

Summary
Type III secretion (T3S) plays a pivotal role in the colonization of ruminant hosts by Enterohemorrhagic Escherichia coli (EHEC). The T3S system translocates effector proteins into host cells to promote bacterial attachment and persistence. The repertoire and variation in prophage regions underpins differences in the pathogenesis and epidemiology of EHEC strains. In this study, we have used a collection of deletions in cryptic prophages and EHEC O157 O-islands to screen for novel regulators of T3S. Using this approach we have identified a family of homologous AraC-like regulators that indirectly repress T3S. These prophage-encoded secretion regulator genes (psr) are found exclusively on prophages and are associated with effector loci and the T3S activating Pch family of regulators. Transcriptional profiling, mutagenesis and DNA binding studies were used to show that these regulators usurp the conserved GAD acid stress resistance system to regulate T3S by increasing the expression of GadE (YhiE) and YhiF and that this regulation follows attachment to bovine epithelial cells. We further demonstrate that PsrA and effectors encoded within cryptic prophage CP933-N are required for persistence in a ruminant model of colonization.
Introduction
Pathogenesis in bacteria requires controlled expression of virulence factors that are often acquired by horizontal gene transfer and brings with it the issue of appropriate regulation in the context of the new host genetic background (Dorman, 2007). Acquired factors may come from a related organism and so much of the regulation may already be appropriate, for example integration into conserved environmental sensing networks (Fass & Groisman, 2009, Laaberki et al., 2006). More issues are likely to arise because of competition between horizontally-acquired virulence factors, especially those that have to share their localization on the surface of the organism or an export or delivery system. Attrition and addition to the virulence gene regulatory repertoire is already understood to play a significant role in niche adaptation (Mandel et al., 2009) and evolution of virulence (Dorman, 2007, Gomez-Duarte & Kaper, 1995, Perez & Groisman, 2009, Zhang et al., 2004), and appropriate co-ordination between multiple horizontally-acquired virulence factors is also likely to be important for host colonization and be under selective pressure.
Enterohemorrhagic E. coli (EHEC) are a group of pathogens associated with serious gastrointestinal disease in humans that can be life threatening as a result of the systemic effects of released Shiga toxins. The predominant serotype linked with human infections is O157:H7 and strains persist and evolve in ruminant hosts with humans as an incidental host. Several EHEC genomes have now been sequenced and it is proposed that the EHEC pathotype has arisen multiple times by independent acquisition of virulence factors on mobile genetic elements (Ogura et al., 2009). These horizontally-acquired factors add over 1 Mb of extra genetic information to the ‘core’ genome present in non-pathogenic E. coli K-12 and encode colonization factors including a type III secretion (T3S) system, associated secreted effector proteins, fimbriae and Shiga toxins (Hayashi et al., 2001, Ogura et al., 2009, Perna et al., 2001). Integrated prophages account for a large proportion of the extra genetic information present in EHEC strains and variation in this prophage repertoire also accounts for much of the genetic diversity between strains (Ohnishi et al., 2002, Zhang et al., 2007).
In EHEC O157, the T3S system is encoded by the Locus of Enterocyte Effacement (LEE) and injects a cocktail of effector proteins into host cells to promote attachment and persistence in ruminants. Expression of the T3S system is essential for bovine colonization (Naylor et al., 2005) and T3S proteins are effective in vaccines to limit EHEC O157 shedding from cattle (McNeilly et al. 2010, Potter et al., 2004). At least seven secreted effector proteins are encoded on the LEE, including the translocated intimin receptor (Tir) that is critical to the formation of attaching and effacing lesions on epithelial cells. The T3S effector protein repertoire is further enhanced by prophage elements distributed throughout the chromosome that encode T3S effectors as “morons” (more DNA) integrated into sites within the prophage that will tolerate insertion (Deng et al., 2004, Hendrix et al., 2000, Tobe et al., 2006). Regulatory mechanisms that link the expression of prophage-encoded effectors with the T3S system are now becoming apparent. For example, transcription of an effector can be activated by the LEE-encoded regulator Ler, as is the case for the non-LEE encoded effector nleA (Abe et al., 2008). Alternatively, effector encoding prophages may import their own regulators that promote expression of the T3S system and non-LEE encoded effector proteins, as is the case with the PerC-like regulators Pch (Abe et al., 2008). Recent work on the pleiotrophic post-transcriptional regulator Hfq has also shown that the LEE and non-LEE encoded effectors are both regulated at the post-transcriptional level suggesting further potential for co-ordinate regulation (Shakhnovich et al., 2009). Regulation of the LEE responds to a wide range of environmental and genetic factors and has recently been reviewed (Mellies et al., 2007, Tree et al., 2009b).
EHEC O157 strains vary markedly in their level of T3S and this likely reflects the repertoire of T3S regulators carried by cryptic prophages and other horizontally-acquired elements or O-islands (regions of E. coli O157 genome absent from the ‘core’ E. coli K-12 genome). Here we have screened for O-islands that affect the expression of T3S to identify loci that have evolved regulatory inputs to control this essential colonization factor. This approach has identified an integrated prophage that is important for persistence in the ruminant host and has led to the identification of novel regulators that help down regulate specific T3S operons. Why these negative regulators are associated with prophages encoding T3S effector proteins and Pch regulators is discussed.
Results
Phenotypic screening of O-island deletions
Sequencing and whole genome PCR screening has shown that most of the variation in the EHEC O157 pan-genome can be attributed to the presence or absence of horizontally-acquired elements termed O-islands (OIs) in strain EDL933 or S-loops (Sp) in strain Sakai that include many active and cryptic prophages (Ohnishi et al., 2002, Zhang et al., 2007). The T3S system is an essential colonization factor in cattle and secretion levels, as a result of regulatory variation, differ considerably between EHEC O157 strains. In order to determine if some of the larger OI regions regulate T3S, a published collection of 17 OI deletions in the stx- EDL933 derivative, TUV93-0 were screened for T3S level (Campellone et al., 2004). As a control to demonstrate the protein bands being visualized are a result of T3S, ΔO148A was included in the panel of strains examined as this contains a deletion of the first half of the LEE pathogenicity island (Campellone et al., 2004). As expected this strain demonstrated no detectable T3S compared to the TUV93-0 parent. BSA is added to all bacterial supernatants to aid precipitation of proteins and provides an indication of equivalent preparation steps and loading as there is no indigenous supernatant protein that can be used to standardize the samples. Many of the OI-deletions had no obvious impact on secretion level while some appeared to reduce levels (OI-144 and OI-148C) and others increased levels, OI-50 and OI-80 (Fig. 1A). This study has focused on the increased secretion observed on deletion of OI-50. OI-50 is a cryptic prophage (CP933-N) in the sequenced E. coli O157:H7 strain EDL933 from which TUV93-0 was derived as a Stx phage-negative variant. Confirmation that deletion of the OI-50/CP933N prophage increased T3S was obtained by blotting for the translocated intimin receptor (Tir) and by real time PCR for tir and intimin transcripts (Fig. 1B).
Figure 1.
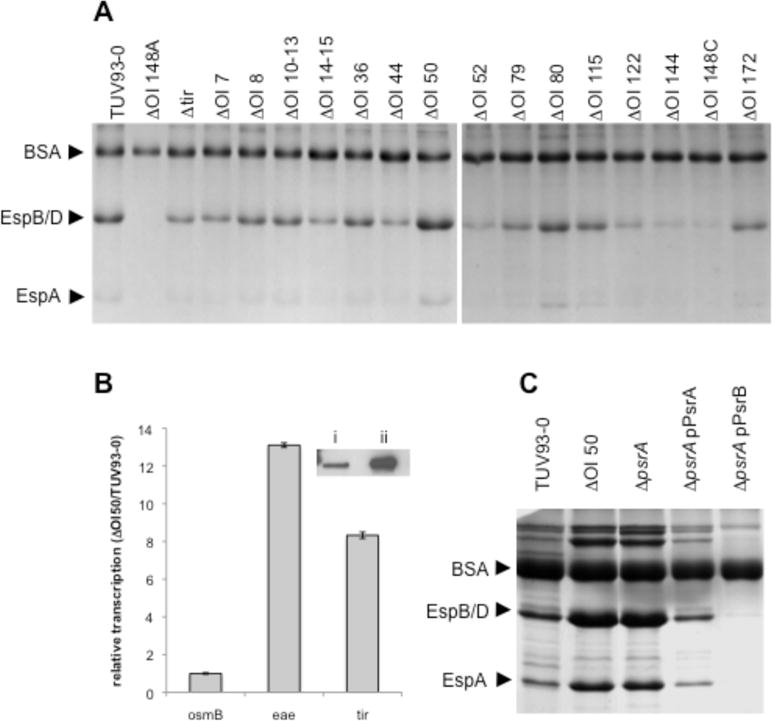
OI-50 encodes a negative regulator of T3S. A: Coomassie stained secreted protein profiles for a collection of O-island deletion strains constructed in the stx- EHEC O157:H7, TUV93-0 (Campellone et al., 2004). ΔOI-148A and ΔOI-148C contain deletions within the LEE and are included as negative secretion controls. Arrows indicate bands corresponding to BSA (added as a precipitation carrier and loading control, 66.4 kDa), EspB/D needle tip proteins (32.6 kDa and 39.1 kDa respectively), and the filament EspA (20.6 kDa). B: RT-qPCR confirmation of an increase in LEE encoded transcripts eae and tir for ΔOI-50 relative to the parental TUV93-0 strain. osmB is included as a negative control. Inset: Western blot analysis of secreted Tir from i) TUV93-0 and ii) ΔOI-50. C. Secreted protein profiles for TUV93-0 and isogenic deletions in OI-50, z1789 (psrA), and z2104 (psrB) complemented with plasmid encoded psrA or psrB (z2104). Arrows indicate bands corresponding to BSA, EspB/D and EspA. D: Graphical representation of OI-50 (CP933-N) (xbase.bham.ac.uk/colibase/). Recognisable modules of the lambdoid phage are indicated below CP933-N. IX, integration/excision; GR, general recombination; rep, repressor/co- repressor; lysis, lysis encoding genes; tail, tail fibres; effector region, contains T3S effector proteins. Red boxes indicate deleted regions in relevant mutants.
Identification of a transcriptional regulator on OI-50 controlling T3S
We tested whether deletion of the genes predicted to encode effector proteins was responsible for the increased T3S displayed by the ΔOI-50 strain and found that this was not the case (data not shown). We then searched for putative transcription regulators amongst the ORFs within OI-50 by BLAST analysis and motif scans. A number of established prophage transcriptional regulators were identified and when these were excluded from the screen a single ORF, z1789, encoding a putative AraC/XylS family regulator was identified downstream of the Q antiterminator in CP933-N (Fig. 1D). The region downstream of the Q antiterminator in lambdoid prophages has previously been shown to tolerate insertion of virulence genes that have been termed ‘morons’ and include the Shiga toxin genes encoded by BP933-W and CP933-V. To determine whether z1789 encodes a repressor, acting directly or indirectly on T3S, z1789 was deleted in TUV93-0 and secreted proteins isolated. Figure 1C shows that deletion of z1789 increased T3S in an analogous manner to deletion of the entire OI-50 region and could be complemented by introduction of z1789 in trans. This demonstrates that this regulator on OI-50 is responsible for repression of T3S under the conditions tested. Based on its activity, we have termed this prophage-encoded secretion regulator A, psrA.
A subset of AraC/XylS family regulators repress T3S
AraC/XylS family regulators commonly recognize AT rich binding sequences and have been associated with de-repression of H-NS silenced genes (Yang et al., 2008). Using psrA as a query for PSI-BLAST we were able to identify 33 putative regulators in the EDL933 genome that contain an AraC/XylS family DNA binding domain. Seven of these putative AraC/XylS family regulators were present on OIs. A phylogenic tree was constructed for these EDL933 sequences using tools available in the ANGIS bioinformatics package (www.angis.org.au). The closest homologue to PsrA was the prophage-encoded regulator z2104 (90% identity) present on OI-57, which we have termed PsrB. PsrB is similarly encoded after the predicted Q antiterminator of OI-57, cryptic lambdoid prophage CP933-O, that encodes a set of T3-secreted effector proteins. Deletion of psrB did not have a significant effect on T3S secretion (data not shown) but was able to repress secretion when provided in trans in the psrA deletion mutant (Fig. 1C) indicating that it was functional but possibly not expressed under our experimental conditions.
We have also cloned 14 of the 33 AraC/XylS family regulators identified by PSI-BLAST, including the other five regulators identified on O-islands, in an effort to identify related regulators that control T3S. Of these, only GadX, AdiY and to a lesser extent YdeO, repressed T3S under our experimental conditions (Fig. S2). All three are encoded in the ‘backbone’ genome of E. coli and induce the acid stress response. GadX and YdeO induce the glutamate dependant acid stress response and AdiY induces the arginine dependant acid stress response. These results support a link between acid stress and repression of T3S that has previously been reported for GadX and YdeO.
Analysis of sequenced pathogenic E. coli isolates identified a number of homologues of psrA, predominately in EHEC isolates (Table 1S). These homologues have been designated PsrB-F. All homologues were encoded within prophage and significantly, of 40 prophage islands carrying a Psr homologue, 32 contained predicted T3-secreted effectors; three of those not encoding effectors were within contigs that were too small to identify the region encoding bacteriophage tail fibres, and one prophage was identified in a meningitis-associated isolate. Furthermore, twenty five (62%) of the Psr encoding prophage also encoded the positive T3S regulators, pchA or pchB (Fig. 2 and Table 1S). These results demonstate that the majority of Psr regulators are encoded on prophages that also express secreted effectors and positive regulators of T3S and these genes are potentially acquired together on a single mobile element.
Figure 2.

Homologues of PsrA are encoded on cryptic prophage and associated with T3S regulators and effectors. Psr homologues were identified within publicly available genome sequences. All identified Psr homologues were identified within integrated prophage and the extremities were delineated by alignment with related prophage or annotated extremities. Prophages presented are representative of combinations of virulence genes found on Psr encoding prophage (an entire list of homologous sequences and encoding prophage are presented in Supp. Table 1). Prophage are indicated in bold (left) and are: CP933-N from EDL933, Sp4 from EHEC O157 Sakai, prophage sequence identified within NMEC O45:K1:H7 strain S88 (see Supp. Table 1), region of CP933-O from EDL933 homologous to Sp11, Sp11 from EHEC O157 Sakai, ECO103_P09 from EHEC O103:H2, ECO111_P12 from EHEC O111:H-. Grey areas between prophage represent related sequences. Genes are coloured: blue, prophage sequence; orange, int (integrase)/ xis (excisionase); pink, prophage regulators; green, T3S regulators; red, putative virulence genes encoded within tail fibres.
PsrA and PsrB indirectly regulate T3S through the GAD acid stress response regulators gadE and yhiF
Transcriptional profiling of TUV93-0 with and without induced psrA or psrB expression was used to identify genes controlled by PsrA and PsrB (GEO accession #GSE17798). As expected, transcription of LEE operons was reduced by the expression of either psrA or psrB (Fig. 3A). Microarray results were confirmed using previously described promoter fusions for LEE1, LEE2 and LEE5 (Fig. 3C). A reasonably small number of transcripts not located on the LEE were also affected by the increased levels of psrA or psrB expression, the majority of these being upregulated transcripts and were involved in the glutamate decarboxylase (GAD) acid stress response (Fig. 3B and Table S2). However, deletion of psrA and/or psrB did not significantly affect the glutamate-dependant acid tolerance of TUV93-0 (Fig. S1).
Figure 3.
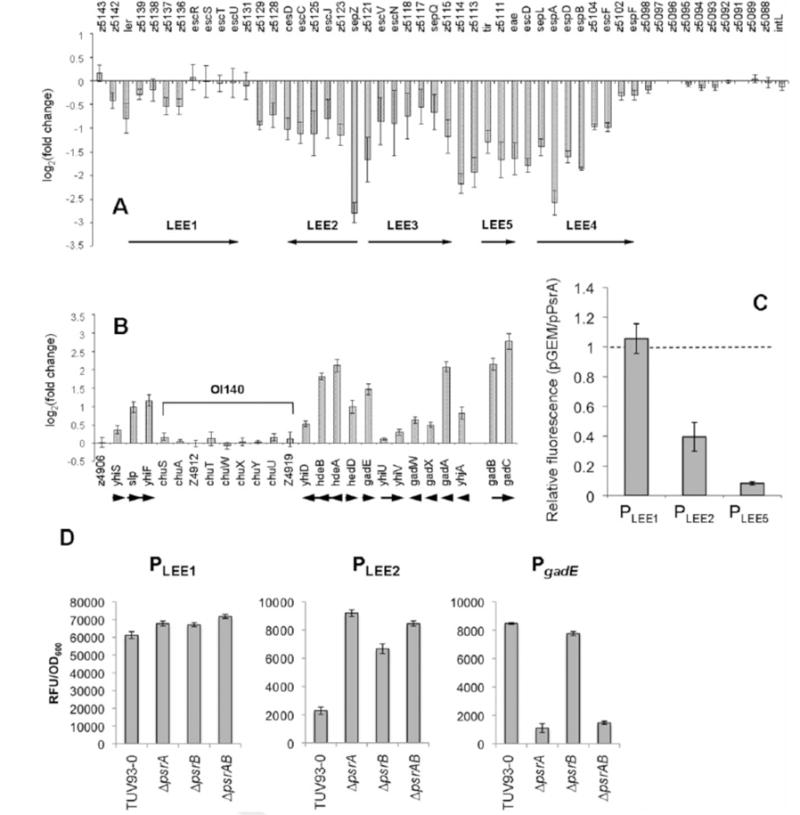
PsrA and PsrB induce the GAD acid stress regulators, gadE/yhiF, and repress the LEE. The PsrA regulon was defined by microarray analysis of TUV93-0 with psrA in trans on a multicopy plasmid. A: Relative transcription of the LEE in TUV93-0 pPsrA (pPsrA/pGEM) and B: Relative transcription of the GAD acid stress island. gadB and gadC are not encoded on the GAD island but have been included as part of the GAD regulon. In EDL933, OI-140 has been inserted into the GAD island and is indicated. Arrows below figure indicate direction of transcription. TUV93-0 pPsrB provided a similar transcriptional profile of the LEE and GAD island and is presented in Supplementary Table 2S. C: eGFP fusions of PLEE1 (pAJR71), PLEE2 (pAJR72), and PLEE5 (pAJR75) were used to confirm repression of the LEE in TUV93-0 pPsrA. Fluorescence of the pPsrA strains are presented as a fraction of the same fusion in a pGEM only background. The dashed line represents equal fluorescence between pGEM and pPsrA containing strains. D: Repression of LEE1 and LEE2 were confirmed in psrA, psrB, and psrAB deletion strains using eGFP fusions described above. Induction of gadE was similarly confirmed using a PgadE fusion to GFP+ (pPgadE.GFP+). Error bars represent standard error.
Previous work by Tatsuno and co-workers (Tatsuno et al., 2003) has shown that the LuxR family regulators YhiE (GadE) and YhiF repress the LEE in a LEE1-independent fashion as no repression of ler transcription was observed by northern dot-blot analysis. These observations are consistent with our transcriptional analyses in which there was only limited repression of LEE1 but more marked repression of LEE2 and 5. This indicated that the repression of T3S demonstrated for psrA/B may be through the induction of gadE and yhiF. Consequently, psrA and psrB repression was examined in deletion mutants of the two LuxR family regulators in TUV93-0. Single deletions of either gadE or yhiF inhibited repression by psrA or psrB, and this requirement was also seen in the double deletion background (Fig. 4A). It was evident that the majority of the psr-based T3S regulation required gadE/yhiF. We note that gadX is induced in our array analysis and identified in our screen of AraC family regulators that repress T3S (Fig. S2). It is possible that a fraction of PsrA/B repression may occur through induction of GadX. While the multi-copy expression of psrA/B is useful to define the potential regulatory network, it can induce artifacts. Therefore LEE1, LEE2, and gadE expression were measured in ΔpsrA, ΔpsrB, and ΔpsrAB backgrounds (Fig 3D). Consistent with our array results, LEE1 was slightly derepressed (10–20%), with more pronounced regulation of LEE2 (de-repressed 3–4 fold) and gadE (repressed up to 7 fold). The regulation is also consistent with the increased levels of T3S demonstrated for both the OI50 and psrA deletions (Fig. 1A & C).
Figure 4.
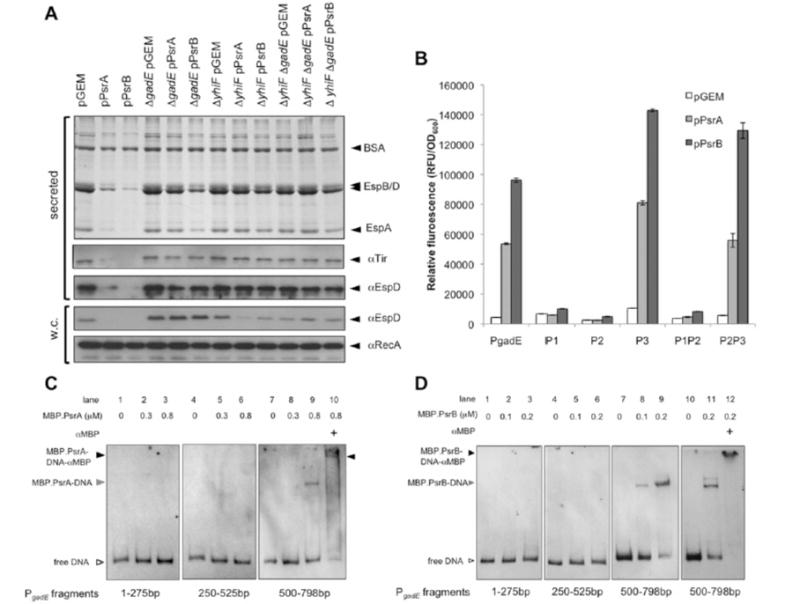
PsrA and PsrB repress the LEE by stimulating transcription of the negative LEE regulators GadE and YhiF. A: (top) Coomassie stained T3S secretion profiles of TUV93-0 and designated gadE and yhiF mutants with psrA or psrB in trans. (below) Western blots of secreted proteins (supernatant) and whole cell fractions (w.c.). Proteins recognized by the primary antibodies are indicated to the right of the blots B: Induction of gadE promoter fusions by pPsrA and pPsrB. TUV93-0 ΔgadE pPsrA, ΔgadE pPsrB, or ΔgadE pGEM were transformed with full length gadE promoter (PgadE), single promoter (P1, P2, and P3), or double gadE promoter (P1P2 and P2P3) fusions to GFP+ (Supplementary Table 1S). Fluorescence was normalized to OD600. Error bars represent standard error. C&D: EMSA was used to assess PsrA (C) and PsrB (D) binding to gadE promoter fragments containing transcriptional start sites. Start and stop positions for each DNA fragment from the gadE start codon are indicated below. Reaction constituents are indicated above. Open arrowhead indicates free labelled DNA, the grey arrowhead indicates MBP.Psr-DNA complexes, and the black arrowhead indicates supershifted MBP.Psr-DNA-antibody complexes.
We further investigated induction of gadE transcription by plasmid expressed psrA using the PgadE transcriptional fusion to GFP+ described above. Previous work (Sayed and Foster, 2009) and our own unpublished data have shown that the gadE promoter is auto-regulated and so we have used the ΔgadE mutant background to monitor GFP+ induction to avoid feed-back loops involving GadE. We have also tested an arabinose inducible PsrA construct (pBADmycHis) to quantify induction of gadE in this alternative expression vector. Induction from the arabinose inducible promoter (0.1% arabinose) results in a 2.8 fold increase in GFP+ fluorescence. A 12.2 fold induction is achieved using pGEM::PsrA (Fig. 4B) and so further experiments were conducted using this construct. Three transcriptional start sites designated P1 (−124), P2 (−324/−317) and P3 (−566) have been reported within the promoter of gadE. Transcriptional fusions of each promoter and combinations thereof were constructed in the GFP+ vector and transformed into ΔgadE pPsrA, ΔgadE pPsrB, or the pGEM control strain. PsrA and PsrB induced expression of the P3 and P2P3 transcriptional fusions indicated that PsrA and PsrB act to induce gadE transcription at P3 (Fig. 4B). Consistent with our phenotypic data showing stronger repression of T3S by PsrB, pPsrB induced P3 to higher levels (70%) than pPsrA.
In an effort to determine whether Psr binds to P3 directly or acts through induction of another regulator, EMSAs were carried out on fragments of the gadE promoter that span each transcriptional start site. Figure 4C & D show that both MBP.PsrA and MBP.PsrB bind the gadE promoter between bases −500 to −798 (containing P3). This complex is further retarded by the addition of polyclonal MBP antibody indicating that MBP.Psr is present in the shifted complex and that PsrA and PsrB can bind directly to the gadE promoter at P3. Consistent with our promoter fusion expression data and T3S profiles, MBP.PsrB does appear to have a higher affinity for P3 in comparison to MBP.PsrA.
GadE directly binds the LEE at PLEE1 and PLEE2/3
Our results show that Psr induces gadE expression and that gadE/yhiF are required for Psr repression of T3S. While GadE binding sites have previously been predicted within the LEE1 and LEE2/3 promoters (Kailasan Vanaja et al., 2009), no physical interactions have been described. We also note a potential additional site matching 14/20 bases of the GadE consensus binding site located divergent to the proposed LEE2/3 binding site (Fig 5, bottom). In order to investigate these predicted interactions, GadE was expressed and purified as an MBP.GadE fusion for EMSA (Experimental procedures). MBP.GadE bound to both PLEE1 and PLEE2/3 indicating that MBP.GadE could directly affect expression at both of these promoters (Fig. 5, lanes 2–4 & 12–14). Sequence specific binding of MBP.GadE to PLEE1 and PLEE2/3 was shown by competing MBP.GadE from the digoxin labeled complex with unlabelled probe or unlabelled PLEE5 (Fig. 5, PLEE1 lanes 5, 9 & 10; PLEE2/3 lanes 15, 19 & 20). Addition of excess PLEE5 did not compete PLEE1 or PLEE2/3 from the MBP.GadE complex indicating that MBP.GadE does not bind the LEE5 promoter and repression of LEE5 is indirect. The presence of MBP.GadE in the digoxin labeled DNA-protein complex was confirmed by further retardation of the complex in the presence of polyclonal MBP antibody (Fig. 5, lanes 8 & 18).
Figure 5.
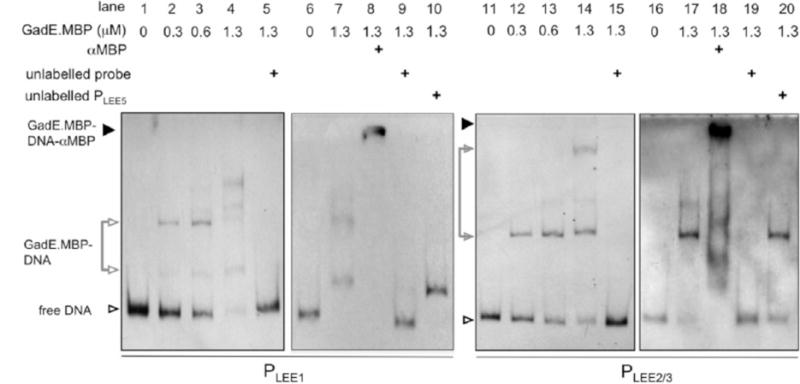
GadE binds the LEE at both PLEE1 and PLEE2/3. Top: Electrophoretic mobility shift assays were used to demonstrate binding of MBP.GadE to the LEE1 and LEE2/3 promoters. Increasing concentrations of MBP.GadE (indicated top) were incubated with 60 fmoles of digoxin end labelled PLEE1 (lanes 1–4) or PLEE2/3 (lanes 9–12). Labelled DNA was competed from the complex using a 1000 fold excess of unlabelled PLEE1 (lane 5) or PLEE2/3 (lane 13). Sequence specific interactions between MBP.GadE and LEE promoter fragments were confirmed by incubating with 50 ng of unlabelled PLEE5 (lanes 10 & 20). The presence of MBP.GadE in the shifted DNA-protein complex was confirmed by supershifting the complex with polyclonal MBP antisera (lanes 8 & 16). Labelled DNA complexes are indicated by arrowheads: free DNA (open), DNA-MBP.GadE (grey), and DNA-MBP.GadE-antibody (black).
gadE is induced on contact with epithelial cells and requires psrA and psrB for full induction
The acquisition of the psr genes on cryptic prophages encoding T3-secreted effectors suggests that the Psr regulators may be important to induce the gadE/yhiF sensor regulon during the T3S-dependent colonization of epithelial cells. To examine this, transcription of gadE was assessed in wild type and psrAB mutant bacteria adhering to cultured epithelial cells using two independent approaches. In the first, a GFP+ transcriptional fusion to PgadE was imaged on cells by fluorescence microscopy and single cell fluorescence levels measured as described in Experimental procedures. In the second, using the wild type and mutant strain without the fluorescent fusion plasmid, levels of the gadE transcript were measured by RT-qPCR.
For the single cell output, fluorescence was measured at hourly intervals for 4hrs and the average pixel intensity for individual adherent bacteria determined (Fig. 6A & B). Initially, PgadE fluorescence was induced in both the wild type and mutant strains and likely reflects induction of gadE by T3S-MEM-HEPES. The psrAB mutant was less induced at the start of the assay consistent with psrA inducing transcription of gadE in vitro as demonstrated previously (Fig. 4B). After 3 hours incubation with bovine epithelial cells (EBLs), PgadE was repressed in both strains (Fig. 6A). PgadE was induced after 4hrs incubation on the epithelial cells in the wild type strain but this induction was significantly reduced in the isogenic psrA/B mutant, indicating that psrA/B are required for full gadE induction on epithelial cells following attaching and effacing lesion formation.
Figure 6.

Transcription of gadE is induced by PsrA and PsrB after attachment to bovine epithelial (EBL) cells. A: TUV93-0 (open bars) and ΔpsrA ΔpsrB (shaded) bacteria containing a transcriptional fusion of the gadE promoter to GFP+ were used to infect EBL cell monolayers. Cells were fixed at indicated times and stained for immunofluorescence microscopy with O157 antibody. GFP+ expression in individual bacteria was measured as average pixel intensity across the area of the bacteria and averages of these intensities for indicated numbers of bacterial cells (n) are shown. A t-test was used to determine significance. Error bars represent standard error. B: Representative images from (A) of GFP+ expressing cells (green) stained with anti-O157 (red), obtained at 1 and 4 hours post-inoculation. C–F: RT-qPCR measurement of relative gadE transcription levels during adhesion to EBL cell monolayers. For clarity, TUV93-0 (open bars) and ΔpsrA ΔpsrB (light grey bars) are presented in (C) and the complemented ΔpsrA ΔpsrB pPsrA strain (dark grey bars) included in (D). E: gapA transcript abundance was also measured during adhesion in the TUV93-0, ΔpsrA ΔpsrB, and ΔpsrA ΔpsrB pPsrA backgrounds. F: Bacteria were visualized 3hr after addition to EBL monolayers. Bacterial cells were stained with anti-O157 antisera (red) and actin-rich pedestals stained with FITC conjugated phalloidin (green).
Direct measurement of gadE transcript levels by RT-qPCR demonstrated a very similar pattern of regulation. Monolayers of EBL cells were inoculated with wild type, ΔpsrAB, or ΔpsrAB pPsrA bacteria and RNA extracted at hourly time points for 6 hours. As measured in the PgadE-GFP fluorescence assays, gadE was initially induced in the wild type and the psrAB mutant but to a lesser extent in the psrAB mutant (Fig. 6C). Additional copies of psrA in trans significantly increased gadE transcription on EBL cells (Fig. 6D). At 3 hours post-inoculation gadE was maximally repressed also correlating well with the time pedestal formation is known to occur under these conditions (Fig. 6F). From 3 to 6 hours post-inoculation gadE was increasingly induced, and these results were mirrored by the complemented ΔpsrAB pPsrA strain although to a much higher degree (Fig. 6C & D). The housekeeping gene gapA was also measured during adhesion to confirm the validity of our normalization (Fig. 6E). These results indicate that psrAB promote gadE induction after intimate attachment of EHEC O157 to epithelial cells.
The effector-encoding locus of CP933-N and psrA contribute to E. coli O157:H7 persistence in ruminants
T3S is an essential virulence factor for colonization of ruminant reservoir hosts by EHEC O157:H7 and likely plays an important role in human colonization (Frankel et al., 1998, Naylor et al., 2005). To assess the contribution of CP933-N (OI-50) to ruminant colonization, wild type TUV93-0 and the isogenic ΔOI-50 strain were competed for colonization in an established ovine model (Best et al., 2009). The ΔOI-50 strain was able to colonize animals and was initially shed at similar levels to the parental strain (Fig. 7A), consistent with our observations that the OI-50 mutant forms pedestals on bovine epithelial cells at an equivalent level to the wild type (data not shown). However, by day 9 post-inoculation, shedding of the mutant strain was only detectable from 1 of the 6 animals compared to 4/6 for the wild type (Fig. 7A). From day 14 shedding of only the wild type strain was detectable, including 3/6 that continued shedding the wild type up to the end of the study at 28 days (and see Fig. 7B for the week 3 cumulative shedding pattern). To determine the relative contribution of the secreted effectors to this reduced persistence, we tested a defined deletion of the effector-containing locus spanning from espX7 to espK (Fig. 1D, boxed red) in the ovine colonization model (Fig. 7D). As these were comparative infection studies we excluded 2 sheep that were not colonized by the wild type for the three weeks of the experiment. On this basis, deletion of espX7-espK significantly reduced persistence in week 3 in the other 4 sheep (p=0.005, Fig. 7E&F) but not weeks 1 and 2 (p>0.075). These results indicate that the reduced persistence phenotype observed in the ΔOI-50 strain is conferred, at least in part, by the effector proteins on this cryptic prophage. We have also used the ovine model to determine the significance of psrA for colonization. Of six sheep colonized with wild type TUV93-0 and the isogenic ΔpsrA strain, colonization of two sheep did not persist for the full 3 weeks of the experiment and were excluded as for the espX7-espK analysis. All 4 remaining sheep demonstrated a reduction in ΔpsrA persistence relative to wild type (ranging from 15% to 99% reduction at week 3). This reduction is statistically significant (p=0.028) and specific for the later stages of colonization as there is no statistically significant reduction in shedding from these animals at weeks 1 and 2 post-inoculation (p>0.130). These results indicate that OI-50 is required for persistence in a ruminant model of colonization and that the effector encoding loci and OI-50 encoded regulator PsrA both contribute to this persistence phenotype.
Figure 7.
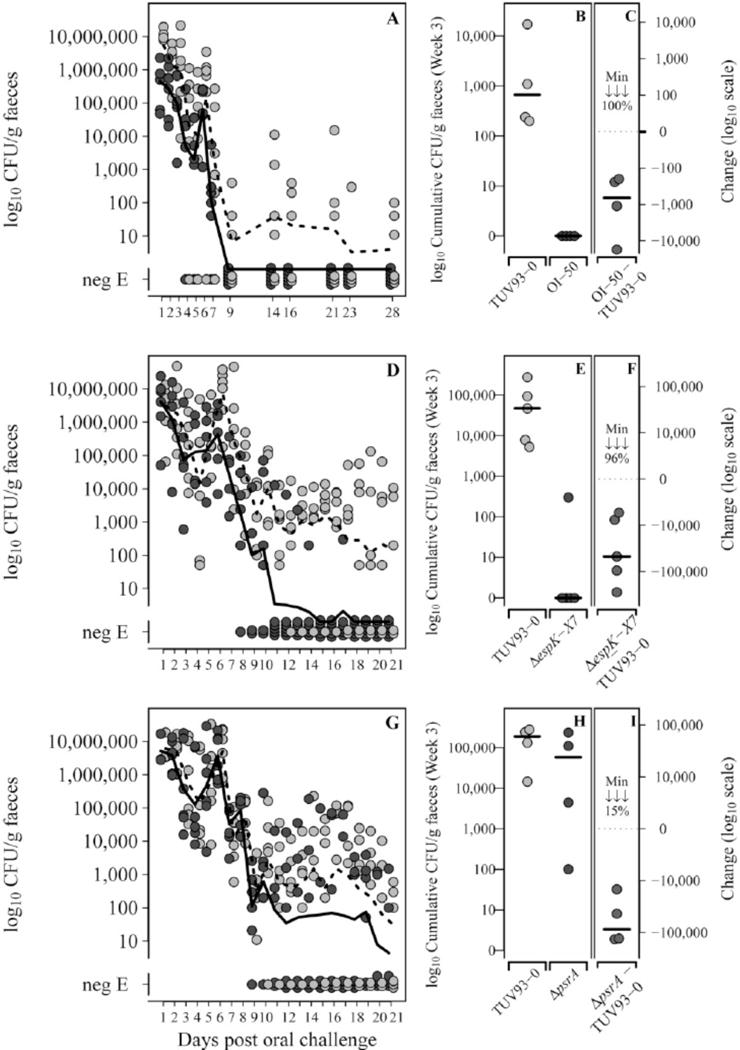
The effector encoding loci and psrA are required for persistence in a ruminant model of colonization. Six sheep were colonized with equal levels of the wild type (WT) and relevant mutant (ΔOI-50, ΔespK-X7, ΔpsrA) and shedding monitored from fecal samples over a three week period as described in Experimental Procedures. Shedding of wildtype TUV93-0 (light grey circles) and (A) the OI-50 mutant (dark grey circles), (D) the espK to espX7 mutant (dark grey circles) and (G) the psrA mutant (dark grey circles). NegE indicates samples that were tested by enrichment but remained negative for detection of the bacteria. Preliminary analysis indicated an effect of the mutations on persistence. To quantify this, cumulative bacterial counts for the individual weeks were compared and data is shown for the 3rd week of this analysis (B,E & H) for the indicated strains. As these are competitive index experiments data is only shown for animals in which the WT strain was still shedding at the end of the three week period. Graphs C, F & I show the level of reduction of the mutant strain vs. the WT. The minimum reduction (%) between the WT and indicated mutant is also shown in each graph.
Discussion
Horizontally acquired genetic elements in bacterial pathogens are often associated with virulence genes that can contribute to niche (host) colonization and persistence. Regulation, particularly between surface expressed factors, is important to prevent obstructive co-expression and limit antigen presentation. Regulation between these loci may also be important to co-ordinate production of factors that require co-expression. A clear example of this is the expression of non-LEE encoded effectors; many are acquired independently on cryptic prophages in ‘effector-encoding loci’ yet secreted by the T3S system of EHEC expressed from the LEE (OI-148). In this case the LEE-encoded regulator Ler, is recruited to activate both LEE and non-LEE encoded genes and similar inter-OI control is achieved by Pch regulators that are encoded on some of the OIs encoding effector proteins. In the present study, horizontally-acquired OIs in EHEC O157:H7 were screened for their capacity to regulate T3S. The initial screen employed a partial OI deletion library that was previously used to identify the effector EspFU in EHEC EDL933 stx- (TUV93-0) (Campellone et al., 2004). T3S profiles were prepared from the deletion mutants and compared with the parent strain TUV93-0, a Stx phage-negative derivative of the sequenced EHEC O157 EDL933 strain. From this analysis (Fig. 1) it was evident that OI-50 (CP933-N) repressed T3S under the conditions tested leading to an increase in T3S following its deletion. OI-50 encodes the cryptic prophage CP933-N and has previously been shown to carry the non-LEE encoded effectors espK, espO1-1, nleB2-2, espN, and espX7 (Tobe et al., 2006). We have demonstrated that this cryptic prophage is important for persistence in a ruminant model and that this persistence phenotype is primarily attributable to these effector proteins (Fig. 7D–F).
Deletion of the OI-50 effector-encoding genes did not alter the level of T3S (data not shown), indicating that the regulatory factor(s) controlling T3S must be present elsewhere on OI-50. Following bioinformatics analyses, a single ORF designated z1789 and encoded downstream of the Q anti-terminator was identified that did not appear to be a common prophage sequence and should encode a protein containing an AraC/XylS family DNA binding domain. Deletion of this gene led to an increase in T3S and this increase was restored to wild type levels by complementation in trans. As a result of this we have termed this: prophage-encoded secretion regulator (psrA). A closely related regulator, z2104 (psrB) was also identified, encoded on OI-57 (CP933-O), although when deleted this had no affect on T3S but was able to repress T3S when supplied in trans. Interestingly, between OI-50 and OI-57, these two cryptic prophages encode eleven putative effector proteins, almost a third of the predicted non-LEE encoded effector repertoire (Tobe et al., 2006).
Transcriptional profiling of wild type TUV93-0 provided with multicopy psrA or psrB was used to define their respective regulons. These were almost identical showing repression of the LEE and induction of the GAD acid stress island in a similar way to that described for the related AraC regulator YdeO (Masuda & Church, 2003). YdeO, and indeed the AraC family regulators GadX and GadW are all able to induce gadE and yhiF despite considerable sequence divergence (Sayed & Foster, 2009, Tramonti et al., 2008). GadE and YhiF are the central regulators of the GAD response and both have been shown to negatively regulate the LEE although no direct evidence for their binding has previously been presented (Tatsuno et al., 2003, Kailasan Vanaja et al., 2009). Deletion of gadE and yhiF abrogated repression of the LEE by multicopy psrA and psrB and this provided additional evidence that the primary mechanism by which psrA and psrB repress T3S is via induction of gadE and yhiF.
Recent work by Sayed and Foster (2009) has shown that there are three transcriptional start sites within the gadE promoter. GadE is thought to autoregulate gadE transcription from P1 (−124) and YdeO, GadX and GadW activate transcription from the P3 (−566) and P2(−324/−317) promoters. Earlier DNA footprinting by Tramonti and co-workers (2008) had shown that the AraC/XylS family regulators GadX and GadW bind −606 to −562 upstream of the gadE start codon which places the binding sites upstream of the P3 transcriptional start. Using GFP+ fusions to P1, P2 and P3 we have been able to show that PsrA also activates P3, further supporting the idea that PsrA regulators usurp the GAD acid stress response in an analogous fashion to the endogenous AraC/XylS family regulators YdeO, GadX and GadW. It appears that the P3 promoter of gadE is induced by at least 5 AraC/XylS family regulators in EHEC O157. We have been able to purify small quantities of MBP.PsrA and MBP.PsrB and these proteins bind the gadE promoter between −500 and −798 bases consistent with direct binding upstream of the P3 transcriptional start.
We have also purified MBP.GadE and performed EMSA to validate the predicted PLEE1 and PLEE2/3 binding. MBP.GadE bound both PLEE1 and PLEE2/3 in our semi-quantitative assay and we did not observe significant differences in binding affinity of the MBP.GadE preparation in vitro suggesting that preferential repression of LEE2/3 is not due to preferential binding. We note that the transcriptional activity of these promoters is clearly different; PLEE1 induction is routinely >5 fold higher than PLEE2 using transcriptional fusions (Fig. 5D and unpublished results) and may indicate that this promoter has stronger activating signals that may impede repression. The mechanisms of repression also appear to be different. Repression at PLEE1 is suggested by the predicted overlapping GadE binding site and -35 element of P2LEE1, and this mechanism does not seem to be shared by the predicted divergent PLEE2/3 sites.
To better understand the role of PsrA and PsrB we searched for homologues within the publicly available genome sequences and identified six closely related homologues among EHEC, EPEC and NMEC isolates. All six homologues are encoded on prophage and inserted downstream of the predicted Q anti-terminator. One of the most striking features of the pan-genome analysis was the association of Psr regulators with T3S effectors and Pch alleles. Of 40 prophage elements carrying Psr homologues, all except three encoded T3S effectors and more than half encoded a Pch allele. This association strongly suggests that Psr genes somehow benefit prophage elements carrying effectors, as would a Pch allele. Intriguingly, Pch has an almost inverse activity to Psr in that it represses gadE and yhiF transcription and promotes expression of the LEE (Abe et al., 2008). This led us to investigate whether there would be temporal regulation of these regulators to avoid simultaneous expression, with Psr potentially reversing the repression of gadE/yhiF by Pch. Transcriptional profiling of EHEC O157 adhering to red blood cells has shown that gadE and yhiF are induced after 5hrs contact and suggests that gadE induction occurs on cells (Dahan et al., 2004). This induction of gadE on cells has been confirmed in our study using both fluorescence microscopy and RT-qPCR (Fig. 6). Deletion of psrA/B led to a significant reduction in gadE induction on cells, indicating that they contribute to gadE activation on cells but are not solely responsible for it.
A hierarchy of effector secretion into cells has been demonstrated in EPEC and Salmonella (Mills et al., 2008, Winnen et al., 2008) and this is thought to be largely dependant on affinity of the effector for the T3S apparatus and concentration of effector within the cytoplasm (with some exceptions such as Tir that directly interacts with SepL to inhibit secretion of effectors). We suggest that Psr may function to induce gadE and repress further transcription of LEE-encoded effectors after biogenesis of the T3SS and secretion of ‘first wave’ effectors. This would allow the relative concentration of non-LEE effectors to increase and compete for secretion. Interestingly, YdeO has already been identified in a screen for genes controlling inhibition of fibroblast cell spreading, a phenotype that is T3S dependant in EPEC, but not conferred by LEE encoded effectors (Nadler et al., 2006). This may indicate that YdeO (and by extension GadE) plays a role in non-LEE effector secretion after adhesion of EPEC. In this respect, Psr homologues may have been acquired in EHEC to bolster induction of gadE after adhesion. Using an ovine model of colonization we have shown that OI-50 is required for persistence of EHEC. This phenotype was largely attributable to the effector-encoding locus in OI-50 (Fig.7), consistent with studies examining the functions of EspK and EspO1-1 (Kim et al., 2009, Vlisidou et al., 2006). We have also found that psrA contributes to prolonged colonization and is consistent with the idea that the Psr-GadE pathway facilitates non-LEE effector secretion. While direct effects of the Psr-GadE pathway on non-LEE effector secretion remains to be shown, it is possible that PsrA mediates persistence by inducing gadE after pedestal formation (as in Fig. 6), leading to repression of LEE-encoded effector transcription, which favours secretion of non-LEE encoded effectors such as those encoded within OI-50.
AraC/XylS family regulators are one-component sensor regulators that commonly bind small molecules or proteins to an N-terminal sensor domain (Gallegos et al., 1997, Plano, 2004). Substrate binding leads to conformational changes that can alter the activity of the regulator from inactive or repressive to a positive transcriptional regulator. The closest homologues of Psr are the acid stress regulators GadW and YdeO, and so it is conceivable that these newly identified regulators also monitor ion changes, potentially in response to host cell contact or the opening of a conduit with the host cell, for example changes in Na+, Ca2+, or bicarbonate (Michiels et al., 1990, Richard & Foster, 2007, Yang et al., 2008). Recent work by Yu and co-workers (2010) has indicated that the T3S system of Salmonella senses a pH change on contact with the host cytoplasm that induces effector secretion and suggests that there are changes sensed within the bacterial cell on contact with host cells (Yu et al., 2010).
H-NS repression of the LEE and the GAD acid stress island are well documented (Hommais et al., 2001, Bustamante et al., 2001) and fit with the proposed role of H-NS as a generalized xenogeneic silencer that functions to repress expression of foreign (AT rich) DNA (Dorman, 2007, Lucchini et al., 2006, Navarre et al., 2006). The identification of Psr control means there are at least three competing layers of regulation (H-NS, Pch, and Psr/YdeO) controlling and co-ordinating GAD and LEE expression. Recent work has suggested that the unusually long gadE promoter may constitute a ‘sensory integration region’ in E. coli allowing multiple environmental signals to feed into gadE transcription (Sayed & Foster, 2009). Future work will investigate the signals that activate gadE transcription via PsrA/B and whether this induction, that occurs after pedestal formation, aids translocation of non-LEE effectors.
Experimental procedures
Bacterial strains, cell culture and media
E. coli O157:H7 TUV93-0 is a shiga toxin negative derivative of the sequenced isolate O157:H7 EDL933 and has been previously described with the isogenic O-island deletion collection (Campellone et al., 2004). For genetic manipulations strains were grown in LB broth or plates supplemented with ampicillin (100μg/ml), kanamycin (50μg/ml), or chloramphenicol (25 μg/ml) where appropriate. For T3S secretion profiles cultures were inoculated 1/100 from overnight LB broth into MEM-HEPES (Sigma) supplemented with 250nM Fe(NO3)2 and 0.1% glucose (here termed “T3S-MEM-HEPES”). Bovine epithelial lung (EBL) cells were cultured in MEM-HEPES supplemented with 10% bovine fetal calf serum, glutamate, penicillin and streptomycin (here termed “supplemented MEM-HEPES”).
Cloning and construction of deletion strains
Complementation plasmids were constructed using PCR products generated with Phusion polymerase (Finnzymes) and cloned into pGEM-T Easy cloning vector as per manufacturer’s instructions. Primer pairs are detailed in Table 3S. Deletion of psrA, psrB and the effector encoding region of CP933-N was achieved using lambda Red mutagenesis and three-way PCR to generate linear PCR products with 500bp of flanking sequence (Beloin et al., 2004, Datsenko & Wanner, 2000). gadE and yhiF deletions were constructed using allelic exchange as previously described (Merlin et al., 2002). Mutants were verified by PCR and products were sequenced to confirm deletion and insertion. Where required, kanamycin cassettes were removed using pCP20 (Datsenko & Wanner, 2000).
Ovine colonization studies
Six 6-week-old crossbred lambs were housed in biosecure containment level 2 accommodation and supplied with food and water ad libitum. Prior to commencing the studies all lambs were confirmed to be free of EHEC O157 by enrichment and O157 immunomagnetic separation. Lambs were dosed orally with 1 × 1010 CFU of WT E. coli O157:H7 and mutant bacteria (5 × 109 CFU of each). Inocula were delivered in a 10 ml volume (resuspended in 10 ml of PBS pH 7.4) using a worming gun (Novartis Animal Health) ensuring that the whole inoculum was delivered directly to the pharynx.
Approximately 24 h after the dosing, and as required thereafter for the duration of the study, rectal fecal samples from each lamb were collected for direct plating onto sorbitol-MacConkey (Oxoid) plates supplemented with appropriate antibiotics. Samples that were negative on direct plating were enriched in buffered peptone water for 6 h at 37°C and then plated onto sorbitol-MacConkey plates supplemented with the appropriate antibiotic. Representative colonies were confirmed to be E. coli O157 by latex agglutination (Oxoid). All animal experiments were performed in accordance with the Animals Scientific Procedures Act (1986) and were approved by the local ethical review committee.
Differences in the weekly cumulative bacterial counts between the WT and the respective 3 mutant strains (OI-50, espX7-espK and psrA) were compared with paired t-tests of the log10 transformed cumulative values. Only those sheep that were still colonised by the WT for the three weeks of the experiment were included. Statistical significance was set to P<0.05 and all analyses were carried out in R (v2.10.1 © The R Foundation for Statistical Computing).
Adhesion and pedestal formation on bovine epithelial lung cells
For cell binding and actin pedestal formation by TUV93-0 and mutants, EBL cells were seeded at 5×104 cells/well into 8-well chamber slides (Becton, Dickinson and Company) and cultured overnight. Cells were washed two times with pre-warmed MEM-HEPES to remove the antibiotics before addition of bacteria. Subconfluent monolayers of EBL cells were infected with approx. 2 × 106 bacteria in MEM-HEPES for 3 hrs, washed three times with PBS and fixed for 20 min with PBS containing 2.5% paraformaldehyde (PFA). Cells were permeabilized for 5 min with PBS containing 0.1% Triton X-100 and washed and further three times with PBS and treated with anti-O157 rabbit antibody (MAST ASSURE) diluted 1:50 for 30 min. Monolayers were then washed three times with PBS, incubated with Alexa-568 conjugated anti-mouse IgG (Molecular Probes) at a dilution of 1:1000 for 30 min. For detection of actin-pedestals, chamber slides were then washed three times and stained with 1 μg/ml FITC labeled phalloidin (Sigma) for 30 min, washed two times with PBS and examined by fluorescent and light microscopy. The cell-binding frequencies of EHEC strains were quantified by measuring the number of bacteria (identified by anti-O157 staining) associated with randomly chosen fields of view. Twenty fields of view were examined for cell binding and actin pedestal formation assays. Pedestal formation frequencies were quantified by measuring the number of localized F-actin pedestals (identified by intense phalloidin staining) on randomly chosen EBL cells.
Sequence alignments and phylogenetic tree construction
Homologues of PsrA were identified using three iterations of PSI-BLAST and verified using InterPro Scan to identify AraC/XylS family DNA binding domains (www.ebi.ac.uk). Sequences of homologues were obtained from Genbank and multiple alignments were performed with ClustalW. Phylogenetic tree construction was performed using Protpars and bootstrapped using Seqboot, Protpars and Consense (www.angis.org.au). Regions of DNA carrying homologues of PsrA were identified by BLASTp. Regions carrying a homologue of PsrA were aligned with CP933-N, CP933-O, Sp4, Sp11, or lambda phage to identify potential phage sequences using Artemis Comparison Tool (ACT) (Carver et al., 2008). Representations of these regions were constructed using DNAplotter and overlaid on alignments generated with ACT.
Type III secretion assays and immunoblot detection of secreted proteins
Type III secretion (T3S) assays were performed essentially as previously described (Roe et al., 2003). In brief, overnight LB broth cultures were diluted 1/100 in T3S-MEM-HEPES. Cultures were grown to and OD600 of 0.8 and supernatants collected by centrifugation at 4000 rpm for 30min. Pellets were resuspended in 1ml of 1X Lamelli loading buffer and appropriate volumes used to quantify whole cell RecA and EspD. Supernatants were syringe filtered through 0.45 μm filters and secreted proteins precipitated overnight at 4°C with 10% TCA and 100μg of BSA. Secreted proteins were resuspended in 150μl of loading buffer and analysed by SDS-PAGE. Immunoblotting for RecA, EspD and Tir was performed with antibodies diluted 1/2000 in PBS containing 5% skim milk powder.
Microarray analysis of RNA
Microarray analysis was performed as previously described (Tree et al., 2009a). In brief, RNA was extracted from cultures grown as for T3S assays. Cells were harvested at OD600 0.8 and RNA extracted using an RNeasy kit (Qiagen). Test and reference RNAs were reverse transcribed and labeled with Cy3 and Cy5 using a CyScribe Post-Labelling Kit (GE Life Sciences). Labeled cDNA was hybridized using a Maui hybridization machine (Maui) and microarrays scanned using an Axon 4100A autoloader (Axon) and GenePix software. Data were analysed using Genespring GX 7.3.1. Microarray data are deposited at GEO (www.ncbi.nlm.nih.gov/geo/) under the accession number GSE17798.
Electrophoretic mobility shift assay (EMSA)
PsrA, PsrB and GadE were prepared as an N-terminal MBP5 fusion by cloning into pMAL-c5X. Fusions were expressed and purified as per manufacturer’s instructions excepting PBS was used in lieu of Tris buffer. Purified proteins were found to contain a significant fraction of free MBP that obscured accurate quantification of fusion proteins. The relative fraction of fusion protein in the total protein preparation was estimated from coomassie stained gels using densitometry and final fusion protein concentrations reflect the predicted amount of fusion in the preparation. Fusion proteins were incubated with the indicated concentrations of ddUTP-11-DIG (Roche) end labeled DNA in binding buffer for 30min at 25°C and loaded onto 5% nondenaturing polyacrylamide gels in 0.5X TBE. Binding buffer contained 10mM Tris (pH 8.0), 50mM KCl, 0.5% EDTA, 200μg/ml BSA, 100ng/ml poly d(I-C), 50mM glutamate and 5% glycerol. For competition experiments, 50ng of unlabelled DNA was added to the binding reaction. Labeled DNA was transferred to nylon membrane by electro-transfer and developed with AP conjugated anti-DIG antibody (Roche) as per manufacturer’s instructions. Supershift assays were performed as per standard EMSA excepting 1μl of polyclonal MBP antisera was added to the binding reaction before incubation.
Fluorescent reporters of LEE expression in vitro
LEE promoter eGFP fusions, pAJR70, pAJR71, pAJR72, and pAJR75, were used to measure LEE1, 2, and 5 expression in T3S-MEM-HEPES media and have previously been described (Roe et al., 2003). Transcriptional fusions to the gadE promoter and fragments thereof (previously described by Sayed and Foster, 2009) were constructed in pKC26 using primer sequences described in Supplementary Table 3S. Fluorescence values were corrected for growth differences by dividing by OD600. Fluorescence values were obtained by subtracting background fluorescence (obtained from pAJR70 or pKC26 vector only controls).
gadE transcription during adhesion to EBL cells
Embryonic bovine lung cells (German Collection of Microorganisms and Cell Cultures, no.21 ACC192) were cultured in supplemented MEM-HEPES media (Sigma) in 8 well chamber slides as previously described (Roe et al., 2004). Monolayers were washed 3 times with PBS and incubated in unsupplemented MEM-HEPES for 1hr before they were infected with 10μl of OD600 0.6 bacteria cultured in T3S-MEM-HEPES. Bacteria were forced onto the monolayer by centrifugation at 400 rpm for 5min. Monolayers were washed 3 times with PBS every hour and fresh unsupplemented MEM-HEPES media replaced in an effort to limit unattached bacteria growing in the media. Bacteria and EBL cells were fixed by incubating in 4% paraformaldehyde for 30min and washed 3 times with PBS. Bacteria were visualized using anti-O157 sera as previously described (Tree et al., 2009a). GFP images of bacteria were standardised by using a fixed exposure of 500ms and avoiding bleaching of the GFP channel. GFP+ levels in bacteria were quantified in individual bacteria using Openlab 4.0 software. Bacteria were identified in corresponding images using the anti-O157 channel and average GFP intensity recorded across the area of the bacteria and a background measurement for each image was subtracted from this average bacteria intensity. The number of individual bacteria measured for each time point is recorded in Fig. 6A.
For RT-qPCR measurements of gadE, gapA and 16S rRNA transcript abundance, monlayers of EBL cells were cultured in supplemented MEM-HEPES media in 100mm diameter cell culture dishes (Corning). Bacteria were cultured in T3S-MEM-HEPES media to OD600 0.6 and 5ml of bacteria culture mixed with an equal volume of unsupplemented MEM-HEPES. Monolayers were prepared by washing 3 times with PBS and incubating for 1hr with unsupplemented MEM-HEPES. 10ml of bacterial culture was added to the monolayer (MOI ~2000) and incubated for 1–6 hours. Monolayers were washed 3 times at 2 and 4 hours with PBS and fresh unsupplemented MEM-HEPES added to prevent excess growth of unattached bacteria in the culture media. Prior to harvesting RNA at each time point monolayers were also washed 3 times with PBS to further limit to amount of unattached bacteria represented in the RNA sample. RNA was stablised in 1ml of RNA protect and the monolayer harvested with a cell scrapper. Total RNA was extracted using an RNeasy kit (Qiagen) and reverse transcribed using Affinityscript (Stratagene) and random primers. qPCR was performed using PowerSybr mastermix (Applied Biosystems) and MxPro 3000 qPCR machine (Stratagene). qPCR primers was are presented in Table 3S. Transcript abundance was normalized to 16SrRNA and relative transcription calculated using MxPro software (Stratagene). gapA was used to verify normalized levels of transcript.
Supplementary Material
Acknowledgments
DLG, JJT, AJR, AM and SM would like to acknowledge funding from DEFRA under the Veterinary Training and Research Initiative (VT0102) and core support from the BBSRC at the Roslin Institute. The work was supported by grants from the Biotechnology and Biological Sciences Research Council (BB/G011389/1) and Medical Research Scotland (ref. 223 ORG G 0709) to AJR. AF is supported by a studentship from the College of Medicine and Veterinary Medicine at the University of Edinburgh. XXu is supported by a studentship from the China Scholarship Council. We would like to thank Prof. Trinad Chakraborty at the University of Giessen for his generous supply of monoclonal antibodies, and Kenneth Campellone at UC Berkley for helping construct the OI deletion collection.
References
- Abe H, Miyahara A, Oshima T, Tashiro K, Ogura Y, Kuhara S, Ogasawara N, Hayashi T, Tobe T. Global regulation by horizontally transferred regulators establishes the pathogenicity of Escherichia coli. DNA Res. 2008;15:25–38. doi: 10.1093/dnares/dsm033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D, Haagensen JA, Molin S, Prensier G, Arbeille B, Ghigo JM. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol. 2004;51:659–674. doi: 10.1046/j.1365-2958.2003.03865.x. [DOI] [PubMed] [Google Scholar]
- Best A, Clifford D, Crudgington B, Cooley WA, Nunez A, Carter B, Weyer U, Woodward MJ, La Ragione RM. Intermittent Escherichia coli O157:H7 colonisation at the terminal rectum mucosa of conventionally-reared lambs. Vet Res. 2009;40:9. doi: 10.1051/vetres:2008047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante VH, Santana FJ, Calva E, Puente JL. Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: Ler antagonizes H-NS-dependent repression. Mol Microbiol. 2001;39:664–678. doi: 10.1046/j.1365-2958.2001.02209.x. [DOI] [PubMed] [Google Scholar]
- Campellone KG, Robbins D, Leong JM. EspF(U) is a translocated EHEC effector that interacts with Tir and N-WASP and promotes nck-independent actin assembly. Developmental Cell. 2004;7:217–228. doi: 10.1016/j.devcel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Carver T, Berriman M, Tivey A, Patel C, Bohme U, Barrell BG, Parkhill J, Rajandream MA. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan S, Knutton S, Shaw RK, Crepin VF, Dougan G, Frankel G. Transcriptome of enterohemorrhagic Escherichia coli O157 adhering to eukaryotic plasma membranes. Infect Immun. 2004;72:5452–5459. doi: 10.1128/IAI.72.9.5452-5459.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O’Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci USA. 2004;101:3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman CJ. H-NS, the genome sentinel. Nat Rev Microbiol. 2007;5:157–161. doi: 10.1038/nrmicro1598. [DOI] [PubMed] [Google Scholar]
- Fass E, Groisman EA. Control of Salmonella pathogenicity island-2 gene expression. Curr Opin Microbiol. 2009;12:199–204. doi: 10.1016/j.mib.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel G, Philips AD, Novakova M, Batchelor M, Hicks S, Dougan G. Generation of Escherichia coli intimin derivatives with differing biological activities using site-directed mutagenesis of the intimin C-terminus domain. Mol Microbiology. 1998;29:559–570. doi: 10.1046/j.1365-2958.1998.00950.x. [DOI] [PubMed] [Google Scholar]
- Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev. 1997;61:393–410. doi: 10.1128/mmbr.61.4.393-410.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Duarte OG, Kaper JB. A plasmid-encoded regulatory region activates chromosomal eaeA expression in enteropathogenic Escherichia coli. Infect Immun. 1995;63:1767–1776. doi: 10.1128/iai.63.5.1767-1776.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Makino K, Ohnishi M, Kurokawa K, Ishii K, Yokoyama K, Han CG, Ohtsubo E, Nakayama K, Murata T, Tanaka M, Tobe T, Iida T, Takami H, Honda T, Sasakawa C, Ogasawara N, Yasunaga T, Kuhara S, Shiba T, Hattori M, Shinagawa H. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001;8:11–22. doi: 10.1093/dnares/8.1.11. [DOI] [PubMed] [Google Scholar]
- Hendrix RW, Lawrence JG, Hatfull GF, Casjens S. The origins and ongoing evolution of viruses. Trends Microbiol. 2000;8:504–508. doi: 10.1016/s0966-842x(00)01863-1. [DOI] [PubMed] [Google Scholar]
- Hommais F, Krin E, Laurent-Winter C, Soutourina O, Malpertuy A, Le Caer JP, Danchin A, Bertin P. Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS. Mol Microbiol. 2001;40:20–36. doi: 10.1046/j.1365-2958.2001.02358.x. [DOI] [PubMed] [Google Scholar]
- Kailasan Vanaja S, Bergholz TM, Whittam TS. Characterization of the Escherichia coli O157:H7 Sakai GadE regulon. J Bacteriol. 2009;191:1868–1877. doi: 10.1128/JB.01481-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, Koyama T, Nagai S, Lange A, Fassler R, Sasakawa C. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature. 2009;459:578–582. doi: 10.1038/nature07952. [DOI] [PubMed] [Google Scholar]
- Laaberki MH, Janabi N, Oswald E, Repoila F. Concert of regulators to switch on LEE expression in enterohemorrhagic Escherichia coli O157:H7: interplay between Ler, GrlA, HNS and RpoS. Int J Med Microbiol. 2006;296:197–210. doi: 10.1016/j.ijmm.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2006;2:e81. doi: 10.1371/journal.ppat.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel MJ, Wollenberg MS, Stabb EV, Visick KL, Ruby EG. A single regulatory gene is sufficient to alter bacterial host range. Nature. 2009;458:215–218. doi: 10.1038/nature07660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda N, Church GM. Regulatory network of acid resistance genes in Escherichia coli. Mol Microbiol. 2003;48:699–712. doi: 10.1046/j.1365-2958.2003.03477.x. [DOI] [PubMed] [Google Scholar]
- McNeilly TN, Mitchell MC, Rosser T, McAteer S, Low JC, Smith DG, Huntley JF, Mahajan A, Gally DL. Immunization of cattle with a combination of purified intimin-531, EspA and Tir significantly reduces shedding of Escherichia coli O157:H7 following oral challenge. Vaccine. 2010;28:1422–1428. doi: 10.1016/j.vaccine.2009.10.076. [DOI] [PubMed] [Google Scholar]
- Mellies JL, Barron AMS, Carmona AM. Enteropathogenic and enterohemorrhagic Escherichia coli virulence gene regulation. Infect Immun. 2007;75:4199–4210. doi: 10.1128/IAI.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin C, McAteer S, Masters M. Tools for characterization of Escherichia coli genes of unknown function. J Bact. 2002;184:4573–4581. doi: 10.1128/JB.184.16.4573-4581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michiels T, Wattiau P, Brasseur R, Ruysschaert JM, Cornelis G. Secretion of Yop Proteins By Yersiniae. Infect Immun. 1990;58:2840–2849. doi: 10.1128/iai.58.9.2840-2849.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills E, Baruch K, Charpentier X, Kobi S, Rosenshine I. Real-time analysis of effector translocation by the type III secretion system of enteropathogenic Escherichia coli. Cell Host Microbe. 2008;3:104–113. doi: 10.1016/j.chom.2007.11.007. [DOI] [PubMed] [Google Scholar]
- Nadler C, Shifrin Y, Nov S, Kobi S, Rosenshine I. Characterization of enteropathogenic Escherichia coli mutants that fail to disrupt host cell spreading and attachment to substratum. Infect Immun. 2006;74:839–849. doi: 10.1128/IAI.74.2.839-849.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, Porwollik S, Wang YP, McClelland M, Rosen H, Libby SJ, Fang FC. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science. 2006;313:236–238. doi: 10.1126/science.1128794. [DOI] [PubMed] [Google Scholar]
- Naylor SW, Roe AJ, Nart P, Spears K, Smith DGE, Low JC, Gally DL. Escherichia coli O157:H7 forms attaching and effacing lesions at the terminal rectum of cattle and colonization requires the LEE4 operon. Microbiology. 2005;151:2773–2781. doi: 10.1099/mic.0.28060-0. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Ooka T, Iguchi A, Toh H, Asadulghani M, Oshima K, Kodama T, Abe H, Nakayama K, Kurokawa K, Tobe T, Hattori M, Hayashi T. Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc Natl Acad Sci U S A. 2009;106:17939–17944. doi: 10.1073/pnas.0903585106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi M, Terajima J, Kurokawa K, Nakayama K, Murata T, Tamura K, Ogura Y, Watanabe H, Hayashi T. Genomic diversity of enterohemorrhagic Escherichia coli O157 revealed by whole genome PCR scanning. PNAS. 2002;99:17043–17048. doi: 10.1073/pnas.262441699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JC, Groisman EA. Evolution of transcriptional regulatory circuits in bacteria. Cell. 2009;138:233–244. doi: 10.1016/j.cell.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna NT, Plunkett G, 3rd, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. Genome sequence of enterohemorrhagic Escherichia coli O157:H7. Nature. 2001;409:529–533. doi: 10.1038/35054089. [DOI] [PubMed] [Google Scholar]
- Plano GV. Modulation of AraC family member activity by protein ligands. Mol Microbiol. 2004;54:287–290. doi: 10.1111/j.1365-2958.2004.04306.x. [DOI] [PubMed] [Google Scholar]
- Potter AA, Klashinsky S, Li Y, Frey E, Townsend H, Rogan D, Erickson G, Hinkley S, Klopfenstein T, Moxley RA, Smith DR, Finlay BB. Decreased shedding of Escherichia coli O157:H7 by cattle following vaccination with type III secreted proteins. Vaccine. 2004;22:362–369. doi: 10.1016/j.vaccine.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Richard H, Foster JW. Sodium regulates Escherichia coli acid resistance, and influences GadX- and GadW-dependent activation of gadE. Microbiology. 2007;153:3154–3161. doi: 10.1099/mic.0.2007/007575-0. [DOI] [PubMed] [Google Scholar]
- Roe AJ, Naylor SW, Spears KJ, Yull HM, Dransfield TA, Oxford M, McKendrick IJ, Porter M, Woodward MJ, Smith DG, Gally DL. Co-ordinate single-cell expression of LEE4- and LEE5-encoded proteins of Escherichia coli O157:H7. Mol Microbiol. 2004;54:337–352. doi: 10.1111/j.1365-2958.2004.04277.x. [DOI] [PubMed] [Google Scholar]
- Roe AJ, Yull H, Naylor SW, Woodward MJ, Smith DGE, Gally DL. Heterogeneous surface expression of EspA translocon filaments by Escherichia coli O157:H7 is controlled at the posttranscriptional level. Infect Immun. 2003;71:5900–5909. doi: 10.1128/IAI.71.10.5900-5909.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed AK, Foster JW. A 750 bp sensory integration region directs global control of the Escherichia coli GadE acid resistance regulator. Mol Microbiol. 2009;71:1435–1450. doi: 10.1111/j.1365-2958.2009.06614.x. [DOI] [PubMed] [Google Scholar]
- Shakhnovich EA, Davis BM, Waldor MK. Hfq negatively regulates type III secretion in EHEC and several other pathogens. Mol Microbiol. 2009;74:347–363. doi: 10.1111/j.1365-2958.2009.06856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuno I, Nagano K, Taguchi K, Rong L, Mori H, Sasakawa C. Increased adherence to Caco-2 cells caused by disruption of the yhiE and yhiF genes in enterohemorrhagic Escherichia coli O157:H7. Infect Immun. 2003;71:2598–2606. doi: 10.1128/IAI.71.5.2598-2606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobe T, Beatson SA, Taniguchi H, Abe H, Bailey CM, Fivian A, Younis R, Matthews S, Marches O, Frankel G, Hayashi T, MJ P. An extensive repertoire of type III secretion effectors in Escherichia coli O157 and the role of lambdoid phages in their dissemination. Proc Natl Acad Sci U S A. 2006;3:14941–14946. doi: 10.1073/pnas.0604891103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramonti A, De Canio M, De Biase D. GadX/GadW-dependent regulation of the Escherichia coli acid fitness island: transcriptional control at the gadY-gadW divergent promoters and identification of four novel 42 bp GadX/GadW-specific binding sites. Mol Microbiol. 2008;70:965–982. doi: 10.1111/j.1365-2958.2008.06458.x. [DOI] [PubMed] [Google Scholar]
- Tree JJ, Wang D, McInally C, Mahajan A, Layton A, Houghton I, Elofsson M, Stevens MP, Gally DL, Roe AJ. Characterization of the effects of salicylidene acylhydrazide compounds on type III secretion in Escherichia coli O157:H7. Infect Immun. 2009a;77:4209–4220. doi: 10.1128/IAI.00562-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tree JJ, Wolfson EB, Wang D, Roe AJ, Gally DL. Controlling injection: regulation of type III secretion in enterohemorrhagic Escherichia coli. Trends Microbiol. 2009b;17:361–370. doi: 10.1016/j.tim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Vlisidou I, Marches O, Dziva F, Mundy R, Frankel G, Stevens MP. Identification and characterization of EspK, a type III secreted effector protein of enterohemorrhagic Escherichia coli O157:H7. FEMS Microbiol Lett. 2006;263:32–40. doi: 10.1111/j.1574-6968.2006.00410.x. [DOI] [PubMed] [Google Scholar]
- Winnen B, Schlumberger MC, Sturm A, Schupbach K, Siebenmann S, Jenny P, Hardt WD. Hierarchical effector protein transport by the Salmonella Typhimurium SPI-1 type III secretion system. PLoS ONE. 2008;3:e2178. doi: 10.1371/journal.pone.0002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Hart E, Tauschek M, Price GD, Hartland EL, Strugnell RA, Robins-Browne RM. Bicarbonate-mediated transcriptional activation of divergent operons by the virulence regulatory protein, RegA, from Citrobacter rodentium. Mol Microbiol. 2008;68:314–327. doi: 10.1111/j.1365-2958.2008.06171.x. [DOI] [PubMed] [Google Scholar]
- Yu XJ, McGourty K, Liu M, Unsworth KE, Holden DW. pH sensing by intracellular Salmonella induces effector translocation. Science. 328:1040–1043. doi: 10.1126/science.1189000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chaudhuri RR, Constantinidou C, Hobman JL, Patel MD, Jones AC, Sarti D, Roe AJ, Vlisidou I, Shaw RK, Falciani F, Stevens MP, Gally DL, Knutton S, Frankel G, Penn CW, Pallen MJ. Regulators encoded in the Escherichia coli type III secretion system 2 gene cluster influence expression of genes within the locus for enterocyte effacement in enterohemorrhagic E. coli O157:H7. Infect Immun. 2004;72:7282–7293. doi: 10.1128/IAI.72.12.7282-7293.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Laing C, Steele M, Ziebell K, Johnson R, Benson AK, Taboada E, Gannon VP. Genome evolution in major Escherichia coli O157:H7 lineages. BMC Genomics. 2007;8:121. doi: 10.1186/1471-2164-8-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.