
Figure 2.
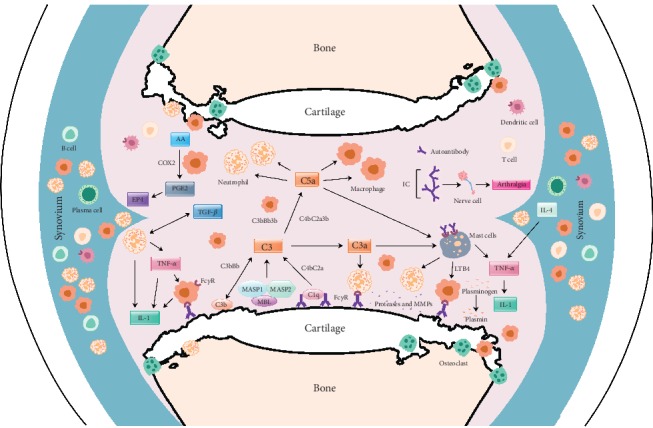
Likely interactions of molecules and factors in the antibody mediated joint inflammation. Upon binding to joint antigens or deposited as immune complexes on the cartilage surface, autoantibodies initiate inflammation-dependent and inflammation-independent activities, which culminate in the direct damage to the cartilage and bone. Activation of complement cascades by autoantibodies leads to the release of anaphylatoxins (C3a, C5a), attracting FcR-bearing immune cells to the inflammation foci, which in turn get more activated and secrete cytokines that can further activate resident nonimmune cells in the joint. All these cells in the inflamed joint secrete more inflammatory mediators and extracellular matrix lysing enzymes that could destroy the cartilage and bone. AA: arachidonic acid; C1q, C2a, C3, C3a, C4b and C5a and B (factor B): complement components; CCL3: chemokine (C-C motif) ligand 3; COX2: cyclooxygenase-2; EP4: prostaglandin receptor; MASP: mannose-associated serine protease; MBL: mannose-binding lectin; IC: immune complex; IL: interleukin; LTB4: leukotriene B4; FcγR: Fc gamma receptors; PGE2: prostaglandin E2; TGF: transforming growth factor; TNF: tumor necrosis factor.