

Abstract
Objective
Systemic sclerosis (SSc) is a connective tissue disease with a significant morbidity and reduced survival of patients. Effective treatment and clinical control of the disease remain challenging. In particular, the development of pulmonary and cardiac fibrosis and pulmonary hypertension are severe complications responsible for excessive mortality. Currently available treatment strategies only alleviate symptoms and slow disease progression. Here, we investigated the therapeutic potential of ibrutinib, a Bruton’s tyrosine kinase (BTK) inhibitor used in B cell malignancies, to alter B cell pathology in SSc in an in vitro model of autoimmunity.
Methods
PBMCs and sorted B cells of 24 patients with SSc were used for functional testing after stimulation with hypomethylated DNA fragments (CpG) to induce an innate immune response. The effects of ibrutinib on cytokine production, autoantibody release, and activation of the transcription factor NFκB were evaluated.
Results
Ibrutinib was able to reduce the production of the profibrotic hallmark cytokines IL-6 and TNF-α mainly from the effector B cell population in patients with SSc. Importantly, small doses of ibrutinib (0.1 μM) preserved the production of immunoregulatory IL-10 while effectively inhibiting hyperactivated, profibrotic effector B cells. In a flow cytometry analysis of phosphorylated NFκB, an important transcription factor in the induction of innate immune responses in B cells, significantly less activation was observed with ibrutinib treatment.
Conclusion
Our data could pave the avenue for a clinical application of ibrutinib for patients with SSc as a novel treatment option for the underlying pathogenetic immune imbalance contributing to disease onset and progression.
Keywords: Systemic sclerosis, B cells, Ibrutinib, Autoimmune disease, Treatment
Introduction
Systemic sclerosis (SSc) is a connective tissue disease that affects the skin, blood vessels, and internal organs. Within a complex and incompletely understood pathogenesis, the defining pathophysiological features include immune dysregulation with the production of autoantibodies, vasculopathy, and chronic activation of fibroblasts. In particular, the involvement of internal organs critically reduces survival of patients [1]. A stratification of patients in risk-associated groups in the recently developed SCleroderma mOrtality p Eustar (SCOpE) score allows for precise prediction of 3-year mortality, estimating the survival of high-risk patients (SCOpE ≥ 15) at 53% [2]. More than half of the patients die from SSc itself, mostly due to cardiopulmonary complications [3]. Thus, currently available treatment options summarized in the updated EULAR recommendations remain insufficient for controlling disease progression in a clinically satisfactory way [4].
The pathogenesis of SSc remains poorly understood. Accumulating evidence suggests that B cells are involved in SSc beyond the mere production of autoantibodies. Alterations to the B cell compartment maintain the hyperreactivity and chronic activation of large portions of the immune system. The sensitive equilibrium between effector B cells and regulatory B cells (Bregs) is disrupted, and immunoregulatory Bregs prove to be numerically and functionally impaired [5]. Furthermore, evidence for an important pathogenetic role for effector B cell-derived profibrotic IL-6 and TNF-α, as well as for protective effects of anti-inflammatory IL-10, has been published recently [6].
Consequently, different approaches have been tested to address the hyperactivation of B cells in SSc. In a phase III study, the IL-6-receptor-α inhibitor tocilizumab failed to reduce skin thickening, but a trend toward improvement of the modified Rodnan Skin Score and pulmonary function was observed [7]. Complete B cell depletion with rituximab showed better efficacy with regard to the reduction of skin fibrosis and respiratory restriction in a case-control study [8]. The clinical improvement in both trials might have been limited by the lack of specificity, as depletion of all B cell subsets eliminates the protective effects conveyed by Bregs in the context of autoimmunity.
The specific inhibition of autoreactive, profibrotic, and chronically activated B cell subsets represents a more promising approach to achieve effective treatment of SSc. The Bruton’s tyrosine kinase (BTK) inhibitor ibrutinib could inhibit hyperactivated B cell subpopulations to counteract underlying pathogenetic mechanisms. First FDA-approved for mantle cell lymphoma in 2013 and chronic lymphocytic leukemia in 2014, a potential application for ibrutinib has been suggested for any autoimmune disease in which B cells play an important role [9, 10]. Here, we report on the potential of this small molecule inhibitor to alter B cell pathology in primary patient samples in order to pave the avenue for a clinical application of ibrutinib in patients with SSc.
Materials and methods
Patients and healthy volunteers
Peripheral blood samples were collected from patients with SSc enrolled at the Centre for Interdisciplinary Clinical Immunology, Rheumatology and Autoinflammatory Diseases at the University Hospital Tuebingen, Germany, from 2017 to 2019. Written consent was obtained from all patients. Human buffy coats from healthy volunteers were obtained from the Center of Clinical Transfusion Medicine Tuebingen. The institutional review board of the Eberhard-Karls-University Tuebingen (IRB approval number 114/2016BO) approved this study to be in accordance with the ethical standards and the Helsinki Declaration of 1975, as revised in 2013.
Magnetic cell separation
B cells were purified from cryopreserved peripheral blood mononuclear cells (PBMCs) of patients with SSc using CD19-Microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), a QuadroMACS™ Separator (Miltenyi Biotec) and LS Columns (Miltenyi Biotec) according to the manufacturer’s instructions. After purification, cells were cultured in a medium consisting of RPMI 1640 GlutaMAX™ Medium (ThermoFisher Scientific, Waltham, USA), 10% human serum (C-C-Pro, Oberdorla, Germany), 100 IU/ml penicillin-streptomycin (Lonza, Basel, Switzerland), 5.5 μM 2-mercaptoethanol (Roth, Karlsruhe, Germany), 0.1 mM non-essential amino acids (Gibco, New York, USA), 10 mM HEPES (Gibco), and 1 mM sodium pyruvate (Gibco).
Cytokine analysis
For cytokine profiling, B cells were preincubated with ibrutinib (Selleckchem, Houston, USA) or 0.1% DMSO (control) for 1 h at a concentration of 2 × 106 cells/ml. B cells were then stimulated with the Toll-like receptor 9 agonist CpG (1 μM, ODN2006, Invivogen, San Diego, USA) and cultivated on a 96-well-plate for 24 h at a concentration of 1 × 106/ml. Supernatants were collected and stored at − 20 °C prior to analysis in a multiplex assay (LEGENDplex™ Mix-and-Match-Panel 5-plex, BioLegend, San Diego, USA) according to the manufacturer’s instructions. For data analysis, the LEGENDplex™ Software v8.0 from BioLegend was used.
Anti-Scl-70-ELISA
Supernatants of B cell cultures were collected after 72 h and stored at − 20 °C until further analysis. Nunc polystyrene plates (ThermoFisher Scientific) were coated with recombinant human DNA topoisomerase (Scl-70, Diarect AG, Freiburg, Germany) overnight. Anti-Scl-70 antibodies were detected in undiluted culture supernatant using peroxidase-conjugated anti-human IgG H + L (goat, Jackson ImmunoResearch Laboratories Inc., West Grove, USA).
Intranuclear staining of PBMCs
Patient PBMCs were thawed and preincubated with ibrutinib (Selleckchem) or 0.1% DMSO (control) for 1 h at a concentration of 2 × 106 cells/ml. The TLR9-agonist CpG (0.1 μM, ODN2006, Invivogen) was added to PBMCs cultivated on a 24-well-plate (1 × 106/well). After 24 h, cells were stained for CD3 (OKT3, BV605, BioLegend) and CD19 (HIB19, BV421, BioLegend). Fixable Viability Dye eFluor™ 780 (eBiosience, Thermo Fisher Scientific) was used for the identification of live cells. The cells were fixed with IC Fixation Buffer (eBiosience, Thermo Fisher Scientific) and permeabilized with 90% ice-cold methanol. To detect phosphorylation levels of NFκB (Ser536), a phospho (p) NFκB antibody (93H1, Cell Signaling, Boston, USA) was stained with a secondary anti-rabbit antibody (PE-Cy7, Cell Signaling). Samples were measured using an LSR II Fortessa flow cytometer (BD Biosciences, Franklin Lakes, USA).
Intracellular cytokine staining
PBMC cultures were performed as described. A cell stimulation cocktail (eBioscience, Thermo Fisher Scientific) was added to the culture for the last 4 h of culturing. After 24 h, cells were stained with Fixable Viability Dye eFluor™ 780 and the following antibodies purchased from BioLegend or BD BioScience: CD3 (HIT3a, PerCP-Cy5.5), CD20 (2H7, BV510), CD24 (ML5, BV650), CD27 (O323, BV421), CD38 (HIT2, PE/Dazzle), and IgD (IA6-2, FITC). Fixation and permeabilization were performed using an IC Fix and Perm Buffer Kit (eBioscience, Thermo Fisher Scientific). Anti-human IL-6 (MQ2-13A5, PE-Cy7) was used to detect intracellular cytokine levels in B cells.
Statistical analysis
Flow cytometry data were analyzed in FlowJo V10 (Tree Star Inc., Ashland, USA). Prism 7.01 (GraphPad Software, La Jolla, USA) was used for further statistical analysis and graphical representation. Experiments were performed in technical duplicates and repeated independently at least three times. Significance (*p < 0.05, **p < 0.01, ***p < 0.001) was calculated using a paired Student’s t test for single comparisons and ANOVA testing for repeated measures for multiple comparisons.
Results
Patient characteristics
In total, samples of 24 patients were used for in vitro testing (Table 1). The median age of the patient cohort was 54 years (range 30–81), and the median disease duration after diagnosis was 8 years (range 1.2–24). Most patients were tested positive for antinuclear antibodies (ANA), and half of the study cohorts were positive for anti-Scl-70 antibodies. The median modified Rodnan skin score was 8. Importantly, 75% of patients received no immunosuppressive treatment at the time of blood draw. Six patients indicated prior intensive immunosuppressive regimens in their patient history.
Table 1.
Patient characteristics
| SSc (n = 24) | |
|---|---|
| Demographics | |
| Age (years) | |
| Median | 54 |
| Range | 30–81 |
| Sex | |
| Female | 15 (63%) |
| Male | 9 (38%) |
| Disease characteristics | |
| SSc subtype | |
| Limited cutaneous SSc | 14 (58%) |
| Diffuse cutaneous SSc | 10 (42%) |
| Disease duration (years) | |
| Median | 8 |
| Range | 1.2–24 |
| Modified Rodnan skin score | |
| Median | 8 |
| Range | 0–44 |
| Autoantibodies | |
| Antinuclear antibodies (ANA) | 20 (83%) |
| Anti-Scl-70 antibodies | 12 (50%) |
| Anticentromer antibodies (ACA) | 6 (25%) |
| Pretreatment | |
| Cyclophosphamide | 4 (17%) |
| Mycophenolate mofetil | 2 (8%) |
| Stem cell transplant (> 10 years before blood draw) | 2 (8%) |
| Immunosuppressive therapy at time of blood draw | |
| None | 18 (75%) |
| Prednisolone | 2 (8%) |
| Mycophenolate mofetil | 2 (8%) |
| Methotrexate | 3 (13%) |
Reduction of proinflammatory cytokines and anti-Scl-70 in B cells through high-dose ibrutinib
Elevated cytokine levels in SSc mirror the ongoing process of chronic inflammation that contributes to fibrosis and organ destruction. We reasoned that ibrutinib would exert meaningful effects on the cytokine profiles of stimulated B cells in SSc. A number of observations support a relevant immuno-activating role for TLR9-activating double-strand self-DNA in SSc [11]. Therefore, B cells were stimulated with the TLR9-agonist CpG robustly inducing the profibrotic cytokines IL-6 and TNF-α.
Cytokine levels were determined in the culture supernatant after 24 h (Fig. 1a). Here, high-dose ibrutinib treatment significantly reduced the production of IL-6 and TNF-α by B cells from 522.7 pg/ml (SEM ± 88.9 pg/ml) to 333.5 pg/ml (SEM ± 51.48 pg/ml, p = 0.003) and from 75.0 pg/ml (SEM ± 13.1 pg/ml) to 33.5 pg/ml (SEM ± 8.8 pg/ml, p = 0.0004), respectively. A similar reduction of cytokine production by B cells with ibrutinib treatment was observed in healthy volunteers (Supplemental Figure 1). Moreover, the effects of ibrutinib on autoantibody production by B cells of anti-Scl-70-positive patients were investigated after 72 h of culture (Fig. 1b). Anti-Scl-70 antibodies, or anti-topoisomerase I-antibodies, are characteristic for SSc and were produced by stimulated B cells in vitro. Ibrutinib reduced the release of anti-Scl-70 significantly from 103.7 (SEM ± 16.0) to 72.0 (SEM ± 17.3, p = 0.002).
Fig. 1.

Effects of high-dose ibrutinib on the release of proinflammatory cytokines and anti-Scl-70. B cells were treated with ibrutinib (10 μM) and stimulated with CpG (1 μM); DMSO was used as control. a Supernatants of B cell cultures were analyzed for cytokine levels in a multiplex assay after 24 h of culture (n = 10). b Supernatants of B cell cultures were analyzed for anti-Scl-70 levels via ELISA after 72 h of culture (n = 5). Bars represent the mean. Error bars indicate SEM. *p < 0.05, **p < 0.01, ***p < 0.001
Dose-dependent modulation of the B cell cytokine profile through ibrutinib
To further determine the clinical applicability of ibrutinib in the context of SSc, alterations to the B cell cytokine profile of patients with SSc were assessed over a spectrum of concentrations whenever allowed by the respective cell count of the samples. The relative changes of cytokine production were analyzed, comparing ibrutinib-treated and control samples (mean concentrations: IL-6 = 575.9 pg/ml, TNF-α = 95.7 pg/ml, IFN-γ = 4.9 pg/ml, IL-10 = 5.7 pg/ml) within each individual patient (Fig. 2). Lower concentrations of ibrutinib exerted effects on B cells that differed strongly from the modulation of cytokine production through high-dose ibrutinib. For high-dose ibrutinib (10 μM), a nonspecific reduction in cytokine production for all detected cytokines (IL-6 − 27.3%, TNF-α − 64.8%, IFN-γ − 9.8%, IL-10 − 33.8%) was observed. In contrast, low concentrations of ibrutinib affected the production of pro- versus anti-fibrotic cytokines in a cytokine-specific manner: the proinflammatory cytokines TNF-α and IL-6 were noticeably reduced at 0.1 μM (TNF-α − 24.6%, IL-6 − 17.9%) and 1 μM (TNF-α − 39.8%, IL-6 − 17.9%), while the immunoregulatory IL-10 remained unchanged at 0.1 μM and was only mildly decreased at 1 μM (− 13.7%). Anti-fibrotic IFN-γ even increased in ibrutinib-treated samples compared to control (0.1 μM + 23.2%, 1 μM + 45.4%). Importantly, B cell viability was not decreased with ibrutinib treatment in experiments with samples of healthy volunteers, nor was apoptosis induced in B cells upon ibrutinib treatment.
Fig. 2.
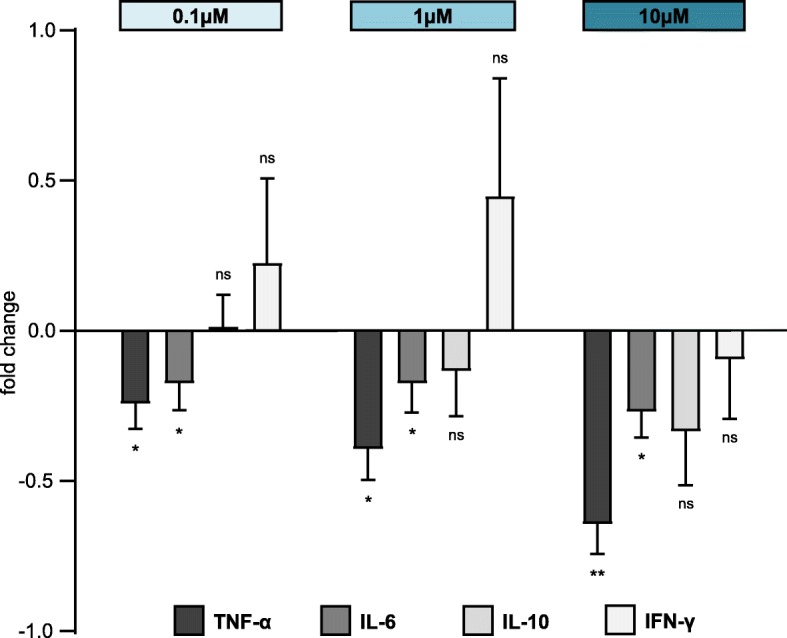
Relative changes of cytokine production under ibrutinib treatment. B cells of patients with SSc (n = 5) were treated with ibrutinib (0.1 μM, 1 μM, 10 μM). The relative change in cytokine production compared to control is depicted. Low concentrations of ibrutinib (0.1 μM, 1 μM) reduce TNF-α and IL-6 significantly but do not alter the level of IL-10. High concentrations of ibrutinib (10 μM) reduce cytokine production nonspecifically. Bars indicate the mean. Error bars show SEM. *p < 0.05, **p < 0.01
Ibrutinib-mediated inhibition of the transcription factor pNFκB
NFκB is a transcription factor with a major role in downstream TLR and B cell receptor (BCR) signaling, conveying survival signals and inducing a proinflammatory response in B cells [12, 13]. Flow cytometry analysis of the phosphorylation of NFκB was performed to measure the transcriptional activation downstream of TLR9 after 24 h. CpG stimulation increased the abundance of pNFκB (Ser536) by 2.82-fold compared to unstimulated B cells (Fig. 3a, b). With ibrutinib treatment, a significant reduction in the level of phosphorylation was observed (0.1 μM, p = 0.047; 1 μM, p = 0.029; 10 μM, p = 0.018).
Fig. 3.

Ibrutinib-mediated inhibition of the transcription factor NFκB. PBMCs of patients with SSc (n = 4) were treated with ibrutinib and stimulated with CpG (0.1 μM) for 24 h; DMSO (0.1%) was used as control. Phosphorylated NFκB (Ser536) was stained after methanol permeabilization. a Levels of pNFκB (MFI) are reduced significantly by ibrutinib. Bars depict the mean. Error bars represent SEM. *p < 0.05. b Representative histograms from one individual SSc patient
Reduction of IL-6+ naïve (CD27−) B cells in SSc PBMCs treated with ibrutinib
To evaluate the effect of ibrutinib on different subpopulations of B cells, patient PBMCs were cultured over 24 h and stained for intracellular IL-6. In our patient cohort, the ratio of naïve (CD27−) to memory (CD27+) B cells was consistently shifted toward naïve B cells (Fig. 4a): in unstimulated samples, naïve B cells accounted for 85.6% (SD ± 5.6%) of all B cells while memory B cells represented only 14.4% (SD ± 5.8%). Neither ibrutinib treatment nor stimulation with the TLR9-agonist CpG altered the ratio of the B cell subpopulations significantly (data not shown). At baseline in unstimulated samples, a higher proportion of memory B cells (7.3%, SD ± 2.6%) was IL-6+ compared to naïve B cells (3.3%, SD ± 1.1%) (Fig. 4b). Ibrutinib (10 μM) showed a significant reduction in the proportion of IL-6+ cells within both naïve and memory B cells. In contrast, 1 μM of ibrutinib significantly decreased the IL-6+ proportion of naïve B cells (Fig. 4b, c) but showed no significant effect on memory B cells (Fig. 4b, d).
Fig. 4.

Subset analysis of IL-6 production via intracellular cytokine staining. PBMCs of patients with SSc (n = 10) were incubated with CpG (0.1 μM) for 24 h under ibrutinib treatment; DMSO (0.1%) was used as control. a Percentage of naïve and memory B cells of all B cells in unstimulated controls. b Ibrutinib treatment reduces the amount of IL-6+ naïve (CD27−) B cells significantly at low dosages, while no significant change was observed in the subset of memory B cells (CD27+). Bars represent mean. Error bars indicate SEM. *p < 0.05, **p < 0.01. c Representative dot plots of naïve (CD27−) B cells. d Representative dot plots of memory (CD27+) B cells
Discussion
Ibrutinib (IC50 = 0.5 nM for BTK inhibition) is a first-in-class, irreversible inhibitor of the BTK that can effectively inhibit BCR signaling by selective active-side binding [14]. The BCR pathway plays an important role in B cell development and survival, as it regulates proliferation, differentiation, and apoptosis [15]. Targeting BCR signaling holds the prospect of significantly altering diseases with pathological B cell activation and proliferation. Consequently, ibrutinib was first tested in the context of B cell malignancies and proved effective in patients with relapsed or refractory non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, or Waldenström macroglobulinemia [16]. Importantly, ibrutinib as a single agent showed proficient tolerability and a low side-effect profile [9, 10, 17]. On the other hand, while a beneficial impact of ibrutinib in autoimmune diseases like rheumatoid arthritis or SSc has been proposed from the very beginning, the transition to a clinical application in this field is incomplete [18].
In SSc, B cells are assumed to be important players in disease onset and progression [19, 20]. B cells of patients with SSc show elevated expression levels of the regulatory surface molecule CD19, which reduces the threshold of BCR signaling, thereby importantly influencing B cell activation and survival [21]. This overexpression of CD19 has been linked to the production of SSc-specific autoantibodies, as well as increased levels of profibrotic cytokines, and contributes to a chronically hyperactivated B cell population [22, 23]. We hypothesize that inhibition of the BTK could be the key to restoring B cell physiology and might, therefore, provide a substantial improvement to SSc treatment.
While the BCR is composed of a unique antigen-specific immunoglobulin binding a certain epitope, TLRs recognize a variety of molecular patterns associated with pathogens or cell damage. Signaling downstream of TLR9, a member of the TLR family recognizing unmethylated single-strand DNA, is known to be augmented in SSc, supporting collagen deposition from fibroblasts and synergizing with BCR signaling for B cell activation and immunoglobulin class-switching [11, 24]. It is suggested that circulating fragments of self-DNA acting as endogenous TLR9-ligands could have a role in SSc development and progression [25, 26]. In our in vitro model, the TLR9-agonist CpG (ODN2006) increased survival of cultured B cells and induced the production of various cytokines and anti-Scl-70-antibodies. While no indications of increased B cell apoptosis were observed, ibrutinib treatment showed convincing potential to counteract the production of key inflammatory cytokines, specifically IL-6 and TNF-α. Both cytokines play pivotal roles in the perpetuation of fibrotic signaling leading to skin thickening and organ fibrotic transformation [27, 28]. As a single agent, ibrutinib combines the effects on IL-6 and TNF-α and could alter the production of more cytokines involved in the pathogenesis of SSc as well. A central transcription factor downstream of TLR9 signaling inducing the production of these inflammatory agents is NFκB. Our flow cytometry analysis of phosphorylated NFκB shows increased activation of TLR9 signaling even 24 h after stimulation. Ibrutinib reduced the abundance of pNFκB significantly, supporting previous findings that describe BTK as a key signaling molecule of the TLR9 pathway [29]. The reduction of anti-Scl-70 under ibrutinib treatment is further encouraging and might be true for other autoantibodies. Even though autoantibodies are more of a diagnostic marker than a prognostic factor in SSc, a contribution to disease development via immune activation cannot be ruled out [30]. In vitro findings suggest that anti-Scl-70-antibodies could be of direct pathogenetic relevance by binding to the cell surface of fibroblasts [31].
In our model, ibrutinib was able to reduce the release of proinflammatory IL-6 and TNF-α at doses even below the effective concentrations achieved in vivo (Fig. 2). Importantly, physiologically applicable doses of ibrutinib showed a biased inhibition of fibrogenic cytokines while maintaining IL-10 and IFN-γ levels. Serum IL-10 levels are generally not decreased in patients with SSc, but a significant reduction in IL-10-producing B cells has been described [5, 32, 33]. The preservation of the anti-fibrotic effects of IL-10 under ibrutinib treatment is considered important in the context of aggravated skin fibrosis in a BIL10−/− mouse model by Matsushita et al. [6] On the other hand, the role of interferon type II (IFN-γ) is controversially discussed in SSc. Some describe a reduction of serum IFN-γ and a pathogenic imbalance of Th1 and Th2 cytokines [33], while others found elevated IFN-γ production without any correlation to clinical outcomes [34]. Functionally, IFN-γ as a Th1-cytokine is categorized to have anti-fibrotic effects and might be reactionarily increased in patients with SSc in an attempt to control fibrotic transformation [35]. Thus, an increase in IFN-γ under ibrutinib treatment could contribute to restore the physiological Th1-Th2-balance.
In a flow cytometric analysis of intracellular cytokines, ibrutinib inhibited the production of IL-6 preferentially in the naïve (CD27−) B cell subpopulation. A reduction of IL-6+ memory (CD27+) B cells was restricted only to high-dose ibrutinib treatment (10 μM). In SSc, the composition of the B cell population is pathologically altered. Memory B cells are reduced and show a high susceptibility for apoptosis-inducing signals, while naïve B cells are numerically increased [20, 36]. Treatment with ibrutinib for 24 h did not change the relative ratio of B cell subsets, but the biased inhibition of IL-6 production represents an indicator for a differential influence of ibrutinib on B cell subpopulations that could translate to meaningful differences between subsets in survival and activation in vivo.
Conclusion
In summary, our in vitro data could pave the avenue for a clinical application of ibrutinib as a novel treatment of the underlying pathogenetic immune imbalance in SSc. Overall, we provide convincing evidence that ibrutinib has the potential to improve B cell pathology contributing to disease onset and progression, encouraging a clinical translation to the benefit of patients with SSc.
Supplementary information
Supplemental Figure 1. Cytokine production by B cells of healthy volunteers with ibrutinib treatment (n = 4). B cells of healthy volunteers were stimulated with CpG for 24 h and treated with DMSO (Control) or ibrutinib (10 μM). With a Legendplex assay, cytokine levels of IL-6, TNF-⍺, IFN-γ and IL-10 were determined in the culture supernatant. Bars represent mean. Error bars indicate SEM. * p < 0.05, ** p < 0.01, *** p < 0.001.
Acknowledgements
We would like to thank the Flow Cytometry Core Facility Berg of the University Hospital Tuebingen as well as Gülay Demirel (Immunopathological Laboratories, University Hospital Tuebingen) for their excellent technical assistance.
Abbreviations
- Anti-Scl-70
Anti-topoisomerase I antibody
- BCR
B cell receptor
- Bregs
Regulatory B cells
- BTK
Bruton’s tyrosine kinase
- CD
Cluster of differentiation
- DGRh
German Society for Rheumatology
- DMSO
Dimethylsulfoxid
- ELISA
Enzyme-linked immunosorbent assay
- FDA
Food and Drug Administration
- SSc
Systemic sclerosis
- IC50
Half-maximal inhibitory concentration
- IFN-γ
Interferon gamma
- IL
Interleukin
- MFI
Mean fluorescence intensity
- ns
Nonsignificant
- PBMCs
Peripheral blood mononuclear cells
- pNFκB
Phosphorylated nuclear factor kappa-light-chain-enhancer of activated B cells
- SD
Standard deviation
- SEM
Standard error of the mean
- Th
T helper
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
Authors’ contributions
JE designed and performed research, analyzed data, and wrote the manuscript. ACP, SDS, HK, and JH acquired the patient samples and performed the experiments. EA, HS, and KAS performed the experiments and analyzed data. ACP, RK, CS, and JH helped interpreting data and assisted in preparing the manuscript. DS designed the experiments, wrote the manuscript, and provided overall guidance. All authors read and approved the final manuscript.
Funding
This work was funded by a Research Grant of the German Society for Rheumatology (DGRh Forschungsfoerderung 2017). JE was supported by the IZKF Postgraduate Program of the Faculty of Medicine Tuebingen (2019-1-18). DS was supported by the Max Eder Research Fellowship Program of the German Cancer Aid (70112548) and the Clinician Scientist Program of the Faculty of Medicine Tuebingen (364-0-0). CS was supported by a Junior Research Group Grant of the Interdisciplinary Centre for Clinical Research (IZKF, 2383-0-0), the Cancer Award of Wuerttemberg and the Clinician Scientist Program of the Faculty of Medicine Tuebingen (390-0-0).
Availability of data and materials
Data are available from the corresponding author on a reasonable request.
Ethics approval and consent to participate
This study was approved by the institutional review board of the Eberhard-Karls-University Tuebingen (IRB approval number 114/2016BO) to be in accordance with the ethical standards and with the Helsinki Declaration of 1975, as revised in 2013.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jakob Einhaus, Email: Jakob.Einhaus@med.uni-tuebingen.de.
Ann-Christin Pecher, Email: Ann-Christin.Pecher@med.uni-tuebingen.de.
Elisa Asteriti, Email: Elisa.Asteriti@med.uni-tuebingen.de.
Hannes Schmid, Email: Hannes.Schmid@gmx.de.
Kathy-Ann Secker, Email: Kathy-Ann.Secker@med.uni-tuebingen.de.
Silke Duerr-Stoerzer, Email: Silke.Duerr-Stoerzer@med.uni-tuebingen.de.
Hildegard Keppeler, Email: Hildegard.Keppeler@med.uni-tuebingen.de.
Reinhild Klein, Email: Reinhild.Klein@med.uni-tuebingen.de.
Corina Schneidawind, Email: Corina.Schneidawind@med.uni-tuebingen.de.
Joerg Henes, Email: Joerg.Henes@med.uni-tuebingen.de.
Dominik Schneidawind, Email: Dominik.Schneidawind@med.uni-tuebingen.de.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13075-020-02153-8.
References
- 1.Rubio-Rivas M, Royo C, Simeón CP, Corbella X, Fonollosa V. Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum. 2014;44:208–219. doi: 10.1016/j.semarthrit.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 2.Elhai M, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 2017;76:1897–1905. doi: 10.1136/annrheumdis-2017-211448. [DOI] [PubMed] [Google Scholar]
- 3.Tyndall AJ, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69:1809–1815. doi: 10.1136/ard.2009.114264. [DOI] [PubMed] [Google Scholar]
- 4.Kowal-Bielecka O, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76:1327–1339. doi: 10.1136/annrheumdis-2016-209909. [DOI] [PubMed] [Google Scholar]
- 5.Matsushita T, Hamaguchi Y, Hasegawa M, Takehara K, Fujimoto M. Decreased levels of regulatory B cells in patients with systemic sclerosis: association with autoantibody production and disease activity. Rheumatology. 2016;55:263–267. doi: 10.1093/rheumatology/kev331. [DOI] [PubMed] [Google Scholar]
- 6.Matsushita T, et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci Adv. 2018;4:eaas9944. doi: 10.1126/sciadv.aas9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khanna D, et al. Efficacy and Safety of Tocilizumab for the Treatment of Systemic Sclerosis: results from a Phase 3 Randomized Controlled Trial [abstract]. Arthritis Rheumatol. 2018;70(suppl 10) https://acrabstracts.org/abstract/efficacy-and-safety-of-tocilizumab-for-the-treatment-of-systemic-sclerosis-results-from-a-phase-3-randomized-controlled-trial/. Accessed 14 Dec 2019.
- 8.Jordan S, et al. Effects and safety of rituximab in systemic sclerosis: an analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015;74:1188–1194. doi: 10.1136/annrheumdis-2013-204522. [DOI] [PubMed] [Google Scholar]
- 9.Byrd JC, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ML, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fang F, et al. Toll-like receptor 9 signaling is augmented in systemic sclerosis and elicits transforming growth factor β-dependent fibroblast activation: TLR9 signaling promotes fibrotic responses. Arthritis Rheumatol. 2016;68:1989–2002. doi: 10.1002/art.39655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. Signaling to NF-κB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Su TT, et al. PKC-β controls IκB kinase lipid raft recruitment and activation in response to BCR signaling. Nat Immunol. 2002;3:780–786. doi: 10.1038/ni823. [DOI] [PubMed] [Google Scholar]
- 14.Honigberg LA, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci. 2010;107:13075–13080. doi: 10.1073/pnas.1004594107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol. 2002;2:945–956. doi: 10.1038/nri955. [DOI] [PubMed] [Google Scholar]
- 16.Advani RH, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31:88–94. doi: 10.1200/JCO.2012.42.7906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coutre SE, et al. Long-term safety of single-agent ibrutinib in patients with chronic lymphocytic leukemia in 3 pivotal studies. Blood Adv. 2019;3:1799–1807. doi: 10.1182/bloodadvances.2018028761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Z, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 19.Brown M, O’Reilly S. The immunopathogenesis of fibrosis in systemic sclerosis. Clin Exp Immunol. 2018. 10.1111/cei.13238. [DOI] [PMC free article] [PubMed]
- 20.Sanges S, et al. Role of B cells in the pathogenesis of systemic sclerosis. Rev Méd Interne. 2017;38:113–124. doi: 10.1016/j.revmed.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 21.Inaoki M, Sato S, Weintraub BC, Goodnow CC, Tedder TF. CD19-regulated signaling thresholds control peripheral tolerance and autoantibody production in B lymphocytes. J Exp Med. 1997;186:1923–1931. doi: 10.1084/jem.186.11.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asano N, et al. B lymphocyte signaling established by the CD19/CD22 loop regulates autoimmunity in the tight-skin mouse. Am J Pathol. 2004;165:641–650. doi: 10.1016/S0002-9440(10)63328-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saito E, et al. CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J Clin Invest. 2002;109:1453–1462. doi: 10.1172/JCI0215078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pone EJ, et al. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-κB pathway. Nat Commun. 2012;3:767. [DOI] [PMC free article] [PubMed]
- 25.Lei W, et al. Abnormal DNA methylation in CD4+ T cells from patients with systemic lupus erythematosus, systemic sclerosis, and dermatomyositis. Scand J Rheumatol. 2009;38:369–374. doi: 10.1080/03009740902758875. [DOI] [PubMed] [Google Scholar]
- 26.Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Reilly S, Cant R, Ciechomska M, van Laar JM. Interleukin-6: a new therapeutic target in systemic sclerosis? Clin Transl Immunol. 2013;2:e4. doi: 10.1038/cti.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rip J, et al. Toll-like receptor signaling drives Btk-mediated autoimmune disease. Front Immunol. 2019;10:95. [DOI] [PMC free article] [PubMed]
- 30.Kayser C, Fritzler MJ. Autoantibodies in systemic sclerosis: unanswered questions. Front Immunol. 2015;6:167. [DOI] [PMC free article] [PubMed]
- 31.Hénault J, Tremblay M, Clément I, Raymond Y, Senécal J-L. Direct binding of anti-DNA topoisomerase I autoantibodies to the cell surface of fibroblasts in patients with systemic sclerosis: antifibroblast autoantibodies in SSc. Arthritis Rheum. 2004;50:3265–3274. doi: 10.1002/art.20515. [DOI] [PubMed] [Google Scholar]
- 32.Mavropoulos A, et al. Breg cells are numerically decreased and functionally impaired in patients with systemic sclerosis: Breg cells in systemic sclerosis. Arthritis Rheumatol. 2016;68:494–504. doi: 10.1002/art.39437. [DOI] [PubMed] [Google Scholar]
- 33.Scala E, et al. Cytokine and chemokine levels in systemic sclerosis: relationship with cutaneous and internal organ involvement. Clin Exp Immunol. 2004;138:540–546. doi: 10.1111/j.1365-2249.2004.02642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gourh P, et al. Plasma cytokine profiles in systemic sclerosis: associations with autoantibody subsets and clinical manifestations. Arthritis Res Ther. 2009;11:R147. doi: 10.1186/ar2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baraut J, Michel L, Verrecchia F, Farge D. Relationship between cytokine profiles and clinical outcomes in patients with systemic sclerosis. Autoimmun Rev. 2010;10:65–73. doi: 10.1016/j.autrev.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 36.Yoshizaki A. B lymphocytes in systemic sclerosis: abnormalities and therapeutic targets. J Dermatol. 2016;43:39–45. doi: 10.1111/1346-8138.13184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Cytokine production by B cells of healthy volunteers with ibrutinib treatment (n = 4). B cells of healthy volunteers were stimulated with CpG for 24 h and treated with DMSO (Control) or ibrutinib (10 μM). With a Legendplex assay, cytokine levels of IL-6, TNF-⍺, IFN-γ and IL-10 were determined in the culture supernatant. Bars represent mean. Error bars indicate SEM. * p < 0.05, ** p < 0.01, *** p < 0.001.
Data Availability Statement
Data are available from the corresponding author on a reasonable request.