

Abstract
Mouse hepatitis virus strain A59 (MHV-A59) triggers various pathologies in several mouse strains, including hypergammaglobulinaemia, hepatitis and thymus involution. We reported previously the presence of autoantibodies (autoAb) to liver and kidney fumarylacetoacetate hydrolase (FAH) in sera from mice infected with MHV-A59.
Long-term MHV-infected mice represented a good model of non-pathogenic autoimmune response since the animals were apparently healthy in spite of the presence of autoAb. The aim of this work was to see whether a severe liver injury, which releases endogenous adjuvants, i.e. danger signals, could elicit a broader spectrum of autoAb and perhaps signs of autoimmune hepatitis. Carbon tetrachloride (CCl4) was injected into mice 30 days after MHV infection, and serum was assayed for autoAb and total IgG 20 days later. The association of MHV infection with the toxic effects of CCl4 resulted in hypergammaglobulinaemia and the production of autoAb to various liver and kidney proteins. Histological examination of liver samples showed tissue damages but without significant differences between the animals submitted to MHV + CCl4 and controls, which were either infected by MHV without CCl4, or poisoned by CCl4 in the absence of MHV infection. Those results show that liver injury after viral infection may lead to the spreading of the immune response and to an increase of serum IgG, suggesting that the procedure used herein could simulate the onset of autoimmune hepatitis.
Keywords: Mouse hepatitis virus, Autoimmune response, Autoimmune hepatitis, Fumarylacetoacetate hydrolase
1. Introduction
Viruses have been long associated with autoimmune diseases [1], [2], [3], [4]. It was proposed that these infectious agents trigger an autoimmune response by diverse mechanisms, including polyclonal B-lymphocyte activation, release of sequestered autoantigens (autoAg), antigenic mimicry, modification of self-antigen, epitope spreading of the anti-viral immune response, enhancement of major histocompatibility complex molecule expression, bystander activation or viral persistence [2], [4], [5], [6].
Mouse hepatitis virus strain A59 (MHV-A59) is a corona virus that causes various mouse pathologies, including hepatitis, thymus involution, IgG2a-restricted hypergammaglobulinaemia and transient demyelination [7], [8]. Its ability to infect hepatocytes and to induce hepatitis correlates with the expression of its cellular membrane receptor, CEACAM-1, previously known as Bgp1a or MHVR, on these cells [7]. We have reported the presence of autoantibodies (autoAb) in sera from various mouse strains after MHV-infection [9]. The autoAb were directed to a 40 kDa protein present in mouse liver and kidney identified as fumarylacetoacetate hydrolase (FAH), a soluble cytosolic enzyme that mediates the hydrolytic formation of fumarate and acetoacetate [9]. We later examined whether the autoimmune response to FAH induced by MHV was based on molecular mimicry and whether the epitope spreading [10], [11], [12] was involved.
Overlapping decapeptides corresponding to the entire mouse FAH sequence were prepared using the PEPSCAN method and their reactivities with sera from MHV-infected mice at different times was determined by ELISA. Results indicated that various regions of the enzyme are recognized as soon as 15 days after infection and that the autoimmune response is not restricted to peptides homologous to viral proteins [13]. As the spectrum of peptides recognized by the autoAb of a given mouse did not change significantly with time, it was likely that the MHV-elicited autoimmune response did not induce an epitope recognition spreading. It was suspected that the induction of the autoAb was not solely related to molecular or structural mimicry, but was mainly due to the tissue damages caused by the MHV infection. In other words, the danger signals [14], also called DAMPs (damage-associated molecular patterns) or alarmins, probably played a key role in the autoimmune process.
To further investigate this hypothesis, we thought that increasing the tissue damages caused by the viral infection should amplify the autoimmune response and perhaps lead to autoimmune hepatitis [15]. Since despite the presence of autoAb against liver and kidney FAH, MHV-infected mice remain apparently healthy [9], we associated the MHV infection with the effects of carbon tetrachloride (CCl4), a strong hepatotoxic agent [16], injected 30 days after MHV inoculation. The sera were then assayed for autoAb and total IgG 20 days later. Briefly, this combination of viral infection and a toxic substance led to the spreading of the immune response and to an increase of serum IgG, marked signs of autoimmune hepatitis.
2. Materials and methods
2.1. Mice
The specific-pathogen-free (SPF) female BALB/c mice from the University of La Plata, Argentina, were used at the age of 8–10 weeks. All animals were maintained in isolators, on standard laboratory chow, under SPF conditions until the end of the experiments, and received care in compliance with international legal requirements.
2.2. Preparation of MHV stock
The NCTC 1469 adherent cell line derived from normal mouse liver was purchased from the American Type Culture Collection. Cells growing in T-75 bottles were inoculated with MHV A59 virus at a multiplicity of 1–5 TCID50/cell. After an adsorption period of 1 h at 37 °C, 15 ml of NCTC 135 medium with 10% fetal calf serum was added to each bottle and incubated at 37 °C. Several cycles of freezing and thawing were used to release the virus 24 h after inoculation. The harvested virus was centrifuged at 400 g for 10 min to removed debris and the supernatant was frozen at − 70 °C for storage.
Virus titration by endpoint method was performed by inoculating serial dilutions of the MHV stock onto cell monolayers in 96-multiwell. After 24 h wells with viral cytopathic effect were counted for each dilution and titer was expressed as 50% tissue infectious doses (TCID50).
2.3. Viral infection and carbon tetrachloride (CCl4) treatment
Mice were inoculated intraperitoneally with 104 TCID50 of MHV-A59 grown in NCTC 1469 cells [5]. Thirty days after the infection, the mice were injected intraperitoneally with 100 µl of mineral oil containing 0.16 µl of CCl4 (dilution: 1:625). The animals were bled 24 h and 20 days after treatment. One group of animals was MHV infected and inoculated with CCl4 (“MHV + CCl4”), a second was only infected (“MHV alone”) and the third received only CCl4 (“CCl4 alone”). Serum aspartate aminotransferase (AST) was determined using the GOT(AST) uni-test (Wiener lab., Rosario, Argentina).
2.4. Immunoglobulin assays
For total IgG determination in mouse serum, microplates (Nunc Maxi-Sorb) were coated with 100 µl of phosphate buffer saline (PBS) containing a 1:500 diluted rabbit antiserum directed against mouse Ig. The plates were blocked 1 h at 37 °C with 0.01 M Tris, 0.13 M NaCl, pH 7.4 (TMS) containing 5% of non-fat milk (TMS-M) and were incubated with serial dilutions of mouse serum in the same medium. After 2 h at 37 °C and washing with PBS containing 0.125 ml of Tween 20/l(PBS-Tween), the plates were incubated 1 h at 37 °C with peroxidase labeled anti-mouse IgG Ab. These donkey IgG Ab (Jackson Immunoresearch Laboratories, Inc., West Grove, PA) were used at a 1:10,000 dilution in TMS-M.
2.5. Preparation of organ lysates
Liver and kidneys from non-infected BALB/c mice were removed, soaked in chilled PBS and ground in a Dounce homogenizer at 4 °C with 5 volumes of PBS containing 10− 3 M phenylmethyl-sulfonyl fluoride (PMSF). The homogenates were centrifuged for 15 min at 400 × g and the clarified extracts kept at − 20 °C until used. A sample of each suspension was solubilized by heating for 30 min at 100 °C in 1 M NaOH and protein concentration was determined by the method of Lowry et al. [17].
2.6. Western-blot analysis
Each organ extract (100 µg of protein) was subjected to 10% SDS-PAGE and then transferred onto nitrocellulose sheets (Amersham, Buckingghamshire, UK). After reversible staining with Ponceau S to check satisfactory transfer, non-specific Ab-binding sites were blocked by incubating the sheets with 5% non-fat milk in 30 mM Tris, 0.14 M NaCl, 0.1% (v/v) Tween 20, pH 8.0 (TBS-M-T) for 1 h at room temperature with shaking. The strips were then incubated overnight at 4 °C with an Ab dilution in TBS-M-T. After several washings with TBS containing 0.1% Tween 20, bound Ab were revealed with peroxidase labeled donkey anti-mouse IgG (Jackson Immunoresearch Laboratories, Inc, West Grove, PA, USA) diluted 1:5000 in TBS-M-T and ECL reagents (Amersham, Buckingghamshire, UK). In every experiment a negative control (pool of naïve sera from the same mice used afterward) as well as a positive control (pool sera from MHV-infected mice) were included. To avoid differences in protein expression the same organ extract was used trough all the work. The apparent molecular mass (kDa) of the detected bands was determined using a wide range protein standard (BDH Laboratory Supplies Poole BH15 1TD, UK).
2.7. Histology
Livers from “MHV + CCl4”, “MHV alone” or “CCl4 alone”were cut into blocks and fixed by immersion into 10% formalin in 0.1 M phosphate buffer, pH 7.4. After fixation, the tissues were dehydrated in graded alcohols, and then embedded in paraffin. 5 µm sections were cut, stained with Harris' hematoxylin for 2 min and counterstained with eosin for 2 min. The sections were then washed with distilled water, dehydrated in graded alcohols and xylene, and mounted with Canada Balsam.
3. Results
We have injected carbon tetrachloride (CCl4) at escalating concentrations into 15 BALB/c mice to find the dose that caused a sublethal liver necrosis. It was found that the administration of 0.16 µl of CCl4 in 100 µl of mineral oil raised the serum levels of aspartate amino transferase (AST) 50–100 times over control 24 h after the injection [16], whereas the animals stayed alive for at least a month (data not shown). Mice were bled 24 h and 20 days after the CCl4 administration, and the sera were assayed for total IgG by ELISA and autoantibodies (autoAb) by Western-blot as described before [9]. Serum IgG did not differ from control values and CCl4 did not induce autoAb to mouse liver or kidney extracts detectable by Western-blot (data not shown).
After 30 days of MHV-infection, as reported previously [9], AST and IgG levels in serum were normal, although autoAb to liver FAH were still present in most samples. Thus, to explore the effect of CCl4 in mice virally infected, we injected CCl4 30 days after the viral inoculation.
One day after the CCl4 injection, IgG values, expressed as serum dilution to give an OD value of 1.0, were slightly higher in the group “MHV alone” (228,000 ± 85,000) than in mice non infected and not exposed to CCl4 (100,000 ± 64,000) or in the group “MHV + CCl4” (118,000 ± 67,000) (Fig. 1 A). By contrast, 20 days after the CCl4 injection, the sera from the group “MHV + CCl4” contained significantly higher levels of IgG (400,000 ± 128,000) than non-infected and non-CCl4-exposed mice (100,000 ± 72,000) or the group “MHV alone” (170 000 ± 78 000) (Fig. 1B). As already mentioned, no increase in IgG concentration was observed after 24 h or 20 days of CCl4 treatment alone (Fig. 1A,B).
Fig. 1.
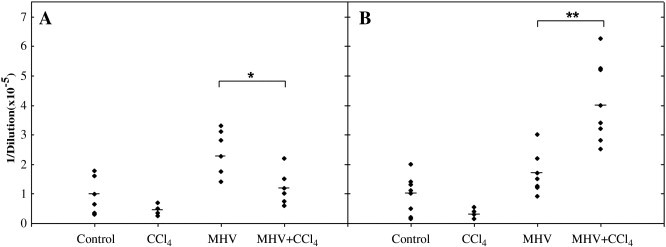
IgG concentration in sera from the three groups of BALB/c mice:controls (non infected and not exposed to CCl4), “CCl4 alone”, “ MHV alone”, “MHV + CCl4”. IgG concentration is expressed as the serum dilution to give an OD = 1.0 in ELISA 24 h (A) or 20 days (B) after the CCl4 injection, i.e., 30 (A) or 50 (B) days after the MHV inoculation. Statistical analysis was performed by the Mann-Whitney U-test. *P < 0.05; **P < 0.001.
Western-blots revealed autoAb to liver and kidney extracts in the group “MHV + CCl4”. The various bands of autoantigens were designated according to their relative mobility (RM) versus the IgG heavy and light chains (values of 1 and 0, respectively) present in the tissue extracts (Fig. 2 A). Representative results from two mice are presented in Fig. 2B. Table 1 presents the RM values for 11 mice “MHV + CCl4” and seven “MHV alone”. Whereas sera from the “MHV + CCl4” group contained autoAb to different liver and kidney antigens, the autoAb of the “MHV alone” group recognized only liver or kidney FAH (Table 1).
Fig. 2.
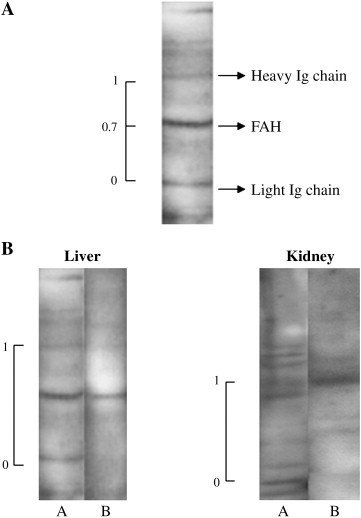
Reactivity of sera from MHV-infected mice, exposed or not to CCl4, with mouse liver and kidney extracts. Tissue lysates were prepared as indicated in Materials and methods and separated by SDS-PAGE in 10% gels, transferred onto nitrocellulose sheets and incubated with 1:50 serum dilution. Bound Ab were revealed by peroxidase-labeled IgG anti-mouse IgG and ECL reagents. (A) Example of the calculation of the Relative Mobility (RM) of the autoantigens. The IgG heavy and light chains already present in tissues were used as conventional markers (values of 1 and 0, respectively) to calculate the RM of the different proteins. In the figure, the FAH position corresponded to a RM of 0.7. (B) Example of Western-blot patterns. A: serum from mouse #3 “MHV + CCl4” (see RM values in Table 1); B: serum from mouse #15 “MHV alone” (see RM values in Table 1). Positions of the IgG heavy and light chains are indicated as 1 and 0, respectively, on the left side of each blot.
Table 1.
Autoantibodies to mouse liver and kidney extracts induced by CCl4 in MHV-infected BALB/c mice.
| Treatment | Serum | Relative mobility (RM) of the autoantigens reacting with the mouse serum(1) |
|
|---|---|---|---|
| Liver(2) | Kidney(2) | ||
| MHV + CCl4 | 1 | 0.4–0.7–1.2 | – |
| 2 | 0.4–0.5–1.8 | ND | |
| 3 | 0.2–0.7–1.1–1.2–1.5 | 0.1–0.3–0.8–1.4–1.6 | |
| 4 | 0.7–1.4 | 1.3 | |
| 5 | 0.1–0.7–1.2 | 0.7–0.9–1.2–1.4 | |
| 6 | 0.1–0.5–0.7 | 0.7–0.9–1.2 | |
| 7 | 0.2–0.3–0.6–0.9 | ND | |
| 8 | – | ND | |
| 9 | – | ND | |
| 10 | 0.7–1.1–1.3–1.6–1.7 | 0.7–0.9–1.2 | |
| 11 | 0.7 | – | |
| MIV | 12 | 0.7 | – |
| 13 | – | 1.2 | |
| 14 | – | 0.5 | |
| 15 | 0.7 | 0.7–1.2 | |
| 16 | – | – | |
| 17 | – | – | |
Tissue extracts, separated by SDS-PAGE in 10% gels, were transferred onto nitrocellulose sheets and incubated with 1:50 serum dilution. Bound Ab were revealed by peroxidase-labeled IgG anti-mouse IgG and ECL reagents (see Materials and methods).
(1) Relative Mobilities (RM) of the autoantigens were calculated as explained in Fig. 2A.
(2) RM values 0.6–0.8 correspond to the FAH protein.
ND: not done.
In the three groups, “MHV + CCl4”, “MHV alone”, and “CCl4 alone”, histology of the liver, 20 days after the CCl4 injection and 50 days after MHV inoculation, showed the following damages: cell swelling, necrosis, and hydropic and hepatocellular degeneration (Fig. 3 ). However, neither infiltrating cells nor fibrosis were detected, and close examination of several liver samples did not find significant differences in the damage extent between the group “MHV + CCl4” and the two other groups, either “MHV alone” or “CCl4 alone” (Fig. 3). Some MHV-infected mice died a few days after the CCl4 injection. It is quite possible that the liver damages were more severe in those mice, but histology was not obtained from these animals.
Fig. 3.
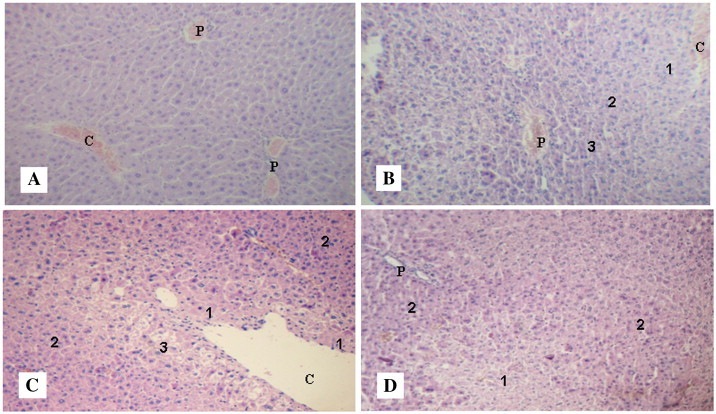
Representative micrographs of hematoxylin-eosin stained liver sections. Magnification: 10×. See the procedures in Materials and methods. (A) Control mice (no treatment). C: central vein; P: portal area. (B) Mice from the group “CCl4 alone”. Centrilobular degeneration (1); mid-zonal degeneration (2); perilobular swelling (3); C: central vein; P: portal area. (C) Mice from the group “MHV alone”. Necrosis (1); cellular swelling (2); hydropic degeneration (3); C: central vein. (D) Mice from the group “MHV + CCl4”. Necrosis (1); cellular swelling (2); P: portal area.
4. Discussion
We have reported that mice infected with MHV produced autoAb to mouse liver and kidney FAH [9], [13]. Competition assays indicated that the autoAb detected both conformational and cryptic FAH epitopes in ELISA but only cryptic determinants in Western-blot assays, whereas anti-MHV Ab were directed to native epitopes of the viral proteins [18].
The Danger model proposes that the immune system is more concerned with damage than with foreignness, and is called into action by alarm signals from injured tissues rather than by the recognition of non-self [14]. Thus, it was suggested that structural features of autoAg, their locations, and catabolism during cell death and their translocation to cells that can present antigens to the immune system could contribute to selection of the autoimmune repertoire [14], [19]. Moreover, there is evidence supporting the concept that the immune system has also evolved mechanisms to sense primary and secondary cell death and respond to it with innate and adaptive immune response [20].
MHV is known to be lymphotropic and to induce diverse alterations of immune responses that depend on the mouse genetic background [5], [20], [21]. Thus, a model of MHV-induced anti-FAH autoAb was based on antigen mimicry – FAH N-terminal sequence is about 50% homologous with a MHV protein – together with the alarm signals released by the MHV-injured liver. In fact, inoculation of susceptible mice with enterotropic virus such as MHV-A59 causes liver lesions as early as 40 h after infection. The injury during the acute phase of hepatitis includes liver focal necrosis, cells undergoing hyaline degeneration and polymorphonuclear leukocytes invasion [22]. Liver regeneration may take place 10–14 days after infection, ranging from complete curing to chronic damage with intermediate stages [22]. On the other hand, CCl4 metabolization by cytochrome P450 in the hepatocytes produces the highly reactive trichloromethyl radical, which leads to lipid peroxidation and membrane damage, as well as inflammatory response and secretion of cytokines that attract and activate neutrophils [23]. Intriguingly, even if both liver injuries produce violent cell death and thus the potential release of endogenous adjuvants (damage-associated molecular patters, or DAMPs) [20], CCl4 did not elicited an autoimmune process, and only autoAb restricted to FAH were detected after MHV infection.
In this report we show that a hepatotoxic drug such as CCl4 inoculated when the MHV infection was resolved, is able to induce a spreading of the autoimmune response elicited by the virus, leading to increased levels of serum IgG (Fig. 1) and the production of a variety of autoAb directed to different liver and kidney proteins (Table 1). Unexpectedly, no enhancement of tissue liver damage compared with each treatment alone was observed (Fig. 3). Thus, although the autoAg recognized by mouse sera were not identified, the autoimmune response shown in this work was somewhat comparable to that described in the various animal models of autoimmune hepatitis (AIH) reported in the literature [15], [24], [25], [26], [27].
In humans, AIH is characterized by the presence of interface hepatitis and portal plasma cell infiltration, hypergammaglobulinemia, and autoAb. It has been proposed that this disorder results from a complex interaction between triggering factors, autoantigens, genetic predisposition, and immunoregulatory networks [28].
Although the liver is known to be a classical immunoprivileged site, it was recently shown to be susceptible to nonspecific activation of innate immunity through Toll-like receptor 3 signaling [29]. Moreover, it was demonstrated that regulatory CD4+CD25+ T cells (Tregs) are defective numerically and functionally in AIH [30] and that protection from autoimmune neuroinflammation induced by the ectopic expression of myelin basic protein (MBP) in the liver was mediated by MBP-specific Tregs [31].
Most animal models to simulate AIH are based on rather complex ways of disease induction, such as xenoimmunization with human antigens [24], [25], the use of adenovirus vectors [15], [27] or transgenic mice [26]. Here we show that a simple and common phenomenon, i.e., a viral infection followed by a toxic liver injury, may induce certain signs of AIH disease. Additionally, current data indicate that some endogenous danger signals, or DAMPs, may be involved in the production of autoAb to FAH induced by MHV infection [Duhalde Vega et al., manuscript in preparation]. Besides, it has been reported that uric acid could augment humoral immune responses [32] and that various Toll-like receptors associated with the modulation of Tregs may bind alarmins – such as heat shock proteins, high-mobility group Box 1 proteins and chromatin-Ig complexes – liberated by injured cells [33], [34]. Thus, it is suggested that endogenous adjuvants liberated by the virus and the hepatotoxic agent could join some Toll-like receptors and inhibit Tregs so inducing some of the AIH signs described in this work.
Acknowledgements
The authors are indebted to Dr. Pierre L. Masson (de Duve Institute, Université catholique de Louvain, Brussels, Belgium) for helpful discussions and critical revision of the manuscript. This work was supported by grants from CONICET, FONCYT and Universidad de Buenos Aires, Argentina.
References
- 1.Cohen A.D., Shoenfeld Y. The viral-autoimmunity relationship. Viral Immunol. 1995;8:1–9. doi: 10.1089/vim.1995.8.1. [DOI] [PubMed] [Google Scholar]
- 2.Lawson C.M. Evidence for mimicry by viral antigens in animal models of autoimmune disease including myocarditis. Cell Mol Life Sci. 2000;57:552–560. doi: 10.1007/PL00000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kohm A.P., Fuller K.G., Miller S.D. Mimicking the way to autoimmunity: an evolving theory of sequence and structural homology. TRENDS Microbiol. 2003;11:101–105. doi: 10.1016/s0966-842x(03)00006-4. [DOI] [PubMed] [Google Scholar]
- 4.Fujinami R.S., von Herrath M.G., Christen U., Whitton J.L. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microb Rev. 2006;19:80–94. doi: 10.1128/CMR.19.1.80-94.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coutelier J.-P., Coulie P.G., Wauters P., Heremans H., van der Logt J.T.M. In vivo polyclonal B-lymphocyte activation elicited by murine viruses. J Virol. 1990;64:5383–5388. doi: 10.1128/jvi.64.11.5383-5388.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rose N.R. The role of infection in the pathogenesis of autoimmune disease. Semin Immunol. 1998;10:5–13. doi: 10.1006/smim.1997.0100. [DOI] [PubMed] [Google Scholar]
- 7.Godfraind C., Coutelier J.-P. Morphological analysis of mouse hepatitis virus A59-induced pathology with regard to viral receptor expression. Histol Histopathol. 1998;13:181–199. doi: 10.14670/HH-13.181. [DOI] [PubMed] [Google Scholar]
- 8.Lardans V., Godfraind C., van der Logt J.T.M., Heessen F.W.A., Gonzalez M.D., Coutelier J.P. Polyclonal B lymphocyte activation induced by mouse hepatitis virus A59 infection. J Gen Virol. 1996;77:1005–1009. doi: 10.1099/0022-1317-77-5-1005. [DOI] [PubMed] [Google Scholar]
- 9.Mathieu P.A., Gómez K.A., Coutelier J.P., Retegui L.A. Identification of two liver proteins recognized by autoantibodies elicited in mice infected with mouse hepatitis virus A59. Eur J Immunol. 2001;31:1447–1455. doi: 10.1002/1521-4141(200105)31:5<1447::AID-IMMU1447>3.0.CO;2-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farris A.D., Keech C.L., Gordon T.P., McCluskey J. Epitope mimics and determinant spreading: pathways to autoimmunity. Cell Mol Life Sci. 2000;57:569–578. doi: 10.1007/PL00000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croxford J.L., Olson J., Miller S.D. Epitope spreading and molecular mimicry as triggers of autoimmunity in the Theiler's virus induced demyelinating disease model of multiple sclerosis. Autoimmun Rev. 2000;1:251–260. doi: 10.1016/s1568-9972(02)00080-0. [DOI] [PubMed] [Google Scholar]
- 12.Liang B., Mamula M.J. Molecular mimicry and the role of B lymphocytes in the processing of autoantigens. CMLS, Cell Mol Life Sci. 2000;57:561–568. doi: 10.1007/PL00000718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duhalde-Vega M., Loureiro M.E., Mathieu P.A., Retegui L.A. The peptide specificities of the autoantibodies elicited by mouse hepatitis virus A59. J Autoimmun. 2006;27:203–209. doi: 10.1016/j.jaut.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matzinger P. The Danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 15.Christen U., Holdener M., Hintermann E. Animal models for autoimmune hepatitis. Autoimmun Rev. 2007;6:306–311. doi: 10.1016/j.autrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Smith C.I., Cooksley W.G.E., Powell L.W. Cell-mediated immunity to liver antigens in toxic liver injury. I. Occurrence and specificity. Clin Expl Immunol. 1980;39:607–617. [PMC free article] [PubMed] [Google Scholar]
- 17.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurements with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 18.Mathieu P.A., Gómez K.A., Coutelier J.P., Retegui L.A. Sequence similarity and structural homologies are involved in the autoimmune response elicited by mouse hepatitis virus A59. J Autoimmun. 2004;23:117–126. doi: 10.1016/j.jaut.2004.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Plotz P.H. The autoantibody repertoire: searching for order. Nat Rev Immunol. 2003;3:73–78. doi: 10.1038/nri976. [DOI] [PubMed] [Google Scholar]
- 20.Kono H., Rock K.L. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–289. doi: 10.1038/nri2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coutelier J.P., Coulie P.G., Wauters P., Heremans H., van der Logt J.T.M. In vivo polyclonal B-lymphocyte activation elicited by murine viruses. J Virol. 1990;64:5383–5388. doi: 10.1128/jvi.64.11.5383-5388.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godfraind C., Havaux N., Holmes K.V., Coutelier J.-P. Role of virus receptor-bearing endothelial cells of the blood-brain barrier in preventing the spread of mouse hepatitis virus-A59 into the central nervous system. J Neurovirology. 1997;3:428–434. doi: 10.3109/13550289709031188. [DOI] [PubMed] [Google Scholar]
- 23.Louis H., Van Laethem J.-L., Wu W., Quertinmont E., Degraef C., Van den Berg K. Interleukin-10 controls neutrophilic infiltration, hepatocyte proliferation and liver fibrosis induced by carbon tetrachloride in mice. Hepatology. 1998;28:1607–1615. doi: 10.1002/hep.510280621. [DOI] [PubMed] [Google Scholar]
- 24.Mori Y., Mori T., Yoshida H., Ueda S., Iesato K., Wakashin Y. Study of cellular immunity in experimental autoimmune hepatitis in mice. Clin Exp Immunol. 1984;57:85–92. [PMC free article] [PubMed] [Google Scholar]
- 25.Lapierre P., Djilali-Saiah I., Vitozzi S., Alvarez F. A murine model of type 2 autoimmune hepatitis: xenoimmunization with human antigens. Hepatology. 2004;39:1066–1074. doi: 10.1002/hep.20109. [DOI] [PubMed] [Google Scholar]
- 26.Voehringer D., Blaser C., Grawitz A.B., Chisari F.V., Buerki K., Pircher H. Break of T cell ignorance to a viral antigen in the liver induces hepatitis. J Immunol. 2000;165:2415–2422. doi: 10.4049/jimmunol.165.5.2415. [DOI] [PubMed] [Google Scholar]
- 27.Holdener M., Hintermann E., Bayer M., Rhode A., Rodrigo E., Intereder G. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J Exp Med. 2008;205:1409–1422. doi: 10.1084/jem.20071859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czaja A.J. Understanding the pathogenesis of autoimmune hepatitis. Am J Gastroenterol. 2001;96:1224–1231. doi: 10.1111/j.1572-0241.2001.03707.x. [DOI] [PubMed] [Google Scholar]
- 29.Lang K.S., Georgiev P., Recher M., Navarini A.A., Bergthaler A., Heikenwalder M. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J Clin Invest. 2006;116:2456–2463. doi: 10.1172/JCI28349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Longhi M.S., Hussain M.J., Mitry R.R., Arora F.K., Mieli-Vergani G., Vergani D. Functional study of CD4+CD25+ regulatory T cells in health and autoimmune hepatitis. J Immunol. 2006;176:4484–4491. doi: 10.4049/jimmunol.176.7.4484. [DOI] [PubMed] [Google Scholar]
- 31.Lüth S., Huber S., Schramm C., Buch T., Zander S., Stadelmannet C. Ectopic expression of neural autoantigen in mouse liver suppresses experimental autoimmune neuroinflammation by inducing antigen-specific Tregs. J Clin Invest. 2008;118:3403–3410. doi: 10.1172/JCI32132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Behrens M.D., Wagner W.M., Krco C.J., Erskine C.L., Kalli K.R., Krempski J. The endogenous danger signal, crystalline uric acid, signals for enhanced antibody immunity. Blood. 2008;111:1472–1479. doi: 10.1182/blood-2007-10-117184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu G., Zhao Y. Toll-like receptors and immune regulation: their direct and indirect modulation on regulatory CD4+CD25+ T cells. Immunology. 2007;122:149–156. doi: 10.1111/j.1365-2567.2007.02651.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marshak-Rothstein A., Rifkin I.R. Immunologically active autoantigens: the role of Toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]