

Highlights
• Massachusetts-type is the most prevalent serotype of infectious bronchitis virus worldwide. • Analysis of genomic sequences provides more information about occurrence, origin and evolution. • Pathogenic, non-pathogenic and recombinant Mass-type IBVs are co-existing in chicken flocks in China. • Data of Mass-type from isolates in China can help to explain the emergence of Mass-type IBV.
Keywords: Avian infectious bronchitis coronavirus, Mass-type, Mutation, Recombination event, Genome
Abstract
Four Massachusetts-type (Mass-type) strains of infectious bronchitis coronavirus (IBV) were compared genetically with the pathogenic M41 and H120 vaccine strains using the complete genomic sequences. The results revealed that strains ck/CH/LNM/091017 and ck/CH/LDL/101212 were closely related to the H120 vaccine, which suggests that they might represent re-isolations of vaccine strains or variants of vaccine strains that have resulted from the accumulated point mutations after several passages in chickens. In contrast, strains ck/CH/LHLJ/07VII and ck/CH/LHLJ/100902 had a close genetic relationship with the pathogenic M41 strain. In addition, molecular markers have been identified that distinguish between field and vaccine (or vaccine-like) Mass-type viruses, which may be able to differentiate between field and vaccine strains for diagnostic purposes. Phylogenetic analysis, and pairwise comparison of full-length genomes and the nine genes, identified the occurrence of recombination events in the genome of strain CK/VH/LHLJ/07VII, which suggests that this virus originated from recombination events between M41- and H120-like strains at the switch site located at the 3′ end of the nucleocapsid (N) genes. To our knowledge, this is the first time that evidence for the evolution and natural recombination under field conditions between Mass-type pathogenic and vaccinal IBV strains has been documented. These findings provide insights into the emergence and evolution of the Mass-type IB coronaviruses and may help to explain the emergence of Mass-type IBV in chicken flocks all over the world.
1. Introduction
In 1931, Schalk and Hawn described “an apparently new respiratory disease of chicks” in North Dakota in the United States, which was considered to be infectious bronchitis (IB) by later researchers of avian respiratory diseases (Schalk and Hawn, 1931). Currently, IB still occurs in nearly all poultry-producing countries; it is a highly contagious, acute, and economically important viral disease of chickens. The etiology of IB, which was first demonstrated by Beach and Schalm (1936), is infectious bronchitis virus (IBV).
IBV is grouped in the genus Gammacoronavirus of the family Coronaviridae in the order Nidovirales (de Groot et al., 2011). The coronavirus genomes are the largest among the known RNA viruses and are polycistronic, generating a nested set of subgenomic RNAs with common 5′ and 3′ sequences (Masters, 2006). Like those of all other coronaviruses, the 5′ two-thirds of the IBV genome consists of two large replicase open reading frames (ORFs), ORF1a and ORF1b. The ORF1a polyprotein (pp1a) can be extended with ORF1b-encoded sequences via a −1 ribosomal frameshift at a conserved slippery site (Brierley et al., 1987), which generates the polyprotein pp1ab, comprising more than 7000 amino acids, which includes the putative RNA-dependent RNA polymerase (RdRp) and RNA helicase (HEL) activity (Ziebuhr et al., 2001). The pp1a and pp1ab of IBV are processed autocatalytically by two different viral proteases, encoded by a papain-like protease (PLP) and a 3C-loke protease (3CLpro) (Lee et al., 1991, Ziebuhr et al., 2000, Ziebuhr et al., 2001). Other putative domains, presumably associated with a 3′-to-5′ exonuclease (ExoN) activity, a poly(U)-specific endo-RNase (XendoU) activity, and a 2′-O-methyltransferase (2′-O-MT) activity, have been predicted in pp1ab (Ivanov et al., 2004, Snijder et al., 2003). The 3′ end of a coronavirus genome includes the viral structural and accessory protein genes: a spike (S) glycoprotein gene; an envelope (E) protein gene; a membrane (M) glycoprotein gene; a nucleocapsid (N) phosphoprotein gene; and several ORFs that encode putative non-structural accessory proteins (Masters, 2006). Of the virus-encoded proteins, the S1 subunit of the S protein carries virus-neutralizing activity, determines the serotype of IBV and is responsible for viral attachment to cells. It is also a major determinant of cell tropism in culture (Casais et al., 2003). The accumulation of point mutations, deletions, insertions and recombination events that have been observed in multiple structural genes, especially the S1 gene, of IBV recovered from naturally occurring infections have been considered to contribute to the genetic diversity and evolution of IBV, and consequently, to a number of IBV serotypes (Cavanagh, 2007).
The occurrence and emergence of multiple serotypes of the virus have complicated control by vaccination because many serotypes and variants do not confer complete cross-protection against each other (Cavanagh and Gelb, 2008). The originally discovered Massachusetts (Mass) type of IBV was identified in the United States, beginning in the 1950s (Fabricant, 2000, Johnson and Marquardt, 1975, Mondal et al., 2001). Mass-type strains have been isolated in Europe and Asia since the 1950s and up to the present day (Cavanagh and Gelb, 2008), together with dozens of other serotypes that have been isolated in Africa, Asia, India, Australia, Europe, and South America (Cavanagh, 2001, Cavanagh, 2003, Cavanagh, 2005). The first Mass-type “H” vaccines were developed in about 1960. They include H120 and H52 (Bijlenga et al., 2004), and are used very commonly and widely around the world. However, virus of this type is occasionally isolated from Massachusetts-vaccinated and -unvaccinated flocks with respiratory clinical signs. Some of the viruses have shown close genetic relationships with pathogenic Mass-type, rather than vaccine, strains by S1 gene analysis. However, conclusions based on the genetic analysis of a single gene sequence, and sometimes even a partial gene sequence, require caution because the true phylogeny can only be demonstrated by analyzing complete genomic sequences. Herein, we sequenced the complete genome of four IBV Mass-type strains that showed S1 gene diversity (Liu et al., 2009, Ma et al., 2012, Sun et al., 2011), and we present evidence for in-field recombination between pathogenic and vaccinal strains. Furthermore, we characterized the molecular variability of the four Mass-type strains to gain insight into the emergence and evolution of these viruses.
2. Materials and methods
2.1. Virus strains
Four Mass-type IBV strains were used for complete genomic sequence comparison and analysis in this study. Strain ck/CH/LHLJ/07VII was isolated in 2007 from the kidney of a layer hen vaccinated with H120 and 4/91 in Heilongjiang province, China (Liu et al., 2009). Strain ck/CH/LNM/091017 was isolated in 2009 from the swollen proventricular tissues of a broiler vaccinated with H120 in Neimenggu province, China (Sun et al., 2011). Strains ck/CH/LDL/101212 and ck/CH/LHLJ/100902, both of which were isolated in 2010, were obtained from laying hens in Dalian and Heilongjiang provinces, respectively, in China; the birds were suffering from nephropathogenic lesions and respiratory signs, respectively. In addition, the diseased birds in both flocks were suffering from proventriculitis (Ma et al., 2012). All of the IBV strains have been associated with various IB outbreaks in recent years in China and were assigned to the Mass-type strains by S1 sequence analysis. To avoid the possible mutation in the viral genome after serial passages in specific-pathogen-free (SPF) embryonated chicken eggs, the first passage of each original virus stock was used and purified once by propagating in 9- to 11-day-old SPF chicken eggs with a dose of l04-fold dilutions per egg, and the presence of viral particles in the allantoic fluids of inoculated eggs was confirmed with a negative contrast electronic microscope (JEM-1200, EX) and reverse transcriptase-polymerase chain reaction (RT-PCR) as described previously (Han et al., 2011). In addition, since these viruses were isolated from chickens vaccinated with H120, it is possible that mixed IBV infections are present in one chicken flock. To exclude this, nine clones of S1 gene of each virus obtained from three independent PCR reactions were sequenced and compared. Sequences of each virus identical to the previously results were obtained (Liu et al., 2009, Ma et al., 2012, Sun et al., 2011).
2.2. Eggs
Fertile White Leghorn SPF chicken eggs were obtained from the Laboratory Animal Center, Harbin Veterinary Research Institute, the Chinese Academy of Agricultural Sciences, China.
2.3. RNA extraction and RT-PCR
To determine the full-length genomic sequences of the four viruses, 15 pairs of overlapping primers encompassing the entire genome were used. The primers were designed in regions that are conserved among most of the IBV strains available in the GenBank database. The sequences and locations of the primers used in RT and PCR in this study are presented in Table 1 .
Table 1.
Oligonucleotide primers used for IBV genome amplification and their positions in the genome.
| Primer | Sensea | Sequence (5′ to 3′) | Position in genomeb |
|---|---|---|---|
| IBV-366 | + | AACCCAAAAGATTACGCTGATGCTT | 640–664 |
| IBV-367 | − | CCAGAATTCAAAAGACTTTTCAGTA | 1887–1911 |
| IBV-368 | + | GATGTCTTGAAGCTGTTTCAATC | 1828–1850 |
| IBV-369 | − | AAGAATGTGCTTGTAGATGGCGT | 3817–3839 |
| IBV-370 | + | TATGTTAAGAAACATGGGCCAC | 3685–3706 |
| IBV-371 | − | TACATTTATGTCTCCTGAAGTTGCTGG | 6405–6431 |
| IBV-372 | + | GTAAGAGACATAATTGGTATTG | 6353–6374 |
| IBV-373 | − | GGTGCAGTTTTAATTTTACAAACTGC | 9052–9077 |
| IBV-377 | + | CTTAATCTTGCTAATAATCATGAG | 8971–8994 |
| IBV-378 | − | GTGTACAACTGTTGTTTTAATGCAGC | 10,686–10,711 |
| IBV-379 | + | GTGTGGGAAGTCTTTTCGACAAATATAC | 10,588–10,615 |
| IBV-380 | − | GGACGTTGTCAATTTAAAGGTTC | 12,100–12,122 |
| IBV-381 | + | CTCCTGATCAGGATTCTTATGGAGGAGC | 12,011–12,038 |
| IBV-382 | − | CCTGTGGCTGTTATGGAGCGTTATAT | 14,835–14,860 |
| IBV-383 | + | CATTTTGGGTGCATGTGTTTTTGTAGATG | 14,789–14,817 |
| IBV-384 | − | GACACCATGTGACAAACACTACACAATCTG | 17,407–17,436 |
| IBV-375 | + | CCTTGGCATGTTATAAGACCAAGGATAG | 17,346–17,373 |
| IBV-376 | − | CGCTCCATCTTTTAATGGCATACCATTCTG | 18,903–18,932 |
| IBV-374 | + | GTTCTGTATGATGATAGATATGGTGATTACC | 18,780–18,810 |
| IBV-105-1 | − | CCAAAGTGCCTTTAGACCGGCTG | 20,388–20,412 |
| IBV-257 | + | TATTGATTAGAGATGTGG | 20,356–20,373 |
| IBV-275 | − | GTTACAGATGAGTACATAC | 22,147–22,165 |
| IBV-167 | + | GCTTCTTGAGAA(T/C)CAGTTTTA | 21,921–21,941 |
| IBV-168 | − | AGACGATCAACTTGTGCATCTG | 22,952–22,973 |
| IBV-182 | + | GACATTTAC(C/G)(A/C)GCAACTTGA | 22,921–22,940 |
| IBV-281 | − | GCCACTGACC(C/A)TCACAATAAAG | 24,955–24,976 |
| IBV-280 | + | CCC(C/A)GAATCTAATGCCGTAGG | 24,846–24,866 |
| IBV-171 | − | AACCAAGATGCATTTCCAGA | 25,960–25,979 |
| N(+) | + | GACGCCCCAGCGCCAGTCATTAAA | 25,903–25,926 |
| N(−)c | − | ACGCGGAGTACGATCGAGGGTACA | 27,484–27,507 |
Negative-sense (−) or positive-sense (+) primer.
Nucleotide positions corresponded to the sequence of the IBV Beaudette genome, GenBank accession number M95169.
Primer N(−) was used for RT.
Viral RNA was extracted from 200 μl of infectious allantoic fluid using TRIzol reagents (Invitrogen, Grand Island, USA), following the manufacturer’s protocol. Complementary DNA (cDNA) was synthesized using 80 μl of the first strand mixture (Invitrogen) containing 20 μM of primers N (−), 0.5 mM each of dNTP (TaKaRa, Dalian, China) and 40 μl of total RNA. The mixture was incubated at 70 °C for 5 min and then quick-chilled on ice for 2 min. The RT master mix was composed of 16 μl 5× RT buffer (Invitrogen), 8 μl 10 mM DTT, 200 U of M-MLV Reverse Transcriptase (Invitrogen), and 20 U RNAse inhibitors (Invitrogen). This RT master mix was incubated at 37 °C for 3 h. The reaction was terminated by heating at 70 °C for 10 min then chilling on ice for 5 min.
The PCR was performed in a 25 μl reaction containing 2 μl first strand cDNA; 15 nmol each of downstream and upstream primers; 5 μl of 10× PCR buffer (Mg2+ Plus, TaKaRa); 4 μl of 2.5 mmol dNTPs; 2 U Taq polymerase (TaKaRa); and 18 μl of water. The reaction was conducted at 95 °C for 5 min, and 30 cycles of 94 °C for 1 min; 50 °C for 1 min; 72 °C for 2 min, and a final extension step of 72 °C for 10 min. A product, detectable by ethidium bromide staining, of the expected size was generated.
2.4. The 5′- and 3′-ends of the genome
A cDNA clone representing the 5′ and 3′ ends of the genome of the four IBV strains was synthesized according to the 5′ RACE and 3′ RACE System for rapid amplification of cDNA ends (TaKaRa). PCR was performed according to the instructions accompanying the kits. The sense and antisense primers used to amplifying the 5′- and 3′-ends of the genome had been designed on the basis of the sequences obtained above that were constant in the four IBV strains, respectively. The outer and inner primers used to amplify the 5′-end of the four IBV strains were 5′-CAGCTATGGCAATGCGCAG-3′ and 5′-CATCTTTGGTGTCTCA/TCC-3′, respectively. The primer used to amplify the 3′-end was 5′-GAGGAGAGGAACAATGCACA-3′.
2.5. DNA cloning and sequence determination
The DNA generated by PCR amplification was cloned using a T-tailed vector, pMD18-T (TaKaRa), and transformed using JM109 competent cells (TaKaRa) according to the manufacturer’s instructions. At least five clones of each fragment in each strain were sequenced and the consensus sequence was determined. The sequences were analyzed using the Sequencher 4.5 sequence analysis program, and a single contiguous sequence comprising the entire IBV genome of each of the four IBV strains was constructed.
2.6. Phylogenetic relationships and sequence comparison
The nucleotide and amino acid sequences of the entire genome of the four IBV strains were assembled, aligned, and compared with those of other reference IBV and Turkey coronavirus (TCoV) strains using the MEGALIGN program in DNAStar (version 7, Lasergene Corp, Madison, WI). The ORFs were determined using the Gene Runner program version 3.00 (http://www.generunner.com) by comparison with those of other reference IBV and TCoV strains. A total of 39 IBV and 7 TCoV reference strains, for which entire genomic sequences were available in GenBank database, were selected for phylogenetic analysis of full-length genomes. The selected avian coronavirus reference strains and their accession numbers are provided in Table 2 . Phylogenetic analysis, accurate estimation and comparison of the 5′-UTR, Gene 1, S1, S2, Gene 3, M, Gene 5, N and 3′-UTR of the four IBV strains was conducted with those of the Mass-type strains selected in this study using the Clustal V method of DNAStar software and MEGA4 (Liu et al., 2009), and the alignments were edited manually and adjusted to remove mistakes. Deletion, insertion and gene recombination were determined according to the results of the phylogenetic analysis and pairwise comparisons.
Table 2.
IBV and TCoV strains used for genome sequence comparison in the present study.
| Virus | GenBank accession Nos. | Virus | GenBank accession Nos. |
|---|---|---|---|
| A2 | EU526388 | CK/CH/LDL/97I | AF345267 |
| SAIBK | DQ288927 | Gray | GU393334 |
| SC021202 | EU714029 | FL18288 | GU393333 |
| BJ | AY319651 | Conn46 1991 | FJ904719 |
| CK/CH/LSD/05I | EU637854 | DY07 | HM245923 |
| Partridge/GD/S14/2003 | AY646283 | GX-YL 5 | HQ848267 |
| Peafowl/GD/KQ6/2003 | AY641576 | CQ04 | HM245924 |
| ZJ971 | EU714028 | GX-YL 9 | HQ850618 |
| TW2575/98 | DQ646405 | JMK | GU393338 |
| H52 | EU817497 | Cal 1995 | FJ904714 |
| H120 | FJ888351 | Georgia 1998 Vaccine | GQ504723 |
| Mass 41 | AY851295 | Mass41 1965 | FJ904720 |
| California 99 | AY514485 | Delaware 072 | GU393332 |
| Beaudette | NC_001451 | Arkansas DPI | GQ504720 |
| ArkDPI11 | EU418976 | ck/CH/LZJ/111113 | JX195176 |
| Iowa 97 | GU393337 | Sczy3 | JF732903 |
| TCoV/MG10 | NC_010800 | Holte | GU393336 |
| TCoV/TX/1038/98 | GQ427176 | ck/CH/LDL/091022 | JX195175 |
| TCoV/IN/517/94 | GQ427175 | YN | JF893452 |
| TCoV/TX/GL/01 | GQ427174 | Cal56b | GU393331 |
| TCoV/VA/74/03 | GQ427173 | ck/CH/LHB/100801 | JF330898 |
| TCoV/540 | EU022525 | ITA/90254/2005 | FN430414 |
| TCoV/ATCC | EU022526 | NGA/A116E7/2006 | FN430415 |
2.7. GenBank accession numbers
The full genomic sequences of the four Mass-type IBV strains described in this report have been deposited in the GenBank database with accession numbers ck/CH/LNM/091017 JF330899, CK/CH/LHLJ/07VII JF274479, ck/CH/LDL/101212 JF828981 and CK/CH/LHLJ/100902 JF828980.
3. Results
3.1. Complete genomic sequence and organization of the four Mass-type IBV strains
Four Mass-type IBV strains were subjected to genome sequencing and phylogenetic analysis in this study. The sequences of each the four strains were assembled into one contiguous sequence to represent the entire viral genomes. Sequences of 27630, 27678, 27630 and 27473 nucleotides were obtained from strains ck/CH/LNM/091017, ck/CH/LHLJ/07VII, ck/CH/LDL/101212 and ck/CH/LHLJ/100902, respectively, excluding the polyadenylation tail at the 3′ end. The genomes of the viruses were similar overall in their coding capacity and genomic organization to those of other IBVs. The genome of each of the viruses contained two large slightly overlapping ORFs in the 5′ two-thirds of the genome and multiple additional ORFs in the 3′ one-third of the genome. Both termini were flanked with untranslated regions (UTRs). Ten ORFs were identified within the genome. Gene 1 contained motifs common to all coronaviruses, including ribosomal frameshifting and slippery sequences, because ORF1b is translated in the −1 frame. The typical coronavirus structural genes encoding the S, E, M and N proteins were identified following Gene 1 (Fig. 1 ). The genome organization was determined to be as follows: 5′-UTR-Gene 1 (ORF1a, 1b)-S-Gene 3 (ORFs 3a, 3b, E)-M-Gene 5 (ORFs 5a, 5b)-N-UTR-3′.
Fig. 1.
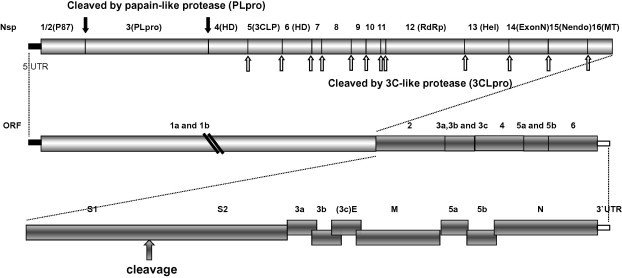
Genome organization of four Mass-type IBV isolates. Diagrammatic representation of the genome organization shows the predicted genes and their relative sizes and positions along the IBV genome. S, spike glycoprotein gene; 3a, 3b, and 3c (E), tricistronic gene 3; M, membrane protein gene; 5a and 5b, bicistronic gene 5; N, nucleocapsid protein gene; UTR, untranslated region. Top: expanded representation of the two ORFs (ORF1a and ORF1b) comprising the polycistronic Gene 1 and the likely cleavage products and cleavage sites after proteolytic processing of the 1a/1b polyprotein. Bottom: expanded representation of the 3′-end structural and accessory protein genes.
3.2. Strains ck/CH/LNM/091017 and ck/CH/LDL/101212 are closely related to the H120 vaccine
The analysis of the complete genome showed that strains ck/CH/LNM/091017 and ck/CH/LDL/101212 possessed 99.9% and 99.8% nucleotide identity with H120, respectively. However, they shared 91.3% and 91.4% identity with M41, respectively. Phylogenetic analysis using the full-length genome and the 5′-UTR, Gene 1, S1, S2, Gene 3, M, Gene 5, N and 3′-UTR showed that the IBV strains ck/CH/LNM/091017 and ck/CH/LDL/101212 consistently formed the same clade with vaccine-related strains of Mass-type (Fig. 2, Fig. 3 ). The analysis of the S1 gene showed that strains ck/CH/LNM/091017 and ck/CH/LDL/101212 had high nucleotide identities (99.8% and 99.8%, respectively) with H120, while they had 97.7% and 97.7% identity with M41. Multiple alignments revealed that there were 2 and 1 nucleotide mutations within the S1 gene between strains ck/CH/LNM/091017 and ck/CH/LDL/101212 and H120; however, there were 36 and 39 mutations between strains ck/CH/LNM/091017 and ck/CH/LDL/101212 and M41. All these results suggest that strains ck/CH/LNM/091017 and ck/CH/LDL/101212 are closely related to the H120 vaccine strain.
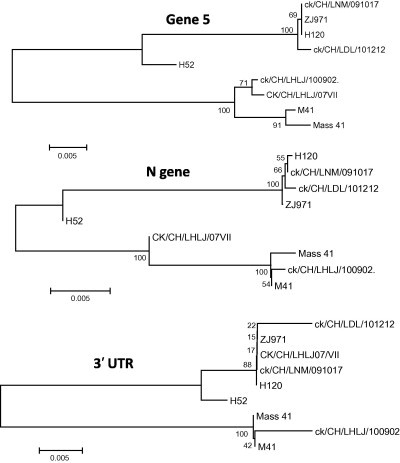
Fig. 2.

Phylogenetic tree constructed on the basis of the full-length genomes of the IBV isolates and Turkey coronavirus (TCoV) reference strains using the neighbor-joining method. IBV strains ck/CH/LNM/091017, ck/CH/LHLJ/07VII, ck/CH/LDL/101212 and ck/CH/LHLJ/100902 are highlighted in bold. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch.
Fig. 3.
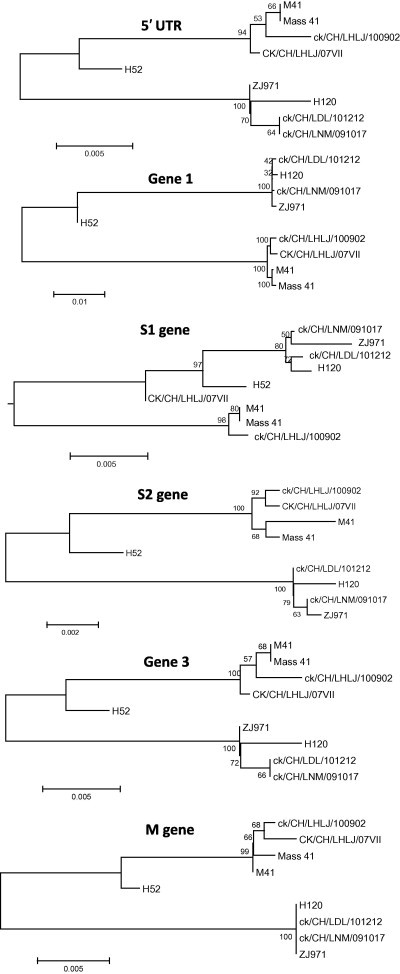
Neighbor-joining tree of non-overlapping portion of the 5′-UTR, Gene 1, S1, S2, Gene 3, M, Gene 5, N and 3′-UTR of nine Massachusetts strains. Numbers on the branches represent the percentage of 1000 bootstrap samples supporting the branch: only values >50% are shown.
3.3. Strain ck/CH/LHLJ/100902 showed a close genetic relationship with pathogenic M41
The percent nucleotide similarity between strain ck/CH/LHLJ/100902 and H120 for the full-length genomes was 91.3%; however, the percent similarity was up to 99.7% between strain ck/CH/LHLJ/100902 and M41. In addition, in all of the nine trees constructed for the 5′-UTR, Gene 1, S1, S2, Gene 3, M, ORF5, N and 3′-UTR, the strain ck/CH/LHLJ/100902 constantly fell into the same clusters as the pathogenic M41 strain, and both belonged to the Mass-type. Pairwise comparison of the S1 protein gene revealed that strain ck/CH/LHLJ/100902 had 3 and 41 nucleotide mutations with respect to M41 and H120, respectively. Taken together, these results demonstrate that strain ck/CH/LHLJ/100902 exhibits a close genetic relationship to pathogenic M41.
3.4. Markers were found that distinguish between pathogenic and vaccinal Mass-type strains
Pathogenic and non-pathogenic Mass-type strains were clustered into different clades by phylogenetic analysis of full-length genomic sequences and the 5′-UTR, Gene 1, S1, S2, Gene 3, M, Gene 5, N and 3′-UTR. In addition, insertions and deletions were also observed that distinguished between the genomes of pathogenic and non-pathogenic Mass-type strains, as illustrated in Fig. 4 and Supplementary material 1. In non-pathogenic strains, five deletions: of 31 nucleotides, 5 nucleotides, 9 nucleotides, 10 nucleotides, and 9 nucleotides, respectively, were observed to be located in the nsp3 of Gene 1. They were found to occur between genomic positions 2897–2929, 3219–3225, 3241–3251, 3257–3268, and 3398–3407, respectively, by comparing the sequences with the homologous regions of pathogenic strains. In contrast, a 3-nucleotide and a 9-nucleotide insertion were found in nsp6 and between the M gene and gene 5, respectively. Additionally, a cluster of insertions was found at the 3′-UTR region in the non-pathogenic strains. These changes might not only account, at least partly, for viral fitness when the pathogenic virus has become adapted to egg embryos (Cavanagh et al., 2005, Hewson et al., 2012), but may act also as molecular markers, able to differentiate between vaccine and field strains, for diagnostic purposes.
Fig. 4.
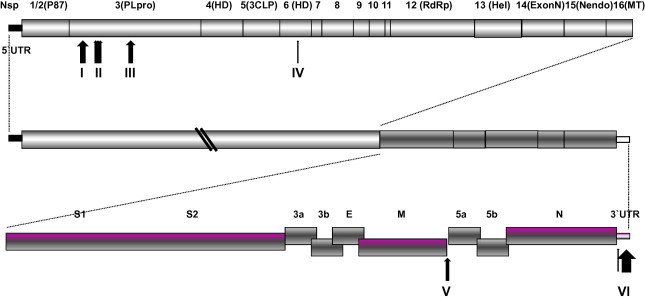
Deletions or insertions in the IBV genomes of Mass-type strains. The sites at which the deletions or insertions are located are indicated by respective arrows and numbered.
3.5. Strain CK/CH/LHLJ/07VII is a mosaic
Comparative sequence analysis based on full-length genomic sequences and the sequences of the 5′-UTR, Gene 1, S1, S2, Gene 3, M, Gene 5 and N showed that strain CK/CH/LHLJ/07VII clustered with pathogenic Mass-type strains. The exceptions were the trees constructed using the 3′-UTR and S1 gene, in which CK/CH/LHLJ/07VII was grouped with non-pathogenic strains; this suggests that a possible recombination event may have occurred. Thus the N and 3′-UTR of CK/CH/LHLJ/07VII were carefully compared pairwise with those of strains ck/CH/LNM/091017, ck/CH/LDL/101212, ck/CH/LNM/091017, H120 and M41. Parallel to the result of the phylogenetic analysis, ck/CH/LHLJ/07VII showed high similarity with M41 at the 5′-end of the N gene; however, it showed high similarity with vaccine strain H120 at the 3′-end of the N gene (Supplementary material 2). The data strongly suggest that CK/CH/LHLJ/07VII arose from a homologous RNA recombinant event that involved a template switch between Massachusetts pathogenic M41-like and non-pathogenic H120-like strains. We located the switch site at the 3′-end of the N gene (Supplementary material 2), which implies that the template switch occurred within the N gene.
3.6. Strain CK/CH/LHLJ/07VII has experienced evolution over time
The percent nucleotide similarity between strain CK/CH/LHLJ/07VII and H120, and CK/CH/LHLJ/07VII and M41, for the full-length genomes was 92.5% and 99.4%, respectively. Alignment revealed that a 3-nucleotide insertion was located in nsp6 of the CK/CH/LHLJ/07VII strain between genomic positions 24009 and 24013 (Supplementary material 3). In addition, the S1 gene of strain CK/CH/LHLJ/07VII showed extensive mutations by pairwise comparison (Supplementary material 4) though it was grouped with H120 by S1 gene phylogenic analysis. These and our previous results (Liu et al., 2009) showed that, with the exception of the occurrence of recombination events, CK/CH/LHLJ/07VII has experienced multiple mutations and deletions in the genome over time.
4. Discussion
Understanding the evolution of Mass-type IBV is important because not only is this virus circulating worldwide but information on virus genomics will aid our understanding of the evolution and emergence of IBV with infectious potential in vaccinated chicken flocks. In this study, we focused on the full-length genomic sequences of four IBV isolates which had been shown to be of the Mass-type by S1 gene analysis (Liu et al., 2009, Ma et al., 2012, Sun et al., 2011). Based on the high degree of similarity in the full genomic sequence, it could be concluded that two IBV strains, ck/CH/LNM/091017 and ck/CH/LDL/101212, were very similar to the vaccine strain H120. They might therefore represent re-isolations of vaccine strains, although they were isolated from vaccinated chickens with respiratory disease. Similarly, IBV strains that showed a very close relationship to the H120 vaccine strain have been isolated from unvaccinated broiler flocks in Slovenia with respiratory problems (Krapež et al., 2010). Alternatively, these strains might be variants of vaccine strains that have resulted from accumulated point mutations after several passages in chickens. A few key mutations in the S1 subunit of the spike protein might result in a change to a new serotype, which is defined as a lack of cross-neutralization with specific sera against different IBV serotypes (Cavanagh et al., 1992). The point mutations found in the genome that distinguish between the two isolates and vaccine strain H120 might be the result of adaptive evolution driven by the host immune response when the vaccine strain was transmitted among chickens. Adaptive evolution is the process by which genetic changes in the viral genome leading to a more fit virus population become fixed over time, and it has been reported to occur in many coronaviruses (Hasoksuz et al., 2007, Lee and Jackwood, 2001, Shi et al., 2006, Tang et al., 2009, Zhang et al., 2006).
As shown in this study, and as also occurs in other countries (Dolz et al., 2008, Rimondi et al., 2009, Roussan et al., 2009), the isolation of Mass-type IBV is expected, because attenuated vaccine strains are used extensively in chicken flocks in China. However, vaccination is not likely to be the only explanation for the circulation of Mass-type virus, because ck/CH/LHLJ/100902 and CK/CH/LHLJ/07VII were most closely related to a Massachusetts pathogenic type strain, M41. The isolation of a Massachusetts pathogenic strain from H120-vaccinated chicken flocks may be due to vaccination failure in these flocks (Han et al., 2011). Alternatively, molecular studies have shown that only a few changes in the amino acid composition of the S1 spike protein can result in immune failure, even when the majority of the virus genome remains unchanged (Cavanagh et al., 1992). Our findings showed that mutations had occurred in the genomes of both ck/CH/LHLJ/100902 and CK/CH/LHLJ/07VII, especially in the S1 genes of CK/CH/LHLJ/07VII though it was in the same group with H120 strain in the phylogenetic analysis, implicating that strain CK/CH/LHLJ/07VII has experienced evolution over time. It has been reported that amino acid changes may result from immunological pressure caused by the widespread use of vaccines (Cavanagh et al., 1988, Cavanagh et al., 2005).
The occurrence of recombination events is another process that allows new strains to emerge, and this has been well documented in IBV (Hughes, 2011, Jia et al., 1995, Kottier et al., 1995, Kusters et al., 1990, Wang et al., 1993) and other coronaviruses (Fu and Baric, 1992, Fu and Baric, 1994, Makino et al., 1986). It is believed that the conditions for recombination amongst IBV strains in the field are as follows: an extremely large number of chickens, most maintained at high density; the ease of spread of the virus; and serotype co-circulation, including proof of co-infection with more than one serotype in a given flock (Cavanagh, 2007). In China, intensive chicken farms are concentrated in many provinces, including Heilongjiang, where ck/CH/LHLJ/07VII was isolated (Liu et al., 2009). Almost all the chickens in China receive Mass-type vaccines at a very young age and subsequently receive this vaccine a couple more times during the rest of their life span. Therefore the vaccine virus exists constantly in chickens; it may persist in various internal organs for 163 days or longer (Cavanagh and Gelb, 2008). Generally, vaccination using the H120 vaccine provides full protection against pathogenic Mass-type pathogenic IBVs and prevents the same type of pathogenic strain from being replicated and spreading in the flocks. However, a single amino acid substitution at position 63 of the S1 subunit of the spike has resulted in escape mutants of Mass 41 (Cavanagh et al., 1988). This may have occurred in the case of the ck/CH/LHLJ/07VII S1 gene (Liu et al., 2009), which might have made it possible for both pathogenic and vaccine strains to co-exist in a given flock, leading to the occurrence of recombination. Similarly, an escape mutant could be a result of adaptive evolution driven by the host immune response. Consequently, it is likely that genetic changes due to adaptive evolution and recombination both contributed to the origin and evolution of strain ck/CH/LHLJ/07VII: it is possible that adaptive evolution created a mutant, followed by recombination between Mass 41- and H120-like strains to create a novel virus.
The recombination events from which the CK/CH/LHLJ/07VII virus resulted can be explained by a scenario in which the recombination may have involved two parental viral strains, with initiation of RNA replication in a M41-like template of either negative or positive polarity (Liao and Lai, 1992). This would be followed by switching of the polymerase-nascent cRNA complex to an H120-like virus template. The switch may have occurred at the 3′-end of the N gene. In general, for a recombinant virus to emerge and establish itself in the field, it must be viable and have selective advantages. It has been reported that uptake of canine coronavirus (CCV) sequences by type II feline coronavirus (FCoV) may have led to increased viral fitness when compared with type I FCoV (Herrewegh et al., 1998). Recombination can also result in the emergence of new strains with distinct characteristics, such as pathogenicity and tissue tropism (Worobey and Holmes, 1999). In addition, in the CoV genome, as with most RNA viruses, the 3′-UTRs usually harbor important structural elements that are involved in replication and/or translation (Chang et al., 1994, Raman et al., 2003, Raman and Brian, 2005, Goebel et al., 2006, Züst et al., 2008). In IBV, the 3′-UTR-binds to the N protein, which is essential for the synthesis of negative-strain viral RNA. Perhaps the acquisition of the 3′ end of the N gene and the 3′-UTR from H120-like virus by an M41-like virus (e.g. CK/CH/LHLJ/07VII) can alter the efficiency of viral replication. This alteration may in turn affect pathogenicity. However, it remains unknown whether this is the true origin of CK/CH/LHLJ/07VII, and therefore this strain is of particular importance to the surveillance of IBV in China. It will be of equal importance to examine future outbreaks of IBV in chickens by full-length genomic sequence analysis in the context of novel recombination events among IBV strains. Furthermore, investigations using reverse genetic systems might provide further insight into these issues and increase our understanding of IBV pathogenesis.
Acknowledgements
This work was supported by grants from the China Agriculture Research System (No. CARS-41-K12) and Special Fund for Agro-Scientific Research in the Public Interest (No. 201203056).
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.meegid.2012.09.016.
Appendix A. Supplementary data
Deletions or insertions in the IBV genomes of Mass-type strains. Multiple sequence alignments surrounding the numbered regions in A. The numbers on the right of each alignment show the nucleotide positions in the genome of each virus. The short lines indicate the deleted nucleotides.
Multiple alignment of the N gene among ck/CH/LNM/091017, ck/CH/LHLJ/07VII, ck/CH/LDL/101212, ck/CH/LHLJ/100902, Z971, H52, Massachusetts, Mass 41 and H120. In the nine IBV strains, only the nucleotides identical to those in strain CK/CH/LHLJ/07VII are highlighted in bold. The putative template switch site is boxed. The initial and stop codons of the N gene are indicated by ∗∗∗.
Alignment of a gene region in which the deletions in the sequence of CK/CH/LHLJ/07VII are indicated. The numbers on the right of each alignment show the nucleotide positions in the genome of each virus. The short lines indicate the deleted nucleotides.
Alignment of the S1 genes and flanking sequences of H120, CK/CH/LHLJ/04VII and M41. Nucleotide mutations that are different among the three sequences are indicated. The ATG that encodes the start codon of S1 is also indicated.
References
- Beach J.R., Schalm O.W. A filterable virus, distinct from that of laryngotracheitis, the cause of a respiratory disease of chicks. Poult. Sci. 1936;15:199–206. [Google Scholar]
- Bijlenga G., Cook J.K., Gelb J., Jr., deWit J.J. Development and use of the H strain of avian infectious bronchitis virus from the Netherlands as a vaccine: a review. Avian Pathol. 2004;33:550–557. doi: 10.1080/03079450400013154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley I., Boursnell M.E., Binns M.M., Bilimoria B., Blok V.C., Brown T.D., Inglis S.C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 1987;6:3779–3785. doi: 10.1002/j.1460-2075.1987.tb02713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casais R., Dove B., Cavanagh D., Britton P. Recombinant avian infectious bronchitis virus expressing a heterologous spike gene demonstrates that the spike protein is a determinant of cell tropism. J. Virol. 2003;77:9084–9089. doi: 10.1128/JVI.77.16.9084-9089.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Commentary: a nomenclature for avian coronavirus isolates and the question of species status. Avian Pathol. 2001;30:109–115. doi: 10.1080/03079450120044506. [DOI] [PubMed] [Google Scholar]
- Cavanagh D. Severe acute respiratory syndrome vaccine development: experiences of vaccination against avian infectious bronchitis coronavirus. Avian Pathol. 2003;32:567–582. doi: 10.1080/03079450310001621198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D. Coronaviruses in poultry and other birds. Avian Pathol. 2005;34:439–448. doi: 10.1080/03079450500367682. [DOI] [PubMed] [Google Scholar]
- Cavanagh D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007;38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Cook J.K.A., Li D., Kant A., Koch G. Location of the amino acid differences in the S1 spike glycoprotein subunit of closely related serotypes of infectious bronchitis virus. Avian Pathol. 1992;21:33–43. doi: 10.1080/03079459208418816. [DOI] [PubMed] [Google Scholar]
- Cavanagh D., Davis P.J., Mockett A.P.A. Amino acids within hypervariable region 1 of avian coronavirus IBV (Massachusetts serotype) spike glycoprotein are associated with neutralization epitopes. Virus Res. 1988;11:141–150. doi: 10.1016/0168-1702(88)90039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh D., Gelb J.B. Infectious bronchitis. In: Saif Y.M., Fadly A.M., Glisson J.R., McDougald L.R., Nolan L.K., Swayne D.E., editors. Diseases of Poultry. 12th ed. Wiley-Blackwell Publishing; Iowa: 2008. pp. 117–135. [Google Scholar]
- Cavanagh D., Picault J.P., Gough R.E., Hess M., Mawditt K.L., Britton P. Variations in the spike protein of the 793/B type of infectious bronchitis virus in the field and during alternate passage in chickens and embryonated eggs. Avian Pathol. 2005;34:20–25. doi: 10.1080/03079450400025414. [DOI] [PubMed] [Google Scholar]
- Chang R.Y., Hofmann M.A., Sethna P.B., Brian D.A. A cis-acting function for the coronavirus leader in defective interfering RNA replication. J. Virol. 1994;68:8223–8231. doi: 10.1128/jvi.68.12.8223-8231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groot, R.J., Baker, S.C., Baric, R., Enjuanes, L., Gorbalenya, A., Holmes, K.V., Perlman, S., Poon, L., Rottier, P.J., Talbot, P.J., Woo, P.C., Ziebuhr, J., 2011. Coronaviridae. In: King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J. (Eds.), Virus Taxonomy, Classification and Nomenclature of Viruses, Ninth Report of the International Committee on Taxonomy of Viruses, International Union of Microbiological Societies, Virology Division, Elsevier Academic Press, pp. 806–828.
- Dolz R., Pujols J., Ordonez G., Porta R., Majo N. Molecular epidemiology and evolution of avian infectious bronchitis virus in Spain over a fourteen-year period. Virology. 2008;374:50–59. doi: 10.1016/j.virol.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabricant L. The early history of infectious bronchitis. Avian Dis. 2000;42:648–650. [PubMed] [Google Scholar]
- Fu K., Baric R.S. Evidence for variable rates of recombination in the MHV genome. Virology. 1992;189:88–102. doi: 10.1016/0042-6822(92)90684-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K., Baric R.S. Map locations of mouse hepatitis virus temperature sensitive mutants: confirmation of variable rates of recombination. J. Virol. 1994;68:7458–7466. doi: 10.1128/jvi.68.11.7458-7466.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel S.J., Miller T.B., Bennett C.J., Bernard K.A., Masters P.S. A hypervariable region within the 3′ cis-acting element of the murine coronavirus genome is nonessential for RNA synthesis but affects pathogenesis. J. Virol. 2006;81:1274–1287. doi: 10.1128/JVI.00803-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z., Sun C., Yan B., Zhang X., Wang Y., Li C., Zhang Q., Ma Y., Shao Y., Liu Q., Kong X., Liu S. A 15-year analysis of molecular epidemiology of avian infectious bronchitis coronavirus in China. Infect. Genet. Evol. 2011;11:190–200. doi: 10.1016/j.meegid.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasoksuz M., Alekseev K., Vlasova A., Zhang X., Spiro D., Halpin R., Wang S., Ghedin E., Saif L.J. Biologic, antigenic, and full-length genomic characterization of a bovine-like coronavirus isolated from a giraffe. J. Virol. 2007;81:4981–4990. doi: 10.1128/JVI.02361-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrewegh A.A.P.M., Smeenk I., Horzinek M.C., Rottier P.J.M., de Groot R.J. Feline coronavirus type II strains 79–1683 and 79–1146 originate from a double recombination between feline coronavirus type I and canine coronavirus. J. Virol. 1998;72:4508–4515. doi: 10.1128/jvi.72.5.4508-4514.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewson K.A., Scott P.C., Devlin J.M., Ignjatovic J., Noormohammadi A.H. The present of viral subpopulations in an infectious bronchitis virus vaccine with differing pathogenicity – a preliminary study. Vaccine. 2012;30:4190–4199. doi: 10.1016/j.vaccine.2012.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A.L. Recombinational histories of avian infectious bronchitis virus and turkey coronavirus. Arch. Virol. 2011;156:1823–1829. doi: 10.1007/s00705-011-1061-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov K.A., Thiel V., Dobbe J.C., van der Meer Y., Snijder E.J., Ziebuhr J. Multiple enzymatic activities associated with severe acute respiratory syndrome coronavirus helicase. J. Virol. 2004;78:5619–5632. doi: 10.1128/JVI.78.11.5619-5632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W., Karaca K., Parrish C.R., Naqi S.A. A novel variant of infectious bronchitis virus resulting from recombination among three different strains. Arch. Virol. 1995;140:259–271. doi: 10.1007/BF01309861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R.B., Marquardt W.W. The neutralizing characteristics of strains of infectious bronchitis virus as measured by the constant virus variable serum methods in chicken tracheal cultures. Avian Dis. 1975;19:82–90. [PubMed] [Google Scholar]
- Kottier S.A., Cavanagh D., Britton P. Experimental evidence of recombination in coronavirus infectious bronchitis virus. Virology. 1995;213:569–580. doi: 10.1006/viro.1995.0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapež U., Slavec B., Barlič-Maganja D., Rojs O.Z. Molecular analysis of infectious bronchitis viruses isolated in Slovenia between 1990 and 2005: a retrospective study. Virus Genes. 2010;41(3):414–416. doi: 10.1007/s11262-010-0528-x. [DOI] [PubMed] [Google Scholar]
- Kusters J.G., Jager E.J., Niesters H.G.M., Zeist B.A.M. Sequence evidence for RNA recombination in field isolates of avian coronavirus infectious bronchitis virus. Vaccine. 1990;8:605–608. doi: 10.1016/0264-410X(90)90018-H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.W., Jackwood M.W. Origin and evolution of Georgia 98 (GA98), a new serotype of avian infectious bronchitis virus. Virus Res. 2001;80:33–39. doi: 10.1016/s0168-1702(01)00345-8. [DOI] [PubMed] [Google Scholar]
- Lee H.J., Shieh C.K., Gorbalenya A.E., Koonin E.V., La Monica N., Tuler J., Bagdzhadzhyan A., Lai M.M. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and RNA polymerase. Virology. 1991;180:567–582. doi: 10.1016/0042-6822(91)90071-I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C., Lai M.M.C. RNA recombination in a coronavirus: recombination between viral genomic RNA and transfected RNA fragments. J. Virol. 1992;66:6117–6124. doi: 10.1128/jvi.66.10.6117-6124.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Zhang X., Wang Y., Li C., Han Z., Shao Y., Li H., Kong X. Molecular characterization and pathogenicity of infectious bronchitis coronaviruses: complicated evolution and epidemiology in China caused by cocirculation of multiple types of infectious bronchitis coronaviruses. Intervirology. 2009;52:223–234. doi: 10.1159/000227134. [DOI] [PubMed] [Google Scholar]
- Ma H., Shao Y., Sun C., Han Z., Liu X., Guo H., Liu X., Kong X., Liu S. Genetic diversity of avian infectious bronchitis coronavirus in recent years in China. Avian Dis. 2012;56:15–28. doi: 10.1637/9804-052011-Reg.1. [DOI] [PubMed] [Google Scholar]
- Makino S., Keck J.G., Stohlman S.A., Lai M.M. High-frequency RNA recombination of murine coronaviruses. J. Virol. 1986;57:729–737. doi: 10.1128/jvi.57.3.729-737.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006;66:193–292. doi: 10.1016/S0065-3527(06)66005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.P., Lucio-Martinez B., Naqi S.A. Isolation and characterization of a novel antigenic subtype of infectious bronchitis virus serotype DE072. Avian Dis. 2001;45:1054–1059. [PubMed] [Google Scholar]
- Raman S., Bouma P., Williams G.D., Brian D.A. Stem-loop III in the 5′ untranslated region is a cis-acting element in bovine coronavirus defective interfering RNA replication. J. Virol. 2003;77:6720–6730. doi: 10.1128/JVI.77.12.6720-6730.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S., Brian D.A. Stem-loop IV in the 5′ untranslated region is a cis-acting element in bovine coronavirus defective interfering RNA replication. J. Virol. 2005;79:12434–12446. doi: 10.1128/JVI.79.19.12434-12446.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondi A., Craig M.I., Vagnozzi A., Konig G., Delamer M., Pereda A. Molecular characterization of avian infectious bronchitis virus strains from outbreaks in Argentina (2001–2008) Avian Pathol. 2009;38:149–153. doi: 10.1080/03079450902737821. [DOI] [PubMed] [Google Scholar]
- Roussan D.A., Khawaldeh G.Y., Shaheen I.A. Infectious bronchitis virus in Jordanian chickens: seroprevalence and detection. Can. Vet. J. 2009;50:77–80. [PMC free article] [PubMed] [Google Scholar]
- Schalk A.F., Hawn M.C. An apparently new respiratory disease of chicks. J. Am. Vet. Med. Assoc. 1931;78:413–422. [Google Scholar]
- Shi P., Yu L., Fu Y.X., Huang J.F., Zhang K.Q., Zhang Y.P. Evolutionary implications of avian infectious bronchitis virus (AIBV) analysis. Cell Res. 2006;16:323–327. doi: 10.1038/sj.cr.7310041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder E.J., Bredenbeek P.J., Dobbe J.C., Thiel V., Ziebuhr J., Poon L.L.M., Guan Y., Rozanov M., Spaan W.J.M., Gorbalenya A.E. Unique and conserved features of genome and proteonome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003;331:991–1004. doi: 10.1016/S0022-2836(03)00865-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C., Han Z., Ma H., Zhang Q., Yan B., Shao Y., Xu J., Kong X., Liu S. Phylogenetic analysis of infectious bronchitis coronaviruses newly isolated in China, and pathogenicity and evaluation of protection induced by Mass serotype H120 vaccine against strains of the LX4-type (QX) Avian Pathol. 2011;40:43–54. doi: 10.1080/03079457.2010.538037. [DOI] [PubMed] [Google Scholar]
- Tang X., Li G., Vasilakis N., Zhang Y., Shi Z., Zhong Y., Wang L.F., Zhang S. Differential stepwise evolution of SARS coronavirus functional proteins in different host species. BMC Evol. Biol. 2009;9:52. doi: 10.1186/1471-2148-9-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Junker D., Collisson E.W. Evidence of natural recombination within the S1 gene of infectious bronchitis virus. Virology. 1993;192:710–716. doi: 10.1006/viro.1993.1093. [DOI] [PubMed] [Google Scholar]
- Worobey M., Holmes E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999;80:2535–2543. doi: 10.1099/0022-1317-80-10-2535. [DOI] [PubMed] [Google Scholar]
- Zhang C.Y., Wei J.F., He S.H. Adaptive evolution of the spike gene of SARS coronavirus: changes in positively selected sites in different epidemic groups. BMC Microbiol. 2006;6:88. doi: 10.1186/1471-2180-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J. Gen. Virol. 2000;81:853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- Ziebuhr J., Thiel V., Gorbalenya A.E. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J. Biol. Chem. 2001;276:33220–33232. doi: 10.1074/jbc.M104097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züst R., Miller T.B., Goebel S.J., Thiel V., Masters P.S. Genetic interactions between an essential 3′ cis-acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J. Virol. 2008;82:1214–12128. doi: 10.1128/JVI.01690-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Deletions or insertions in the IBV genomes of Mass-type strains. Multiple sequence alignments surrounding the numbered regions in A. The numbers on the right of each alignment show the nucleotide positions in the genome of each virus. The short lines indicate the deleted nucleotides.
Multiple alignment of the N gene among ck/CH/LNM/091017, ck/CH/LHLJ/07VII, ck/CH/LDL/101212, ck/CH/LHLJ/100902, Z971, H52, Massachusetts, Mass 41 and H120. In the nine IBV strains, only the nucleotides identical to those in strain CK/CH/LHLJ/07VII are highlighted in bold. The putative template switch site is boxed. The initial and stop codons of the N gene are indicated by ∗∗∗.
Alignment of a gene region in which the deletions in the sequence of CK/CH/LHLJ/07VII are indicated. The numbers on the right of each alignment show the nucleotide positions in the genome of each virus. The short lines indicate the deleted nucleotides.
Alignment of the S1 genes and flanking sequences of H120, CK/CH/LHLJ/04VII and M41. Nucleotide mutations that are different among the three sequences are indicated. The ATG that encodes the start codon of S1 is also indicated.