
Fig. 1.
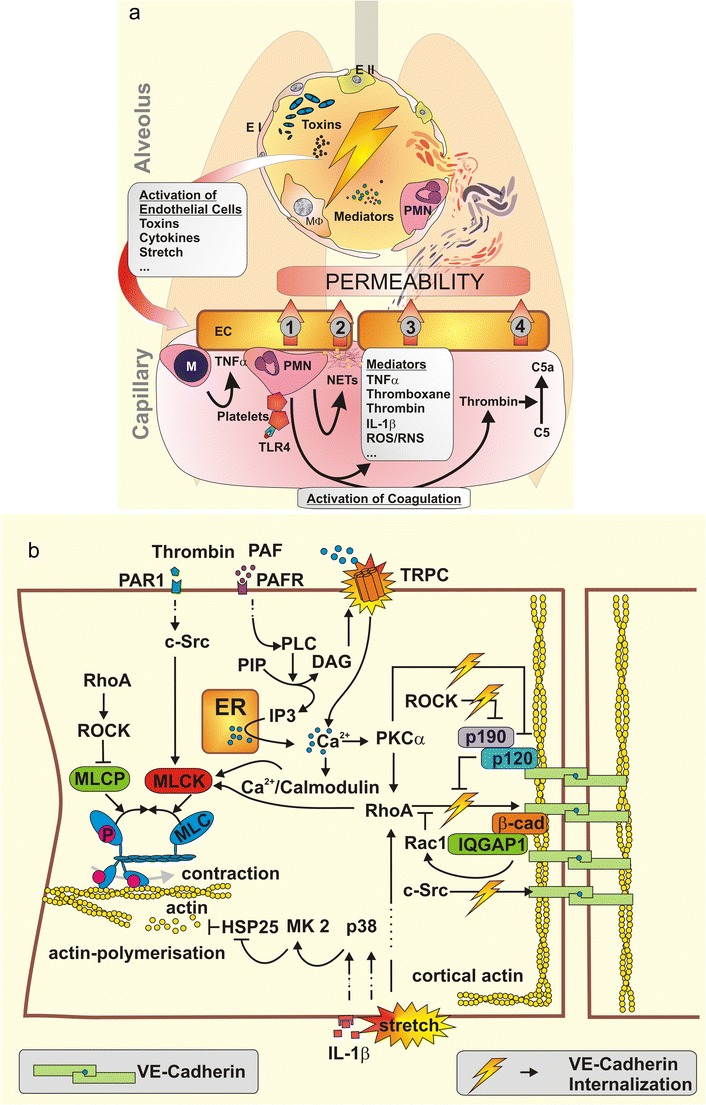
a Airspace-derived activation of the endothelium by mediators, bacterial toxins or physical stress due to mechanical ventilation starts a complex interplay of various inflammatory cascades resulting in vascular permeability. Monocytes (M) are recruited to the endothelium (EC) and facilitate its further activation by secretion of TNFα, thereby augmenting the recruitment of neutrophils (PMN). Activated platelets stimulate PMN. Endothelium-PMN contact leads to permeability (1). Upon stimulation PMN undergo netosis, liberating neutrophil extracellular traps (NETs) consisting in DNA and histones that cause endothelial toxicity and barrier breakdown (2). Specific soluble mediators also increase permeability (3). Neutrophil-platelet complexes activate blood coagulation. Central effector proteases like thrombin directly mediate vascular permeability. Further, thrombin activates complement factor C5 to C5a—a permeability increasing anaphylatoxin (4). TNF tumor necrosis factor; IL-1β Interleukin-1β; ROS/RNS reactive oxygen and nitrogen species. b Intracellular signalling regulates endothelial permeability. Endothelial contraction results from actin myosin interaction after MLC-phosphorylation, which is regulated by myosin light chain kinase (MLCK) and myosin light chain phosphatase (MLCP). MLCP is inhibited by RhoA–ROCK signalling while MLCK is activated by c-Src, RhoA and Ca2+/Calmodulin. Ca2+ enters the cytosol from endoplasmatic reticulum (ER) or extracellular space. Downstream of platelet activating factor (PAF) and PAF receptor (PAFR), phospholipase C (PLC) hydrolyses posphatidyl inositol bisphosphate (PIP) into inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 mediates Ca2+ liberation from the ER while DAG opens transient receptor potential canonical (TRPC) channels in the cellular membrane. The resulting increase of intracellular Ca2+ leads to the activation of protein kinase C (PKC) α, to further RhoA activation and to Ca2+/calmodulin complexes, altogether finally leading to MLCK activation. Actin polymerisation forms stress fibres associated with endothelial contraction. Various stimuli like IL-1β or mechanical force activate mitogen-activated protein kinase (MAPK) p38 (p38), which activates MAPK activated protein kinase 2 (MK2), which phosphorylates heat shock protein 25 (HSP25) leading to actin polymerisation. Adherence junctions (AJ) are mandatory for the sealing of intercellular contacts. VE-cadherin is anchored in peripheral cortical actin to the cytoskeleton. VE-cadherin phosphorylation leads to VE-cadherin internalisation and thereby to increased endothelial permeability. RhoA and c-Src phosphorylate VE-cadherin. Rac-1 and p190RhoAGAP (p190) functionally antagonise RhoA activity. p190RhoAGAP is recruited to the AJ by p120-catenin (p120), which itself inhibits VE-cadherin internalisation. ROCK inhibits p190RhoAGAP and PKCα inactivates p120-catenin thereby augmenting destabilisation of AJ. IQGAP1 recruits and stabilises Rac-1, protecting against VE-cadherin internalisation