
Figure 2.
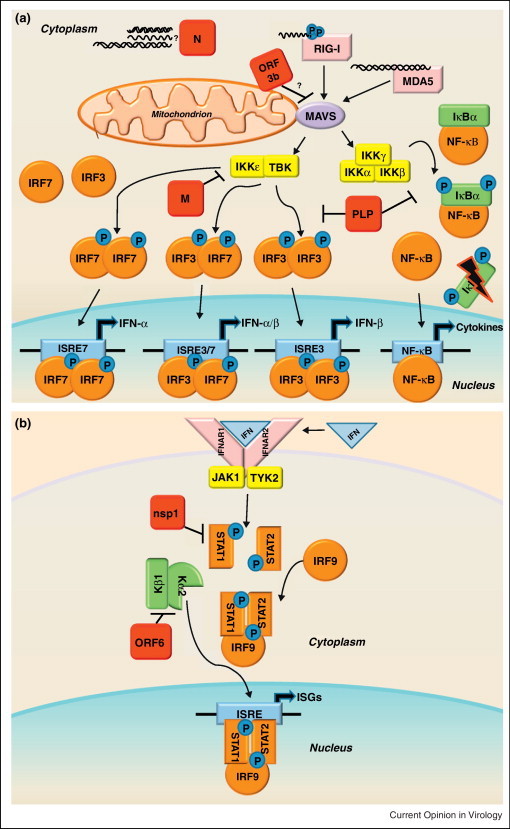
(a) RLR family of innate immune receptors induce Type I interferon. The family of RIG-I Like Receptors (RLRs) contains three cytosolic RNA helicases that recognize non-self RNA species resulting from viral replication [93]. The two signaling sensors within the RLR family are retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated factor 5 (MDA5). The third RLR, laboratory of genetics and physiology 2 (LGP2, not shown), facilitates recognition of viral PAMPs by RIG-I and MDA5, but is dispensable for their signaling [94]. RIG-I recognizes primarily 5′ppp-RNA molecules with secondary motifs of dsRNA or ssRNA of short length [95, 96]. MDA5 recognizes longer dsRNA motifs than RIG-I [97]. Following binding of viral RNAs, RIG-I and MDA5 interact with the mitochondrial membrane bound adaptor molecule MAVS (mitochondrial antiviral signaling protein, also referred to as IPS-1, VISA, or CARDIF) to transduce the signal via complexes of kinases: the IKKɛ/TBK1 complex and the IKKα/IKKβ/IKKγ complex. The IKKɛ/TBK1 kinases phosphorylate the transcription factors IRF3 and IRF7, which then form homodimers or heterodimers. Upon dimerization, the transcription factors enter the nucleus to initiate transcription of Type I IFNs (IFN-α and IFN-β). While IRF3 is nearly ubiquitously expressed in cells, IRF7 is an ISG typically expressed at low levels, so it is thought that IRF3 mediates transcription of the majority of early IFN expression. The IKKα/IKKβ/IKKγ kinases phosphorylate IκBα, targeting this repressor protein of NF-κB for degradation. Activation of NF-κB leads to transcription of proinflammatory cytokines, and NF-kB mediated transcription has also been linked to the pathogenesis of ARDS [46]. SARS-CoV encodes proteins that antagonize RLR family signaling, shown here in red.
(b) Interferon signals through the JAK-STAT pathway to induce interferon stimulated genes. The secretion of IFN-α and IFN-β molecules from an infected cell leads to an autocrine and paracrine signaling through the IFNαβ Receptor (composed of the IFNAR1 and IFNAR2 subunits) resulting in the activation of the JAK-STAT pathway. The JAK/TYK2 kinases phosphorylate the transcription factors STAT1 and STAT2, which form heterodimers complexed with IRF9. The STAT complex translocates to the nucleus leading to the transcription of Interferon Stimulated Genes (ISGs) that establish an antiviral state in the cell. Because neighboring cells can receive IFN stimulation before infection, it is a crucial pathway to preventing viral spread in the host. SARS-CoV also encodes proteins that antagonize the JAK-STAT pathway, shown here in red. Mice deficient in STAT1 showed an increased susceptibility to SARS-CoV infection [98]. Although there were no differences in mice deficient in IFN receptors, STAT1−/− mice showed increased weight loss, viral titer, and lung pathology compared to wild type over the course of MA15-SARS-CoV infection, demonstrating that STAT has important IFN independent role in SARS-CoV infection [80]. Severe lung pathology in STAT1−/− mice infected with MA15-SARS-CoV was associated with the infiltration of immune cells and fibrotic lung response. The STAT1−/− dependent prolonged expression of inflammatory cytokines (IL-1, IL-6, IL-10, IL-12, and TNFα) and chemokines (CCL2, CCL3, CCL4, CCL7, and CCL20), could be a transcriptional regime responsible for fibrotic phenotypes within the lungs. Additionally, ISG responses were significantly lower in STAT1−/− mice compared to wild type or IFNAR−/− mice, leading to the conclusion that STAT1 dependent, IFNAR1 independent ISG expression was protective in these mice [80]. It remains unclear how STAT1 controls ISGs independent of IFNAR expression, or which ISGs have important potential roles in SARS-CoV pathogenesis.