

Abstract
Abnormalities in protein localization, function, and posttranslational modifications are targets of schizophrenia (SCZ) research. As a major contributor to the synthesis, folding, trafficking, and modification of proteins, the endoplasmic reticulum (ER) is well-positioned to sense cellular stress. The unfolded protein response (UPR) is an evolutionarily conserved adaptive reaction to environmental and pathological perturbation in ER function. The UPR is a highly orchestrated and complex cellular response, which is mediated through the ER chaperone protein, BiP, three known ER transmembrane stress sensors, protein kinase RNA-like ER kinase (PERK), activating transcription factor-6 (ATF6), inositol requiring enzyme 1α (IRE1α), and their downstream effectors. In this study, we measured protein expression and phosphorylation states of UPR sensor pathway proteins in the dorsolateral prefrontal cortex (DLPFC) of 22 matched pairs of elderly SCZ and comparison subjects. We observed increased protein expression of BiP, decreased PERK, and decreased phosphorylation of IRE1α. We also observed decreased p-JNK2 and increased sXBP1, downstream targets of the IRE1α arm of the UPR. The disconnect between decreased p-IRE1α and increased sXBP1 protein expression led us to measure sXbp1 mRNA. We observed increased expression of the ratio of sXbp1/uXbp1 transcripts, suggesting that splicing of Xbp1 mRNA by IRE1α is increased and drives upregulation of sXBP1 protein expression. These findings suggest an abnormal pattern of UPR activity in SCZ, with specific dysregulation of the IRE1α arm. Dysfunction of this system may lead to abnormal responses to cellular stressors and contribute to protein processing abnormalities previously observed in SCZ.
Subject terms: Schizophrenia, Neuroscience
Introduction
The onset of schizophrenia (SCZ) occurs during the neurodevelopmental period between adolescence and adulthood, when important neural connections are formed and strengthened to facilitate cognitive maturation [1, 2]. These connections use multiple neurotransmitter systems that have been implicated in SCZ [3–5]. The myriad neurotransmitter receptor abnormalities found in SCZ may suggest dysregulation of central pathways critical for modulation of these multiple systems. The endoplasmic reticulum (ER) is a major intracellular organelle that facilitates the synthesis and processing of many proteins [6], including those associated with neurotransmission. The ER has multiple pathways that are responsible for regulating protein quality control and responding to cellular stress. The expression of these pathways is, in part, regulated by the unfolded protein response (UPR), an evolutionarily conserved biochemical pathway that restores cellular homeostatic mechanisms. The UPR is mediated through three known ER transmembrane stress sensors, protein kinase RNA-like ER kinase (PERK), activating transcription factor-6 (ATF6), and inositol requiring enzyme 1α (IRE1α) [7, 8] (Fig. 1). Under normal conditions, ER stress sensors are maintained in an inactive state within the ER membrane through association with the chaperone protein, glucose regulated protein 78 (Grp78), also known as BiP [9]. In addition to the stress sensors, BiP associates with unfolded and misfolded proteins to facilitate proper protein processing [10–12]. Accumulation of misfolded proteins leads to BiP dissociation from PERK, IRE1α, and ATF6 [13, 14], which can trigger oligomerization, autophosphorylation, and/or translocation of the UPR sensors, leading to activation of the UPR [15, 16]. When activated, ATF6 translocates to the Golgi, where it undergoes cleavage and is then targeted to the nucleus [17–19]. In the nucleus, ATF6 acts as a transcription factor to promote UPR target gene expression [20]. PERK and IRE1α both form homodimers in response to BiP dissociation [21]. Once these complexes are formed they are able to autophosphorylate themselves. Autophosphorylated PERK phosphorylates eukaryotic translation initiation factor 2α (eIF2α) [21]. Phosphorylated eIF2α is no longer able to associate with the translation initiation complex, leading to decreased global protein synthesis and increased expression of activating transcription factor 4 (ATF4) [21]. ATF4 promotes expression of proteins associated with autophagy and apoptosis [22]. Autophosphorylated IRE1α has both kinase and RNAse activity, and a major downstream effect of IRE1α activity is unconventional mRNA splicing of the transcription factor x-box binding protein 1 (XBP1) [23, 24]. The spliced form of XBP1 (sXBP1) acts as a transcription factor and regulates expression of lipid biosynthetic enzymes, proteins responsible for ER quality control (ERQC), and ER associated degradation (ERAD) [25, 26].
Fig. 1.

Signaling pathways from three ER transmembrane stress sensors during unfolded protein response (UPR) in schizophrenia. a ER stress stimulates the activation of the three ER transmembrane stress sensors, protein kinase RNA-like ER kinase (PERK), activating transcription factor-6 (ATF6), and inositol requiring enzyme 1α (IRE1α), that are involved in the unfolded protein response (UPR). a Under basal, unstressed conditions, the UPR sensors are present within the ER membrane. The heat shock protein BiP (Grp78) binds these receptors and prevents them from forming homodimers and from translocating out of the cell. b After dissociation of BiP, ATF6 translocates to the Golgi where it is cleaved and trafficked to the nucleus, while PERK and IRE1α form homodimers and autophosphorylate. Activation of the PERK pathway leads to phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) and increased expression of activating transcription factor 4 (ATF4), while activation of the IRE1α pathway leads to unconventional mRNA splicing of the transcription factor x-box binding protein 1 (XBP1). In addition, IRE1α activation recruits TNF receptor associated factor 2 (TRAF2) and results in phosphorylation of apoptosis signal-regulating kinase 1 (ASK1), which then phosphorylates the transcription factor c-Jun N-terminal kinase (JNK). ATF6, ATF4, and spliced XBP1 (sXBP1) function as transcription factors of UPR target genes, such as ER chaperones, the ER quality control system (ERQC) and ER-associated degradation (ERAD)
The UPR is known to regulate cell differentiation, survival, and death [6, 27, 28]. While activation of the UPR is traditionally associated with increased cellular stress and misfolded proteins, constitutive activity of the UPR is also required in cells with a high secretory load, such as neurons [27, 29]. This leads to increased sensitivity of the brain to abnormalities in the UPR. This is reflected in studies that implicate the UPR in neurodegenerative diseases including Alzheimer’s Disease [30], Parkinson’s Disease [31], amyotrophic lateral sclerosis [32], Huntington’s Disease [33, 34], and Creutzfeldt Jacob Disease [35], as well as in psychiatric disorders including bipolar disorder, major depressive disorder, and SCZ [36–39].
Previous reports demonstrating abnormal N-linked glycosylation, subcellular localization, and expression of receptor associated-proteins of both glutamatergic and GABAergic systems in SCZ brain [40–45] are consistent with abnormal protein processing and subcellular targeting of neurotransmitter receptors. We propose that a potential mechanism that reconciles these findings in SCZ is abnormal synthesis and handling of receptor proteins in the ER. Previous work from our lab has demonstrated abnormalities in various ER-related pathways, including ERAD and glycophosphatidylinositol-associated protein quality control, supporting a role for the ER in SCZ pathophysiology [46, 47]. The central role of the UPR in regulating protein processing led us to hypothesize that abnormalities of the UPR may be associated with ER abnormalities in SCZ brain. To test this, we measured expression of UPR sensors and associated proteins to further define UPR expression and regulation in the dorsolateral prefrontal cortex (DLPFC) in SCZ and matched comparison subjects.
Materials and methods
Human subjects
Samples of dorsolateral prefrontal cortex were obtained from the Mount Sinai/Bronx Veterans Administration Medical Center Department of Psychiatry Brain Collection. Assessment, consent, and postmortem procedures were conducted as required by the Institutional Review Boards of Pilgrim Psychiatric Center [48, 49], Mount Sinai School of Medicine and the Bronx Veterans Administration Medical Center. A total of 22 subjects diagnosed with SCZ based on DSM-III-R criteria were pairwise matched with comparison subjects by sex, age, tissue pH, and postmortem interval (PMI) (Supplementary Table S1). Brains from subjects were removed at autopsy and sliced into 0.8–1 cm slabs in the coronal plane. Full thickness of gray matter from the cortex was blocked into 1 cm3 cubes and stored at −80 °C until use [48, 49].
Antipsychotic treated rats
Male Sprague-Dawley rats were housed in pairs and treated with chronic administration of either 28.5 mg/kg haloperidol decanoate (HAL; N = 10) or vehicle (VEH; N = 10) delivered once every 3 weeks for 9 months via intramuscular injection, for a total of 12 injections. This method of drug delivery, length of treatment, and dose has been previously described [50–52]. Animals were euthanized by rapid decapitation following CO2 administration, frontal cortexes were dissected on ice, then snap frozen and stored at −80 °C until use. The Institutional Animal Care and Use Committee of the University of Alabama at Birmingham approved all procedures using these animals.
Protein expression studies
Tissue preparation
For protein expression studies, tissue samples from each subjects were homogenated on ice in 5 mM Tris–HCl pH 7.5, 0.32 mM sucrose with a protease inhibitor tablet and a phosphatase inhibitor tablet (Complete Mini, EDTA-free and PhosSTOP, both from Roche Diagnostics, Mannheim, Germany) using a glass tissue grinder. Protein concentration was determined using a BCA protein assay kit (Thermo Scientific, Rockford, Illinois). After homogenization, samples were stored at −80 °C until use.
Western immunoblotting
Samples for western blot analyses were prepared with Milli-Q water and sample buffer (6X solution: 4.5% sodium dodecyl sulfate (SDS), 1.5% β-mercaptoethanol, 0.018% bromophenol blue, and 36% glycerol in 170 mM Tris-HCl, pH 6.8) and heated at 70 °C for 10 min. Samples were then loaded on 4–12% Bis-Tris gels (Invitrogen), subjected to SDS-polyacrylamide gel electrophoresis, and transferred to nitrocellulose membranes using standard semi-dry transfer methods. To reduce nonspecific binding, blots were incubated in either 50% Odyssey Blocking buffer (Li-Cor) in Tris-buffered saline (TBS) or 5% bovine serum albumin in TBS for 1 h at room temperature (RT) prior to primary antibody incubation. Conditions for probing were optimized to be within the linear range of detection for each primary antibody (Supplementary Table S2). Antibodies were stored and handled according to the manufacturer’s instructions. The specificity of commercially available antibodies was evaluated by western blotting. Validation of the specificity of commercially available antibodies that were studied are given in Supplementary Fig. F1.TBS + 0.1% Tween-20 was used for wash steps before and after a 1 h incubation at RT with the appropriate secondary antibody, and membranes rinsed with Milli-Q water prior to scanning. Membranes were scanned using a LiCor Odyssey scanner at a resolution of 169 µm and an intensity level of five.
Xbp1 mRNA analysis
Total RNA isolation
RNA was isolated using a RNeasy Mini Kit (cat# 74106 Applied Qiagen®). For each subject, RNA concentration was determined by spectrophotometry (ND1000, Thermo Fischer Scientific, Inc.) at 260 nm and equal amounts of RNA were used to synthesize cDNA using a high capacity cDNA Reverse Transcription Kit (cat# 4368813, Applied Biosystems®). RIN values of all samples were determined. Over 80% of samples had RINs ≥ 6, an integrity value previous reported suitable for RT-qPCR studies in brain tissue [53].
Electrophoretic analysis of Xbp1 mRNA splicing
PCR was performed with Platinum™ Green Hot Start PCR Master Mix (cat#13001012, Thermo Fischer Scientific, Inc), according to the manufacturer’s recommendations. In brief, 1 μM of each primer was mixed with 12.5 μM of Platinum™ Green Hot Start PCR Master Mix, 5 μl of Platinum GC Enhancer and 5 μl of cDNA. To prepare samples for analysis of sXbp1 mRNA, we treated the samples with PstI. Under unmodified conditions, the amount of uXbp1 is much greater than Xbp1. The section removed by IRE1α-mediated splicing is 26 bp, a small enough difference that the excess of uXbp1 masks detection of the spliced form. Therefore, we used PstI, a restriction endonuclease that cleaves Xbp1 at a site only contained within the unspliced form, to specifically digest uXbp1 and unmask sXbp1. Samples (25 μl) were incubated with PstI (1 μl) overnight at 37 °C, followed by preamplification of Xbp1 transcript products using Xbp1 primer sets (forward: 5′-TTACGAGAGAAAACTCATGG-3′ /reverse: 5′-TCTACCCAGAAGGACCCAGT-3′). Samples were then resolved on 2% agarose gels containing ethidium bromide at 60 mV for 1.5 hr before visualizing by UV light. Images were collected with Gel Doc™ XR + Gel Documentation System (Bio-Rad Laboratories, USA) and the identity of the PCR products obtained with the Xbp1 primers was verified by DNA sequencing.
Data and statistical analysis
Image Studio Lite Version 4.0.21 (LiCor, Lincoln, NE, USA) was used to determine integrated intensity values from both western blots and agarose gels. All samples were run in duplicate, and after intralane normalization these values were averaged. For proteins, integrated intensity values were normalized to intralane values of Valosin Containing Protein, which has been reported to not be differentially expressed in SCZ brain [54]. Expression of phosphorylated forms of proteins was normalized to the expression of their total forms. The relative amount of Xbp1 mRNA splicing was defined as the ratio of sXbp1 to uXbp1 mRNA levels. Given that many dependent variables were not normally distributed, primary analyses were performed the nonparametric Wilcoxon matched-pairs signed rank test. To determine the effect of potential covariates, Pearson or Spearman rank correlations between protein expression and subject age, tissue pH, and PMI were performed for all dependent measures. Prior to these correlation analyses, D’Agostino & Pearson omnibus normality tests were performed to determine if data were normally distributed. If a significant Pearson correlation was observed, an ANCOVA was then used to determine the effect of diagnosis when accounting for the relevant covariates. Alternatively, if a significant Spearman correlation was observed, a nonparametric ANCOVA using the Quade method was performed. All statistical analyses were performed in GraphPad Prism 6 (La Jolla, CA, USA) or STATISTICA version 7 (StatSoft, Inc, Tulsa, OK, USA) with α = 0.05.
Results
UPR sensor expression and activation is abnormal in schizophrenia
We measured expression of BiP, the three UPR stress sensors, PERK, IRE1α, and ATF6, and their corresponding activated states, p-PERK, p-IRE1α, and cl-ATF6. We observed increased in SCZ BiP expression (W = 235, p < 0.0001), decreased PERK expression (W = −195, p = 0.0008), and no change in IRE1α or ATF6 expression (Fig. 2, Table 1). In addition, p-IRE1α expression (W = −139, p = 0.0229) was decreased, while p-PERK and cl-ATF6 were unchanged (Fig. 2, Table 1). Analysis of covariates demonstrated a positive correlation between PERK expression and age (r = 0.40, p = 0.007), and a follow-up ANCOVA indicated that when controlling for age, there is no longer a significant effect of diagnosis, suggesting that changes in PERK expression between the subject groups are driven by differences in age rather than illness. In addition, no difference in expression of BiP, PERK, or p-IRE1α was observed in rats treated with haloperidol (Fig. 2).
Fig. 2.

Altered expression level of UPR signaling pathway- related ER transmembrane stress sensor proteins in schizophrenia. Protein expression of UPR signaling pathway-related proteins was measured in paired schizophrenia (SCZ) and comparison (COMP) subjects. a Representative western blots are shown for paired comparison and schizophrenia subjects. b Increased expression was observed for BiP, and decreased expression was observed for PERK, and p-IRE1α, though the ratio of p-IRE1α to total IRE1α was not significantly different. c BiP, PERK, p-IRE1α, and the ratio of p-IRE1α to total IRE1α expression were not significantly different between rats that had received chronic haloperidol (HAL) treatment and those that were vehicle treated (VEH). Data are expressed as means ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001
Table 1.
Quantification of differentially expressed proteins of ER transmembrane stress sensors in schizophrenia and comparison subjects
| Target protein | Comparison | Schizophrenia | W value | p-value |
|---|---|---|---|---|
| BiP | 1.01 (0.75–1.63) | 1.65 (1.23–5.92) | 235 | <0.0001 |
| ATF6 pathway | ||||
| ATF6 | 0.35 (0.23–0.80) | 0.42 (0.21–0.77) | 19 | |
| cl-ATF6 | 0.37 (0.21–0.45) | 0.29 (0.18–0.48) | −29 | |
| cl-ATF6/ATF6 | 0.75 (0.61–1.01) | 0.70 (0.59–0.78) | −109 | |
| PERK pathway | ||||
| PERK | 0.55 (0.47–0.77) | 0.46 (0.35–0.67) | −195 | 0.0008 |
| p-PERK | 0.69 (0.44–1.28) | 0.88 (0.41–1.43) | −25 | |
| p-PERK/PERK | 1.35 (0.78–2.84) | 2.06 (0.65–3.35) | 95 | |
| eIF2α | 0.22 (0.07–0.57) | 0.20 (0.07–1.31) | 57 | |
| p-eIF2α | 2.07 (0.94–8.53) | 1.75 (0.97–8.45) | 39 | |
| p-eIF2α/ eIF2α | 14.45 (9.30–20.75) | 11.17 (9.11–20.12) | 1 | |
| ATF4 | 0.047 (0.030–0.064) | 0.052 (0.040–0.077) | 45 | |
| IRE1α pathway | ||||
| IRE1α | 1.35 (0.87–4.86) | 1.16 (0.98–4.17) | −25 | |
| p- IRE1α | 0.50 (0.25–0.91) | 0.28 (0.14–0.60) | −139 | 0.0229 |
| p- IRE1α/IRE1α | 0.34 (0.19–0.89) | 0.20 (0.09–0.43) | −77 | |
| IRE1α—XBP1 pathway | ||||
| uXBP1 | 0.014 (0.008–0.215) | 0.013 (0.008–0.211) | −55 | |
| sXBP1 | 0.020 (0.017–0.035) | 0.033 (0.020–0.045) | 173 | 0.0037 |
| sXBP1/uXBP1 protein | 1.62 (1.25–2.38) | 2.01 (1.58–2.59) | 143 | 0.0190 |
| sXBP1/uXBP1 mRNA | 0.20 (0.14–0.34) | 0.28 (0.17–0.62) | 169 | 0.0047 |
| IRE1α—JNK pathway | ||||
| ASK1 | 0.28 (0.15–0.45) | 0.42 (0.19–0.65) | 29 | |
| p-ASK1 | 0.58 (0.37–0.89) | 0.44 (0.36–0.70) | −109 | |
| p-ASK1/ASK1 | 1.93 (1.05–3.11) | 1.23 (0.95–2.77) | −65 | |
| JNK1 | 0.045 (0.035–0.078) | 0.052 (0.033–0.069) | −61 | |
| p-JNK1 | 0.13 (0.07–0.21) | 0.10 (0.08–0.13) | −109 | |
| p-JNK1/JNK1 | 2.35 (1.80–3.04) | 2.06 (1.74–2.89) | −69 | |
| JNK2 | 0.064 (0.030–0.084) | 0.069 (0.034–0.102) | 65 | |
| p-JNK2 | 0.13 (0.10–0.20) | 0.11 (0.08–0.14) | −165 | 0.0059 |
| p-JNK2/JNK2 | 2.40 (1.83–3.27) | 2.07 (1.29–2.55) | −177 | 0.0029 |
Data are reported as median (IQR). Data were analyzed using a Wilcoxon matched pairs signed rank test, npair = 22.
Expression and activation of UPR effectors suggests abnormal activation of the IRE1α pathway
In addition to the UPR sensors, we measured expression and activation of UPR downstream effectors. From the PERK pathway, we measured expression of eIF2α, p-eIF2α, and ATF4, and found no differences between comparison and SCZ subjects. In the IRE1α pathway, we measured expression of both the spliced and unspliced forms of XBP1, TRAF2, and both the expression and phosphorylation of ASK1, JNK1, and JNK2. We found a decrease in SCZ of both sXBP1 (W = 173, p = 0.004) and the ratio of sXBP1/uXBP1 (W = 143, p = 0.02). Additionally, both phosphorylation of JNK2 (W = −165, p = 0.006) and the ratio of p-JNK2/JNK2 (W = −177, p = 0.003) was decreased in SCZ (Fig. 3, Table 1). No changes in uXBP1, TRAF2, ASK1, or JNK1 expression were observed, nor was there abnormal phosphorylation of ASK1 or JNK1. None of these dependent measures were significantly correlated with age, pH, or PMI, and were not different between rats treated with vehicle or haloperidol (Fig. 3).
Fig. 3.

IRE1α downstream effectors are abnormally expressed and phosphorylated in schizophrenia. Protein expression and phosphorylation of proteins downstream of p-IRE1α signaling was measured in paired schizophrenia (SCZ) and comparison (COMP) subjects. a p-IRE1α leads to splicing of Xbp1 mRNA and synthesis of the spliced form of XBP1 protein (sXBP1). Representative western blots of sXBP1 and unspliced XBP1 (uXBP1) are shown; /both sXBP1 expression and the ratio of sXBP1/uXBP1 are increased in SCZ. b p-IRE1α also has kinase activity that leads to phosphorylation of protein in the ASK1/JNK signaling cascade. Representative western blots of proteins associated with p-IRE1α kinase activity are shown; both phosphorylation of JNK2 (p-JNK2) and the ratio of p-JNK2/JNK2 are decreased in SCZ. c p-JNK2, sXBP1, and the ratios found changed in SCZ were not significantly different in rats that had received chronic haloperidol (HAL) treatment when compared with vehicle treated animals (VEH). Data are expressed as means ± SEM. *p < 0.05, **p < 0.01
Xbp1 mRNA splicing is increased in schizophrenia
We found decreased p-IRE1α and increased sXBP1 protein expression. Activation of IRE1α leads to an unconventional posttranscriptional splicing of uXbp1 mRNA to produce a spliced form of sXbp1 mRNA that is translated as sXBP1 protein [55, 56]. To determine at what stage the disconnect between IRE1α activation, as measured by p-IRE1α expression, and XBP1 splicing, as measured by sXBP1 protein expression, occurs, we measured mRNA expression of different forms of Xbp1. Similar to protein expression, we found increased expression of sXbp1/uXbp1 mRNA (W = 169, p = 0.005) in SCZ (Fig. 4, Table 1).
Fig. 4.
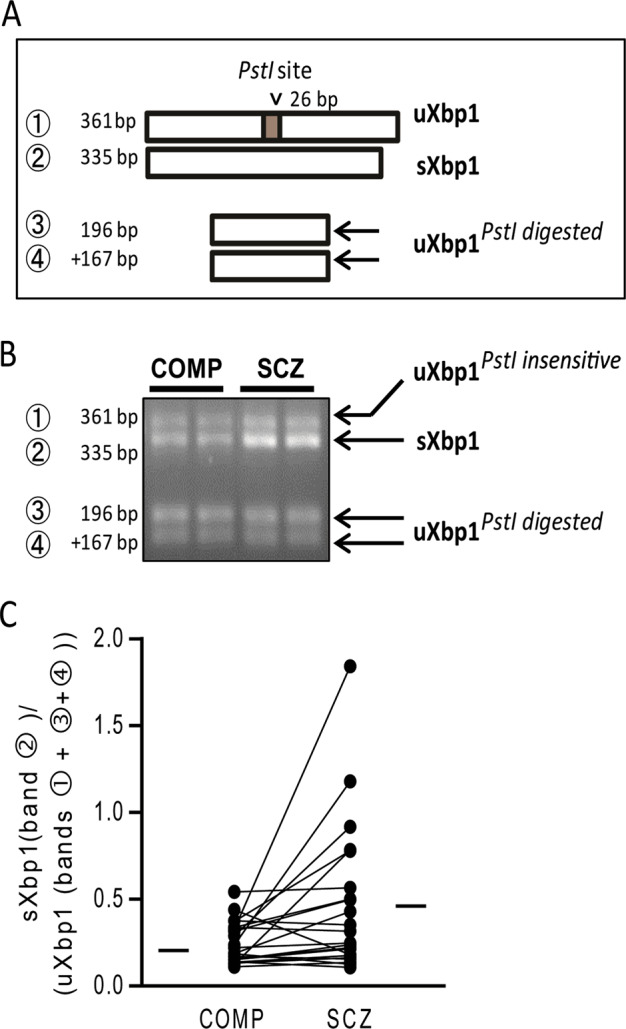
Unconventional Xbp1 mRNA Splicing is changed in schizophrenia. a Diagram illustrating the predicted mRNA products of Xbp1 and their digestion by PstI. uXbp1 (band 1) contain a Pstl site that leads to cleavage into two transcripts (bands 3 and 4), although some of this population appears to be resistant to Pstl digestion. The unique PstI site is removed when Xbp1 is spliced by IRE1α (sXbp1), resulting in this band (2) being unaffected by Pstl digestion. b Representative resultant mRNA digested with PstI follow by emulsion cDNA and PCR amplification, and analyzed by 2% agarose gel electrophoresis to visualize the spliced (s) and unspliced (u) products. c The ratio of sXbp1/ uXbp1 mRNA levels was increased in paired schizophrenia (SCZ) and comparison (COMP) subjects. Data are expressed as means ± SEM. **p < 0.01
Discussion
In this study, we investigated the UPR through protein and activity-marker expression levels in the DLPFC in SCZ. Of the initial stress sensors of the UPR, we observed increased expression of BiP, decreased expression of PERK, and decreased phosphorylated IRE1α. We also measured downstream effectors of both the PERK and IRE1α arms of the UPR. While no change in PERK substrates was found, we observed decreased p-JNK2 and increased sXBP1, downstream targets of the IRE1α arm of the UPR. The disconnect between p-IRE1α, a marker of IRE1α activation, and sXBP1 protein expression was surprising and led us to measure sXbp1 mRNA directly. We observed increased expression of the ratio of sXbp1/uXbp1, suggesting that splicing of Xbp1 mRNA by IRE1α itself has increased and drives upregulation of sXBP1 protein expression rather than a change in sXBP1 degradation or translation. These findings suggest abnormal expression and regulation of the UPR and its downstream targets in SCZ.
These findings support a growing body of evidence for a fundamental deficit in ER protein processing pathways. Increased protein expression of BiP, phosphorylation of IRE1α and PERK, and cleavage of ATF6 are commonly used markers of UPR activity [57]. BiP is a molecular chaperone highly enriched in the ER, and has been found to be dysregulated in several proteomic studies in SCZ [58–61]. Within the lumen of the ER, BiP primarily interacts with unfolded substrates and protein folding machinery, and appears to have an active role in facilitating protein folding [62]. Cellular stress can lead to increased BiP expression, and increased expression of BiP protein has become a marker for ER stress [11, 15, 57, 63]. In this study, we observed increased BiP expression, suggesting a role for cellular stress in SCZ. This is consistent with previous reports that have demonstrated markers of increased oxidative stress, such as increased lipid peroxidation, reactive oxygen species (ROS), and decreased antioxidant levels in postmortem brain in SCZ [64–67].
In contrast to upregulation of BiP, we also observed decreased phosphorylation of IRE1α in SCZ. Of the three sensor arms, the IRE1α pathway is the most evolutionarily conserved signaling branch [24, 68]. IRE1α has both serine/threonine kinase and endoribonuclease (RNase) activities, which are activated upon dissociation of BiP and subsequent dimerization of and autophosphorylation in response to ER stress [6, 27, 28, 69]. Phosphorylation of IRE1α attracts TRAF2, which in turn promotes association of ASK1 [70]. The formation of this complex allows for IRE1α to phosphorylate ASK1, which then phosphorylates JNK [6, 27, 28, 69]. JNK is known to have three distinct isoforms, JNK1, JNK2, and JNK3, and JNK1/2 both upregulate proteins associated with apoptosis [6, 27, 28, 69]. Consistent with reduced p-IRE1α, phosphorylation of JNK2 was also decreased in SCZ. P-ASK1 was not significantly decreased. Decreased p-IRE1α and p-JNK2 suggests that the IRE1α-ASK1-JNK signaling pathway is downregulated, and that apoptotic processes may be attenuated in SCZ. This would be consistent with and may provide a mechanistic explanation for previous observations that there is no evidence of increased cell death despite dysregulation of cellular homeostasis in the brain in SCZ [71, 72]. Phosphorylation of JNK has also been observed in the ACC of SCZ subjects [73], supporting a more widespread role for JNK signaling downregulation in this illness.
A unique target of IRE1α RNase activity is Xbp1 mRNA, from which IRE1α removes a 26-nucleotide intron, resulting in a translational frameshift and the sXBP1 production of the spliced form of Xbp1, sXBP1 acts as a transcription factor and promotes expression of the ER translocon, ER chaperones, proteins that facilitate ERAD, glycosylation enzymes, and phospholipids [6, 27, 28, 69]. Upregulation of these pathways increases resilience of the cell to ER stress, with chaperones and glycosylation enzymes facilitating increased demand for protein folding, ERAD components clearing misfolded proteins, and phospholipids allowing for the expansion of the ER membrane during ER stress [6, 27, 28, 69]. Despite observing decreased p-IRE1α in SCZ, we observed upregulation of both sXbp1 mRNA and protein expression, suggesting upregulation of sXBP1-dependent processes. Supporting this, ERQC and ERAD components have been shown to be upregulated in the DLPFC in SCZ [46]. In addition, our lab has previously reported abnormal immature N-linked glycosylation, an ER-specific protein modification, of components of the glutamate and GABA systems in the frontal cortex in SCZ [40, 45, 74, 75]. Proper regulation of the IRE1- XBP1 pathway is particularly important for neural development [76, 77], and sXBP1 has been shown to specifically impact neurotransmitter systems in a developmental-dependent manner [29, 77]. In Xbp1-/- neurons, Hayashi et al. [77] observed inhibition of BDNF-induced expression of GABAergic markers. In C. elegans, mutants lacking either ire-1 or xbp-1 demonstrated development-dependent inhibition of glutamate receptor trafficking [29]. In SCZ, reduced expression of GABAergic markers has been repeatedly observed, and dysfunction in glutamatergic signaling is believed to be a key component of the illness [78, 79]. As a central regulator of the processes impacting these systems, sXBP1-dependent transcription may contribute to developmental deficits and neurotransmitter dysfunction.
The kinase and RNase activity of IRE1α have been shown to be independently regulated, with certain conformations promoting high kinase activity, others promoting high RNase activity, and some resulting in a combination of the two [80]. A potential mechanism to explain the disconnect between downregulation of p-IRE1α and upregulation of Xbp1 splicing is a selective downregulation of high-kinase IRE1α complexes and preservation of oligomeric complexes with low kinase activity and high RNase activity [80]. Alternatively, Hsp72 has been shown to bind to IRE1α and specifically increase production of sXbp1 without increasing JNK2 phosphorylation [81]. This process appears to be cytoprotective [81]. These mechanisms are not mutually exclusive, and further work to define IRE1α regulation is necessary to fully understand how IRE1α dynamics are impacted in SCZ.
In addition to Xbp1 splicing, IRE1α RNase activity also mediated regulated IRE1-dependent decay of mRNAs (RIDD), in which IRE1α degrades ER-localized transcripts to decrease translation and therefore reduce demand for protein folding and quality processes in the face of cellular stress [82, 83]. Abnormal phosphorylation IRE1α and Xbp1 splicing both suggest that this system is also likely dysregulated. While we are unable to directly demonstrate the impact of IRE1α RNase activity on ER-associated transcripts in this study, this process may contribute to the disconnect between transcript and protein expression in SCZ postmortem brain [84, 85]. In addition to dysregulation of the IRE1α pathway, we also observed decreased PERK expression in SCZ. However, this expression was correlated with age and an ANCOVA indicated that, when controlling for age, the effect of diagnosis on PERK expression is no longer significant. This suggests that what we observed is an effect of aging on the UPR. Age-related neurological disorders have been associated with UPR dysfunction, suggesting that this system is particularly susceptible to stressors during the aging [30–34, 69, 86, 87]. In addition, PERK mRNA has been specifically shown to be reduced in the hippocampus of aged rats [88]. This suggests that the UPR, and the PERK pathway in particular, is sensitive to aging and likely underlies an altered capacity to deal with ER stress in elderly individuals.
This study has several limitations. While our subjects were well-matched, they were elderly and the pathophysiology of subjects who are at later stages of the illness may not reflect abnormalities associated with earlier stages of the disorder. Using postmortem tissue limited us to investigating the UPR pathway in a cross-sectional fashion. It has been previously shown that constitutive UPR activity regulates distinct pathways from ER-stress induced UPR activity [89]. Therefore, while we can conclude that markers of constitutive UPR activity are abnormally expressed, we can only speculate on whether these pathways might respond to cellular stress differently in SCZ. Studies detailing the effects of protein and phosphorylation abnormalities on both constitutive and stress-induced UPR activity would be necessary to elucidate the mechanisms by which these abnormalities are contribute to pathophysiology of SCZ. In addition, the majority of the subjects with SCZ were on antipsychotics for long periods of time. To address this confounding factor we measured the changes we observed in postmortem brain tissue in rats chronically treated with haloperidol, a commonly used first generation antipsychotic, and found no changes due to treatment. This suggests that antipsychotic treatment is not sufficient to induce these changes in brain, making our findings more likely to be due to the illness rather than chronic antipsychotic treatment.
In summary, we examined expression of key elements in the UPR in the DLPFC of patients with SCZ and matched comparison subjects. These findings suggest an abnormal pattern of constitutive UPR activity, with upregulation of cell-protective pathways and downregulation of factors known to promote apoptosis. These abnormalities may contribute to aberrant protein processing and trafficking, and dysfunctional response to cellular stress in SCZ.
Supplementary information
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41380-019-0537-7) contains supplementary material, which is available to authorized users.
References
- 1.Spear LP. Adolescent neurodevelopment. J Adolesc Health. 2013;52(2Suppl 2):S7–13. doi: 10.1016/j.jadohealth.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhlhaas PJ, Singer W. The development of neural synchrony and large-scale cortical networks during adolescence: relevance for the pathophysiology of schizophrenia and neurodevelopmental hypothesis. Schizophr Bull. 2011;37:514–23. doi: 10.1093/schbul/sbr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eggers AE. A serotonin hypothesis of schizophrenia. Med Hypotheses. 2013;80:791–4. doi: 10.1016/j.mehy.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Laruelle M. Schizophrenia: from dopaminergic to glutamatergic interventions. Curr Opin Pharm. 2014;14:97–102. doi: 10.1016/j.coph.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62:1574–83. doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev. 2007;87:1377–408. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 7.Dufey E, Sepulveda D, Rojas-Rivera D, Hetz C. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 1. An overview. Am J Physiol Cell Physiol. 2014;307:C582–594. doi: 10.1152/ajpcell.00258.2014. [DOI] [PubMed] [Google Scholar]
- 8.Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol. 2015;308:C415–425. doi: 10.1152/ajpcell.00334.2014. [DOI] [PubMed] [Google Scholar]
- 9.Lee AS, Delegeane A, Scharff D. Highly conserved glucose-regulated protein in hamster and chicken cells: preliminary characterization of its cDNA clone. Proc Natl Acad Sci USA. 1981;78:4922–5. doi: 10.1073/pnas.78.8.4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, et al. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514:122–8. doi: 10.1016/s0014-5793(02)02289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–75. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang LH, Zhang X. Roles of GRP78 in physiology and cancer. J Cell Biochem. 2010;110:1299–305. doi: 10.1002/jcb.22679. [DOI] [PubMed] [Google Scholar]
- 13.Malhotra JD, Kaufman RJ. The endoplasmic reticulum and the unfolded protein response. Semin Cell Dev Biol. 2007;18:716–31. doi: 10.1016/j.semcdb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schonthal AH. Targeting endoplasmic reticulum stress for cancer therapy. Front Biosci. 2012;4:412–31. doi: 10.2741/s276. [DOI] [PubMed] [Google Scholar]
- 15.Healy SJ, Gorman AM, Mousavi-Shafaei P, Gupta S, Samali A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur J Pharmacol. 2009;625:234–46. doi: 10.1016/j.ejphar.2009.06.064. [DOI] [PubMed] [Google Scholar]
- 16.Manie SN, Lebeau J, Chevet E. Cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. 3. Orchestrating the unfolded protein response in oncogenesis: an update. Am J Physiol Cell Physiol. 2014;307:C901–907. doi: 10.1152/ajpcell.00292.2014. [DOI] [PubMed] [Google Scholar]
- 17.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–99. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, et al. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. 2003;278:31024–32. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- 19.Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/s1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 20.Adachi Y, Yamamoto K, Okada T, Yoshida H, Harada A, Mori K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct Funct. 2008;33:75–89. doi: 10.1247/csf.07044. [DOI] [PubMed] [Google Scholar]
- 21.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 22.Munoz JP, Ivanova S, Sanchez-Wandelmer J, Martinez-Cristobal P, Noguera E, Sancho A, et al. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32:2348–61. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: assembling the IRE1alpha interactome. Mol Cell. 2009;35:551–61. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imagawa Y, Hosoda A, Sasaka S, Tsuru A, Kohno K. RNase domains determine the functional difference between IRE1alpha and IRE1beta. FEBS Lett. 2008;582:656–60. doi: 10.1016/j.febslet.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 25.Braakman I, Bulleid NJ. Protein folding and modification in the mammalian endoplasmic reticulum. Annu Rev Biochem. 2011;80:71–99. doi: 10.1146/annurev-biochem-062209-093836. [DOI] [PubMed] [Google Scholar]
- 26.Jager R, Bertrand MJ, Gorman AM, Vandenabeele P, Samali A. The unfolded protein response at the crossroads of cellular life and death during endoplasmic reticulum stress. Biol Cell. 2012;104:259–70. doi: 10.1111/boc.201100055. [DOI] [PubMed] [Google Scholar]
- 27.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- 28.Samali A, Fitzgerald U, Deegan S, Gupta S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int J Cell Biol. 2010;2010:830307. doi: 10.1155/2010/830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shim J, Umemura T, Nothstein E, Rongo C. The unfolded protein response regulates glutamate receptor export from the endoplasmic reticulum. Mol Biol Cell. 2004;15:4818–28. doi: 10.1091/mbc.E04-02-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, et al. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005;110:165–72. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- 31.Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson’s disease. J Neurosci. 2002;22:10690–8. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–7. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 33.Duennwald ML, Lindquist S. Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity. Genes Dev. 2008;22:3308–19. doi: 10.1101/gad.1673408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum Mol Genet. 2012;21:2245–62. doi: 10.1093/hmg/dds040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 2003;22:5435–45. doi: 10.1093/emboj/cdg537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gold PW, Licinio J, Pavlatou MG. Pathological parainflammation and endoplasmic reticulum stress in depression: potential translational targets through the CNS insulin, klotho and PPAR-gamma systems. Mol Psychiatry. 2013;18:154–65. doi: 10.1038/mp.2012.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nevell L, Zhang K, Aiello AE, Koenen K, Galea S, Soliven R, et al. Elevated systemic expression of ER stress related genes is associated with stress-related mental disorders in the Detroit Neighborhood Health Study. Psychoneuroendocrinology. 2014;43:62–70. doi: 10.1016/j.psyneuen.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.So J, Warsh JJ, Li PP. Impaired endoplasmic reticulum stress response in B-lymphoblasts from patients with bipolar-I disorder. Biol Psychiatry. 2007;62:141–7. doi: 10.1016/j.biopsych.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 39.Hunsberger JG, Machado-Vieira R, Austin DR, Zarate C, Chuang DM, Chen G, et al. Bax inhibitor 1, a modulator of calcium homeostasis, confers affective resilience. Brain Res. 2011;1403:19–27. doi: 10.1016/j.brainres.2011.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bauer D, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Abnormal glycosylation of EAAT1 and EAAT2 in prefrontal cortex of elderly patients with schizophrenia. Schizophr Res. 2010;117:92–98. doi: 10.1016/j.schres.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Funk AJ, Rumbaugh G, Harotunian V, McCullumsmith RE, Meador-Woodruff JH. Decreased expression of NMDA receptor-associated proteins in frontal cortex of elderly patients with schizophrenia. Neuroreport. 2009;20:1019–22. doi: 10.1097/WNR.0b013e32832d30d9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammond JC, McCullumsmith RE, Funk AJ, Haroutunian V, Meador-Woodruff JH. Evidence for abnormal forward trafficking of AMPA receptors in frontal cortex of elderly patients with schizophrenia. Neuropsychopharmacology. 2010;35:2110–9. doi: 10.1038/npp.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kristiansen LV, Patel SA, Haroutunian V, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor subunit and its Tbr-1/CINAP regulatory proteins in postmortem brain suggest altered receptor processing in schizophrenia. Synapse. 2010;64:495–502. doi: 10.1002/syn.20754. [DOI] [PubMed] [Google Scholar]
- 44.Kristiansen LV, Bakir B, Haroutunian V, Meador-Woodruff JH. Expression of the NR2B-NMDA receptor trafficking complex in prefrontal cortex from a group of elderly patients with schizophrenia. Schizophr Res. 2010;119:198–209. doi: 10.1016/j.schres.2010.02.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mueller TM, Haroutunian V, Meador-Woodruff JH. N-Glycosylation of GABAA receptor subunits is altered in Schizophrenia. Neuropsychopharmacology. 2014;39:528–37. doi: 10.1038/npp.2013.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim P, Scott MR, Meador-Woodruff JH. Abnormal expression of ER quality control and ER associated degradation proteins in the dorsolateral prefrontal cortex in schizophrenia. Schizophr Res. 2018;197:484–91. [DOI] [PMC free article] [PubMed]
- 47.Kim P, Scott MR, Meador-Woodruff JH. Abnormal expression of ER quality control and ER associated degradation proteins in the dorsolateral prefrontal cortex in schizophrenia. Transl Psychiatry. 2019;9:6. doi: 10.1016/j.schres.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Powchik P, Davidson M, Haroutunian V, Gabriel SM, Purohit DP, Perl DP, et al. Postmortem studies in schizophrenia. Schizophr Bull. 1998;24:325–41. doi: 10.1093/oxfordjournals.schbul.a033330. [DOI] [PubMed] [Google Scholar]
- 49.Purohit DP, Perl DP, Haroutunian V, Powchik P, Davidson M, Davis KL. Alzheimer disease and related neurodegenerative diseases in elderly patients with schizophrenia: a postmortem neuropathologic study of 100 cases. Arch Gen Psychiatry. 1998;55:205–11. doi: 10.1001/archpsyc.55.3.205. [DOI] [PubMed] [Google Scholar]
- 50.Harte MK, Bachus SB, Reynolds GP. Increased N-acetylaspartate in rat striatum following long-term administration of haloperidol. Schizophr Res. 2005;75:303–8. doi: 10.1016/j.schres.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 51.Kashihara K, Sato M, Fujiwara Y, Ogawa T, Fukuda K, Otsuki S. Effects of intermittent and continuous haloperidol administration on the dopaminergic system in the rat brain. Yakubutsu Seishin Kodo. 1986;6:275–80. [PubMed] [Google Scholar]
- 52.Kippe JM, Mueller TM, Haroutunian V, Meador-Woodruff JH. Abnormal N-acetylglucosaminyltransferase expression in prefrontal cortex in schizophrenia. Schizophr Res. 2015;166:219–24. doi: 10.1016/j.schres.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White K, Yang P, Li L, Farshori A, Medina AE, Zielke HR. Effect of postmortem interval and years in storage on RNA quality of tissue at a repository of the NIH NeuroBioBank. Biopreserv Biobank. 2018;16:148–57. doi: 10.1089/bio.2017.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stan AD, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa KJ, et al. Human postmortem tissue: what quality markers matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chalmers F, van Lith M, Sweeney B, Cain K, Bulleid NJ. Inhibition of IRE1alpha-mediated XBP1 mRNA cleavage by XBP1 reveals a novel regulatory process during the unfolded protein response. Wellcome Open Res. 2017;2:36. doi: 10.12688/wellcomeopenres.11764.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto K, Yoshida H, Kokame K, Kaufman RJ, Mori K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J Biochem. 2004;136:343–50. doi: 10.1093/jb/mvh122. [DOI] [PubMed] [Google Scholar]
- 57.McGuckin MA, Eri RD, Das I, Lourie R, Florin TH. ER stress and the unfolded protein response in intestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2010;298:G820–832. doi: 10.1152/ajpgi.00063.2010. [DOI] [PubMed] [Google Scholar]
- 58.Focking M, Lopez LM, English JA, Dicker P, Wolff A, Brindley E, et al. Proteomic and genomic evidence implicates the postsynaptic density in schizophrenia. Mol Psychiatry. 2015;20:424–32. doi: 10.1038/mp.2014.63. [DOI] [PubMed] [Google Scholar]
- 59.Martins-de-Souza D, Gattaz WF, Schmitt A, Rewerts C, Maccarrone G, Dias-Neto E, et al. Prefrontal cortex shotgun proteome analysis reveals altered calcium homeostasis and immune system imbalance in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2009;259:151–63. doi: 10.1007/s00406-008-0847-2. [DOI] [PubMed] [Google Scholar]
- 60.Martins-de-Souza D, Gattaz WF, Schmitt A, Maccarrone G, Hunyadi-Gulyas E, Eberlin MN, et al. Proteomic analysis of dorsolateral prefrontal cortex indicates the involvement of cytoskeleton, oligodendrocyte, energy metabolism and new potential markers in schizophrenia. J Psychiatr Res. 2009;43:978–86. doi: 10.1016/j.jpsychires.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 61.Sivagnanasundaram S, Crossett B, Dedova I, Cordwell S, Matsumoto I. Abnormal pathways in the genu of the corpus callosum in schizophrenia pathogenesis: a proteome study. Proteom Clin Appl. 2007;1:1291–305. doi: 10.1002/prca.200700230. [DOI] [PubMed] [Google Scholar]
- 62.Dudek J, Benedix J, Cappel S, Greiner M, Jalal C, Muller L, et al. Functions and pathologies of BiP and its interaction partners. Cell Mol life Sci. 2009;66:1556–69. doi: 10.1007/s00018-009-8745-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene. 2013;32:805–18. doi: 10.1038/onc.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boskovic M, Vovk T, Kores Plesnicar B, Grabnar I. Oxidative stress in schizophrenia. Curr Neuropharmacol. 2011;9:301–12. doi: 10.2174/157015911795596595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez-Liencres C, Tas C, Brown EC, Erdin S, Onur E, Cubukcoglu Z, et al. Oxidative stress in schizophrenia: a case-control study on the effects on social cognition and neurocognition. BMC Psychiatry. 2014;14:268. doi: 10.1186/s12888-014-0268-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rajasekaran A, Venkatasubramanian G, Berk M, Debnath M. Mitochondrial dysfunction in schizophrenia: pathways, mechanisms and implications. Neurosci Biobehav Rev. 2015;48:10–21. doi: 10.1016/j.neubiorev.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 67.Reyazuddin M, Azmi SA, Islam N, Rizvi A. Oxidative stress and level of antioxidant enzymes in drug-naive schizophrenics. Indian J Psychiatry. 2014;56:344–9. doi: 10.4103/0019-5545.146516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–55. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 69.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–19. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- 70.Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell Death Differ. 2012;19:310–20. doi: 10.1038/cdd.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, et al. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–66. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- 72.Glantz LA, Gilmore JH, Lieberman JA, Jarskog LF. Apoptotic mechanisms and the synaptic pathology of schizophrenia. Schizophr Res. 2006;81:47–63. doi: 10.1016/j.schres.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 73.Funk AJ, McCullumsmith RE, Haroutunian V, Meador-Woodruff JH. Abnormal activity of the MAPK- and cAMP-associated signaling pathways in frontal cortical areas in postmortem brain in schizophrenia. Neuropsychopharmacology. 2012;37:896–905. doi: 10.1038/npp.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tucholski J, Simmons MS, Pinner AL, McMillan LD, Haroutunian V, Meador-Woodruff JH. N-linked glycosylation of cortical N-methyl-D-aspartate and kainate receptor subunits in schizophrenia. Neuroreport. 2013;24:688–91. doi: 10.1097/WNR.0b013e328363bd8a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tucholski J, Simmons MS, Pinner AL, Haroutunian V, McCullumsmith RE, Meador-Woodruff JH. Abnormal N-linked glycosylation of cortical AMPA receptor subunits in schizophrenia. Schizophr Res. 2013;146:177–83. doi: 10.1016/j.schres.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hayashi A, Kasahara T, Iwamoto K, Ishiwata M, Kametani M, Kakiuchi C, et al. The role of brain-derived neurotrophic factor (BDNF)-induced XBP1 splicing during brain development. J Biol Chem. 2007;282:34525–34. doi: 10.1074/jbc.M704300200. [DOI] [PubMed] [Google Scholar]
- 77.Hayashi A, Kasahara T, Kametani M, Kato T. Attenuated BDNF-induced upregulation of GABAergic markers in neurons lacking Xbp1. Biochem Biophys Res Commun. 2008;376:758–63. doi: 10.1016/j.bbrc.2008.09.059. [DOI] [PubMed] [Google Scholar]
- 78.Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, et al. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008;13:147–61. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011;2011:723184. doi: 10.1155/2011/723184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Joshi S, Rajasekaran K, Hawk KM, Brar J, Ross BM, Tran CA, et al. Phosphatase inhibition prevents the activity-dependent trafficking of GABAA receptors during status epilepticus in the young animal. Epilepsia. 2015;56:1355–65. doi: 10.1111/epi.13098. [DOI] [PubMed] [Google Scholar]
- 81.Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction. PLoS Biol. 2010;8:e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hollien J, Weissman JS. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science. 2006;313:104–7. doi: 10.1126/science.1129631. [DOI] [PubMed] [Google Scholar]
- 83.Harnsberger JD, Hollien H, Martin CA, Hollien KA. Stress and deception in speech: evaluating layered voice analysis. J For Sci. 2009;54:642–50. doi: 10.1111/j.1556-4029.2009.01026.x. [DOI] [PubMed] [Google Scholar]
- 84.Bauernfeind AL, Soderblom EJ, Turner ME, Moseley MA, Ely JJ, Hof PR, et al. Evolutionary divergence of gene and protein expression in the brains of humans and chimpanzees. Genome Biol Evol. 2015;7:2276–88. doi: 10.1093/gbe/evv132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2012;13:227–32. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carnemolla A, Fossale E, Agostoni E, Michelazzi S, Calligaris R, De Maso L, et al. Rrs1 is involved in endoplasmic reticulum stress response in Huntington disease. J Biol Chem. 2009;284:18167–73. doi: 10.1074/jbc.M109.018325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson’s disease. Biochem Biophys Res Commun. 2007;354:707–11. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 88.Paz Gavilan M, Vela J, Castano A, Ramos B, del Rio JC, Vitorica J, et al. Cellular environment facilitates protein accumulation in aged rat hippocampus. Neurobiol Aging. 2006;27:973–82. doi: 10.1016/j.neurobiolaging.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 89.Shen X, Ellis RE, Sakaki K, Kaufman RJ. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet. 2005;1:e37. doi: 10.1371/journal.pgen.0010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.