanism. FEMS Microbiol Rev 33:643–656. doi: 10.1111/j.1574-6976.2009.00164.x. [DOI] [PubMed] [Google Scholar]
- 95.Desai K, Mashburn-Warren L, Federle MJ, Morrison DA. 2012. Development of competence for genetic transformation of Streptococcus mutans in a chemically defined medium. J Bacteriol 194:3774–3780. doi: 10.1128/JB.00337-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang S, Liu X, Liu H, Zhang L, Guo Y, Yu S, Wozniak DJ, Ma LZ. 2015. The exopolysaccharide Psl-eDNA interaction enables the formation of a biofilm skeleton in Pseudomonas aeruginosa. Environ Microbiol Rep 7:330–340. doi: 10.1111/1758-2229.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lennox ES. 1955. Transduction of linked genetic characters of the host by bacteriophage P1. Virology 1:190–206. doi: 10.1016/0042-6822(55)90016-7. [DOI] [PubMed] [Google Scholar]
- 98.Kaiser JC, Omer S, Sheldon JR, Welch I, Heinrichs DE. 2015. Role of BrnQ1 and BrnQ2 in branched-chain amino acid transport and virulence in Staphylococcus aureus. Infect Immun 83:1019–1029. doi: 10.1128/IAI.02542-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Novick RP. 1991. Genetic systems in staphylococci. Methods Enzymol 204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- 100.Vallejo AN, Pogulis RJ, Pease LR. 2008. PCR mutagenesis by overlap extension and gene SOE. CSH Protoc 2008:pdb.prot4861. doi: 10.1101/pdb.prot4861. [DOI] [PubMed] [Google Scholar]
- 101.Cheung AL, Nast CC, Bayer AS. 1998. Selective activation of sar promoters with the use of green fluorescent protein transcriptional fusions as the detection system in the rabbit endocarditis model. Infect Immun 66:5988–5993. doi: 10.1128/IAI.66.12.5988-5993.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bose JL. 2014. Genetic manipulation of staphylococci. Methods Mol Biol 1106:101–111. doi: 10.1007/978-1-62703-736-5_8. [DOI] [PubMed] [Google Scholar]
- 103.Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. 2004. Novel cassette-based shuttle vector system for Gram-positive bacteria. Appl Environ Microbiol 70:6076–6085. doi: 10.1128/AEM.70.10.6076-6085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol 1151:165–188. doi: 10.1007/978-1-4939-0554-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sadykov MR, Olson ME, Halouska S, Zhu Y, Fey PD, Powers R, Somerville GA. 2008. Tricarboxylic acid cycle-dependent regulation of Staphylococcus epidermidis polysaccharide intercellular adhesin synthesis. J Bacteriol 190:7621–7632. doi: 10.1128/JB.00806-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.