
Figure 1.
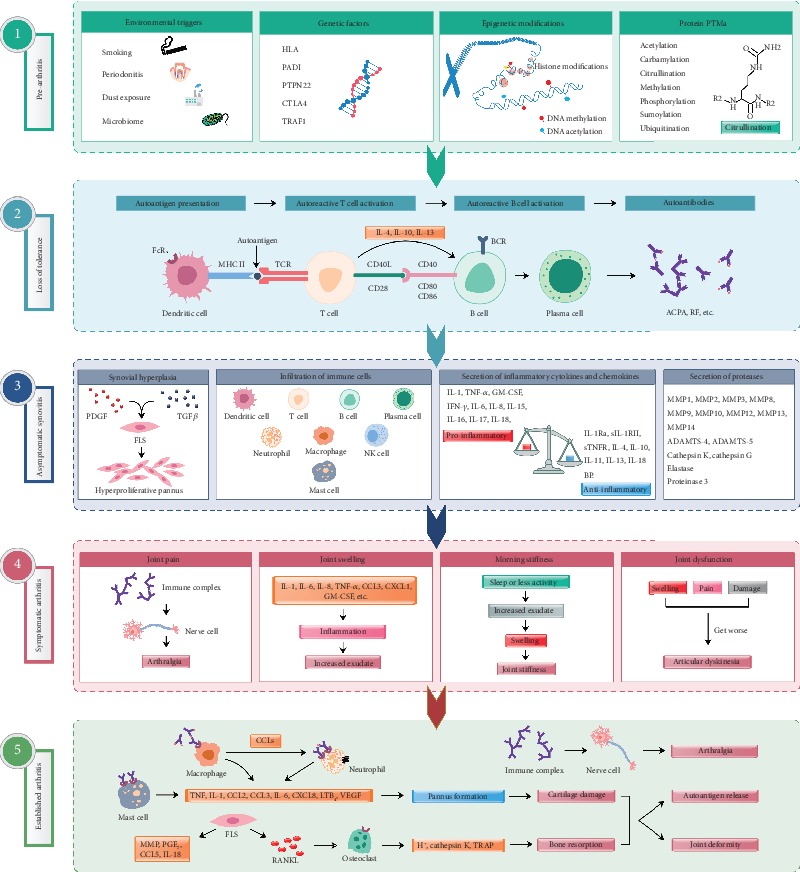
Different phases in RA pathogenesis. (1) Genetic, epigenetic, and environmental factors contribute to arthritis progression. Multiple environmental risk factors (for example, smoking, pollutants, or microbes), when come in contact with the mucosal sites, are most likely responsible for causing local inflammatory events and immune system activation inducing epigenetic modifications and protein posttranslational modifications (PTMs) [59], before crossing the threshold to trigger disease in genetically vulnerable people. (2) Dendritic cells presenting altered self or related peptides to T cells (breakdown of tolerance mechanisms) leads to the activation of T and B cells effectuating synthesis of cytokines and autoantibodies. Progressively, these autoantibodies are produced more and more, which recognize several neoepitopes by the process of epitope spreading, and gets overt during the onset of the clinical disease [1]. (3) Disease development involves autoimmune responses against both posttranslationally modified and unmodified self-antigens, which starts many years before the subclinical synovitis and appearance of clinical symptoms [60]. (4) Autoantibodies induced during this preclinical phase can also be responsible for bone erosion and pain. Before the onset of inflammation, these alterations could reduce overall functions of the joints. After the autoantibodies start binding to different epitopes and form immune complexes, inflammation in the synovium and development of arthritis ensue. (5) Antibody-induced cartilage and bone changes, if minor, resolve without any considerable damages. However, if left untreated or in the presence of continuous external stimuli, these changes can give rise to chronic inflammation, joint destruction, and disability [59]. Arthritis is associated with both local as well as systemic pathological manifestations. ACPA: anticitrullinated protein antibody; ADAMTS: a disintegrin and metalloproteinase with thrombospondin motifs; BCR: B cell receptor; CCL: c-c motif chemokine ligand; CXCL: C-X-C motif chemokine ligand; CTLA4: cytotoxic T-lymphocyte-associated protein 4; FcR: Fc receptor; FLS: fibroblast-like synoviocytes; GM-CSF: granulocyte-macrophage colony-stimulating factor; HLA: human leukocyte antigen; IFN-γ: interferon gamma; IL: interleukin; IL-1Ra: interleukin-1 receptor antagonist; IL-18BP: interleukin-18-binding protein; LTB4: leukotriene B4; MMP: matrix metalloproteinase; MHC II: major histocompatibility complex class II; NK cell: natural killer cell; PADI: peptidyl arginine deiminase; PDGF: platelet-derived growth factor; PGE2: prostaglandin E2; PTPN22: protein tyrosine phosphatase nonreceptor type 22; RANKL: receptor activator of nuclear factor kappa B ligand; RF: rheumatoid factor; sIL-1RII: soluble interleukin 1 receptor II; sTNFR: soluble tumor necrosis factor receptors; TCR: T cell receptor; TGFβ: transforming growth factor β; TNF-α: tumor necrosis factor α; TRAF1: TNF receptor-associated factor 1; VEGF: vascular endothelial growth factor.