

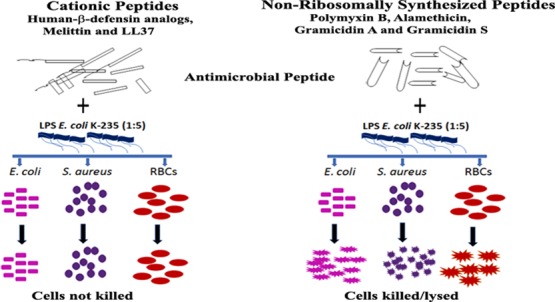
Abstract
Human-β-defensins (HBD1-3) are antibacterial peptides containing three disulphide bonds. In the present study, the effect of Escherichia coli lipopolysaccharide (LPS) on the antibacterial activities of HBD2-3, C-terminal analogues having a single disulphide bond, Phd1-3, and their corresponding myristoylated analogues MPhd1-3 were investigated. The effect of LPS on the activities of linear amphipathic peptides melittin, LL37 and non-ribosomally synthesized peptides, polymyxin B, alamethicin, gramicidin A, and gramicidin S was also examined. The antibacterial activity of HBD 2-3, Phd1-3, and MPhd1-3 in the presence of LPS against E. coli and Staphylococcus aureus was inhibited. While LPS inhibited the antibacterial activity of LL37, the inhibition of melittin activity was partial. The hemolytic activity exhibited by MPhd1, MPhd3, melittin, and LL37 was inhibited in the presence of LPS. HBD2-3, Phd1-3, and MPhd1-3 also showed endotoxin neutralizing activity. The antibacterial and hemolytic activities of polymyxin B, alamethicin, gramicidin A, and gramicidin S were not inhibited in the presence of LPS. Fluorescence assays employing dansyl cadaverine showed that HBD2-3 and defensin analogues bind to LPS more strongly as compared to alamethicin, gramicidin A, and gramicidin S. Electron microscopy images indicated that peptides disintegrate the structure of LPS. The inhibition of the antibacterial activity of native defensins and analogues in the presence of LPS indicates that the initial interaction with the bacterial surface is similar. The native defensin sequence or structure is also not essential, although cationic charges are necessary for binding to LPS. Hydrophobic interaction is the main driving force for association of non-ribosomally synthesized polymyxin B, alamethicin, gramicidin A, and gramicidin S with LPS. It is likely that these peptides rapidly insert into membranes and do not interact with the bacterial cell surface, whereas cationic peptides such as β-defensin and their analogues, melittin and LL37, first interact with the bacterial cell surface and then the membrane. Our results suggest that evaluating interaction of antibacterial and hemolytic peptides with LPS is a compelling way of elucidating the mechanism of bacterial killing or hemolysis.
Introduction
Human-β-defensins (HBDs) are cationic antimicrobial peptides (AMPs) containing three disulfide bonds having a broad spectrum antibacterial and anti-inflammatory activities.1−7 Lipopolysaccharide (LPS), a major constituent of the cell wall of Gram-negative bacteria, is released during bacterial cell division or death on administration of therapeutic antibiotics. It is a potent stimulant of immune response that causes sepsis.8 It has a dual purpose, protecting the bacteria from their surroundings, and as an effector molecule by initiating an immune response against the invading bacteria.9 Several investigations have indicated that AMPs bind to LPS and exhibit antibacterial activity and LPS detoxification activity.10−15 Human enteric α-defensin 5, in the reduced form, is able to suppress the pro-inflammatory effect of bacterial LPS by blocking the interaction between LPS and LPS-binding protein.11 Studies on the structure–activity relationship of anti-endotoxic activity of AMPs and their analogues have been reported.16,17 A higher minimum inhibitory concentration of some AMPs is observed for Gram-negative bacteria because of LPS that is present in the outer membrane as compared to Gram-positive organisms.18 LPS also induced aggregation of AMPs that resulted in their complete inactivation, which is evident in frog skin temporins and Leu/Lys and K12-designed peptides.19−25 Linear membrane active peptides on the interaction with LPS micelles showed distinct α-helical conformation.26−30 This structure is likely to favor insertion of the peptide into the lipid bilayer because of electrostatic interactions with the negatively charged phospholipids in the membrane bilayer of bacteria.27 Biophysical studies on the interaction of AMPs with LPS suggest that the overall biophysical properties required to kill bacteria are not the same as the ones to neutralize LPS. The chemical properties of the peptide that are required for each function are: sequence, cationic charge, an amphipathic structure, affinity for LPS, ability to traverse the LPS layer, and to disaggregate LPS micelles.21 The interaction of AMPs with the bacterial membrane surface is an important step for microbial killing. The cationic and hydrophobic characteristics of peptides and cell wall components of bacteria play important roles in insertion and translocation of peptides toward the cytosolic membrane.31,32 However, the precise mechanism and physicochemical properties of the AMPs required for interactions with bacterial LPS and anti-endotoxin activity remains to be established unequivocally.
AMPs are considered to be very attractive candidates for development as therapeutic agents against drug-resistant bacterial pathogens as well as to neutralize LPS. Several analogues and covalent chemical modifications have been attempted in AMPs that display selectivity for bacterial membranes and to increase their antibacterial potency.31,32 Earlier, we had reported antibacterial and hemolytic activities of C-terminal cationic segments of bovine-β-defensin BNBD2 analogues (P1-4) and mammalian β-defensins HBD1-3 analogues (Phd1-3) having a majority of the cationic residues present in native defensins, containing a single disulfide bond exhibited salt sensitive antibacterial activity without hemolytic activity.33,34 Attempts were made to reduce the salt sensitivity by N-terminal fatty acylation of these analogues; palmitoylation for BNBD2 analogues (P1-4) and myristoylation for HBD1-3 analogues (Phd1-3) that increases the hydrophobicity of the peptides and favors association with the membrane.35,36 Myristoylated HBD1-3 analogues (MPhd1-3) exhibited enhanced antibacterial potency and MPhd1, 3 exhibited hemolytic activity.36 Improved salt sensitive antibacterial activity was observed for MPhd1-2.36 It is of interest to examine the interaction of defensins and their analogues with LPS because these peptides are neither amphipathic nor helical. In the present study, the effect of interaction of LPS with native HBDs HBD2-3, myristoylated, and non-myristoylated C-terminal segments of HBD1-3 (MPhd1-3 and Phd1-3) on their antibacterial and hemolytic activities was investigated. Linear amphipathic host-defense peptides LL37, melittin, and non-ribosomally synthesized antibacterial peptides polymyxin B, alamethicin, gramicidin A, and gramicidin S were also used to delineate the physicochemical properties of peptides that are involved in their selective interaction with the bacterial and RBC membrane in the presence of Escherichia coli LPS.
Results
Peptides investigated in this study are C-terminal segments of HBD1-3, Phd1-3 having positively charged residues at different positions, their N-terminal myristoylated analogues, full-length defensins HBD2-3, and linear amphipathic antibacterial peptides, LL37, melittin, and peptides with noncoded amino acids, polymyxin B, alamethicin, gramicidin A, and gramicidin S (sequences except HBD2-3, are shown in Table 1). The interaction of these peptides with LPS was investigated at their respective minimal bactericidal concentrations (MBC) at which no viable colonies are formed (Table 2), to understand the importance of the sequence, net charge, and structure for antibacterial, hemolytic, and LPS neutralization activities.
Table 1. Sequences of Cationic and Non-Ribosomally Synthesized Antibacterial and Hemolytic Peptidesa.
| peptide | sequence | net charge |
|---|---|---|
| Phd1 | ACPIFTKIQGTYRGKAKCK | +5 |
| Phd2 | FCPRRYKQIGTGLPGTKCK | +5 |
| Phd3 | SCLPKEEQIGKSTRGRKCRRKK | +7 |
| MPhd1 | Myr-ACPIFTKIQGTYRGKAKCK | +4 |
| MPhd2 | Myr-FCPRRYKQIGTGLPGTKCK | +4 |
| MPhd3 | Myr-SCLPKEEQIGKSTRGRKCRRKK | +6 |
| melittin | GIGAVLKVLTTGLPALISWIKRKRQQ-CONH2 | +5 |
| LL37 | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | +5 |
| alamethicin | acetyl-UPUAUAQUVUGLUPVUUEQFol | |
| gramicidin A | formyl-VGALAVVVWLWLWLWG–CH2–CH2–OH | |
| gramicidin S | cyclic (LOVPFdLOVPFd) | +2 |
| polymyxin B | cyclized isooctanoyl BTBB(BFdLBBT) |
Bold underline indicates disulfide bond. Net charge is at neutral pH. Superscript d after the amino acid represents the d-enantiomer; all other amino acids are L-form. Parentheses indicate amino acids that are cyclized. O, ornithine; B, diaminobutyrate; U, α-aminoisobutyric acid; Fol, phenylalaninol. Peptides Phd1-3, MPhd1-3 and LL37 have COOH at the C-terminus. LL37 sequence was taken from ref (3). The sequences of melittin, alamethicin and gramicidin A were taken from ref (32) gramicidin S and polymyxin B were taken from ref (37).
Table 2. Antibacterial Activity of Cationic Peptides and Non-Ribosomally Synthesized Peptidesa.
| MBC
(μM) |
||
|---|---|---|
| peptide | E. coli | S. aureus |
| Phd1 | 19 | 20 |
| MPhd1 | 5 | 5 |
| Phd2 | 19 | 23 |
| MPhd2 | 6 | 5 |
| Phd3 | 16 | 17 |
| MPhd3 | 4 | 4 |
| HBD2 | 8 | 8 |
| HBD3 | 2 | 2 |
| melittin | 2 | 2 |
| LL37 | 2.5 | 2.5 |
| alamethicin | 10 | 16 |
| gramicidin A | 8 | 2 |
| gramicidin S | 2 | 2 |
| polymyxin B | 0.5 | 1 |
The antibacterial activity of Phd1-3 and MPhd1-3, examined at MBC, against E. coli and Staphylococcus aureus in the presence of LPS E. coli 026:B6 is shown in Figure 1. Against E. coli, at a ratio (w/w 1:1), partial inhibition of activity is observed for Phd1, MPhd1, and complete inhibition for Phd2. MPhd2, Phd3, and MPhd3 did not show inhibition in activity at a ratio (w/w 1:1) (Figure 1A). Partial inhibition of activity against S. aureus was observed for Phd1, MPhd1, Phd2, and MPhd2 (Figure 1B). No inhibition was observed for Phd3 and MPhd3. Complete inhibition of activity was observed at a ratio (w/w 1:5) against E. coli and S. aureus.
Figure 1.

Effect of LPS E. coli 026:B6 on antimicrobial activity of Phd1-3 and MPhd1-3. (A) E. coli (B) S. aureus. Ratios (w/w) are indicated in parentheses. Bacteria were incubated with peptides at their MBC in 10 mM sodium phosphate buffer (pH 7.4) containing different concentrations of LPS for 2 h at 37 °C and suitably diluted aliquots were plated on LB agar plates, which were incubated at 37 °C for 18 h. Colonies formed were counted and percentage bacteria killed was determined.
Inhibition of activity in the presence of LPS E. coli K-235 against E. coli and S. aureus is shown in Figure 2A,B. Variation in the activity profile was observed in the presence of LPS. At a ratio of (w/w 1:1), complete inhibition in activity was observed for Phd1 against E. coli and 90% inhibition for Phd2 against S. aureus. Complete inhibition of activity at a ratio (w/w 1:5) against E. coli (Figure 2A) was observed for Phd1 and MPhd1, 3. Inhibition in activity of Phd2, Phd3, and MPhd2 was 90, 80, and 25%, respectively. Complete inhibition of activity in the presence of LPS at a ratio (w/w 1:5) against S. aureus was observed for Phd1, 2 and MPhd2, 3 as shown in Figure 2B, whereas complete inhibition at a ratio (w/w 1:10) of LPS against S. aureus was observed for Phd 3 and MPhd1.
Figure 2.

Effect of LPS E. coli K-235 on antimicrobial activity of analogues Phd1-3 and MPhd1-3. (A) E. coli (B) S. aureus. Ratios (w/w) are indicated in parentheses. Bacteria were incubated with the peptides at their MBC in 10 mM sodium phosphate buffer (pH 7.4) containing different concentrations of LPS for 2 h at 37 °C and suitably diluted aliquots were plated on LB agar plates, which were incubated at 37 °C for 18 h. Colonies formed were counted and percentage bacteria killed was determined.
Antibacterial activity inhibition of native defensins HBD2-3 by LPS E. coli 026:B6 and LPS E. coli K-235 at ratios (w/w 1:1 and w/w 1:5) against E. coli and S. aureus is shown in Figure 3A,B. The activity of HBD2-3 was not inhibited with LPS E. coli 026:B6 at a ratio (w/w 1:1) against E. coli and S. aureus. In the presence of LPS E. coli 026:B6 and LPS E. coli K-235, activity against E. coli was inhibited, while inhibition of activity against S. aureus was more pronounced with HBD2 as compared to HBD3 at a ratio (w/w 1:5).
Figure 3.

Effect of LPS E. coli 026:B6 and LPS E. coli K-235 on the antimicrobial activity of HBD2 and HBD3 (A) E. coli (B) S. aureus. Ratios (w/w 1:1 and w/w 1:5) are indicated in parentheses. Bacteria were incubated with the peptides at their MBC in 10 mM sodium phosphate buffer (pH 7.4) containing LPS for 2 h at 37 °C and suitably diluted aliquots were plated on LB agar plates, which were incubated at 37 °C for 18 h. Colonies formed were counted and percentage bacteria killed was determined.
The activity of polymyxin B, LL37, melittin, alamethicin, gramicidin A, and gramicidin S against E. coli and S. aureus in the presence of LPS E. coli K-235 is shown in Figure 4A,B. LPS did not inhibit activity of LL37 and melittin at a ratio (w/w 1:1). The activity of LL37 was inhibited by LPS against both the strains whereas melittin, showed partial inhibition of activity at a ratio (w/w 1:5). The channel forming peptides alamethicin and gramicidin A, gramicidin S, and polymyxin B did not show the inhibition of antibacterial activity in the presence of LPS at a ratio (w/w 1:5).
Figure 4.

Effect of LPS E. coli K-235 on antimicrobial activity of LL37, melittin (Mel), alamethicin (Ala), gramicidin A (GrA), gramicidin S (GrS), and polymyxin B (Poly). (A) E. coli (B) S. aureus. Ratios (w/w 1:1 and w/w 1:5) are in parentheses. Bacteria were incubated with the peptides at their MBC in 10 mM sodium phosphate buffer (pH 7.4) containing LPS for 2 h at 37 °C and suitably diluted aliquots were plated on LB agar plates, which were incubated at 37 °C for 18 h. Colonies formed were counted and percentage bacteria killed was determined.
The inhibition of hemolytic activity of MPhd1 and MPhd3 at different concentrations of the peptide at a ratio (w/w 1:5) of LPS E. coli K-235 is shown in Figure 5A,B. Significant decrease in hemolytic activity was observed for both peptides in the presence of LPS.
Figure 5.

Hemolytic activity of (A) MPhd1 and (B) MPhd3. Hemolysis (%) was plotted as a function of concentration of MPhd1 and 3. Key: (■) hemolysis (%) by the peptide alone; (●) hemolysis (%) by the peptide upon incubation with LPS E. coli K-235 at (w/w 1:5).
Similarly, the hemolytic activity of peptides LL37, melittin, alamethicin, gramicidin A, and gramicidin S at a given concentration in the absence and presence of LPS E. coli K-235 at a ratio (w/w 1:5) is shown in Figure 6. LPS inhibited the hemolytic activity of LL37 and melittin, whereas no inhibition on the hemolysis of pore forming peptides alamethicin, gramicidin A, and gramicidin S was observed.
Figure 6.

Hemolytic activity of LL37, melittin (Mel), alamethicin (Ala), gramicidin A (GrA), and gramicidin S (GrS). Peptide LPS at a ratio (w/w 1:5) in the presence of LPS E. coli K-235.
LPS E. coli K-235 neutralization activity with HBD2-3, Phd1-3, and MPhd1-3 was assayed by using a commercially available semi-quantitative Limulus amebocyte lysate (LAL) E-TOXATE kit.39 Depending on the LPS concentration in the sample, LAL reagent results could range from increasing the viscosity to the formation of a hard gel. Samples are considered positive for LPS when the formed gel does not collapse after the sample tube is inverted vertically.39 Inverted test tubes used to determine the LPS level are shown in Figure 7. Gel formation was not observed in any of the peptide-treated samples with LPS indicating native defensins as well as analogues have LPS neutralization activity.
Figure 7.

Semi-quantitative LAL assay to estimate endotoxin neutralization following the addition of defensin analogues at a ratio (w/w 1:5). Images show inverted test tubes of endotoxin control and tested defensin analogues. The surface of the generated gels of LPS E. coli K-235 at a concentration (0.2 EU/mL) is shown in the control tubes. Images are representative of 2 independent experiments (photos in this figure were taken by Krishnakumari and Taniya Mary Binny).
The binding affinity of HBD2-3, defensin analogues Phd1-3 and MPhd1-3, amphipathic peptides LL37, melittin, polymyxin B, alamethicin, gramicidin A, and gramicidin S to LPS E.coli K-235 was investigated using dansyl cadaverine (DC) which binds lipid A of LPS and is displaced by molecules which has an affinity for lipid A. The data are shown in Figure 8A,B. The addition of LPS to DC results in the enhancement of the fluorescence intensity accompanied by a blue shift in the emission maximum wavelength which is indicative of interactions of DC with LPS molecules. The addition of the displacer or peptide to the mixture of LPS and DC results in the decrease of fluorescence intensity. The addition of the displacer to DC alone indicates no such effects. This is indicative of the displacement of bound DC from LPS by the displacer. The quenching of the fluorescence intensity as a function of a given peptide concentration is represented as an occupancy factor. Polymyxin B was used as a positive control that has been shown to form stable molecular complexes with the lipid A portion of LPS and its binding involve both electrostatic and hydrophobic interactions.40 The binding data for HBD2-3 and analogues Phd1-3 and MPhd1-3 to LPS are shown in Figure 8A. The control peptides HBD2-3 showed more affinity to LPS compared to their analogues. Phd1 and MPhd1 have more affinity as compared to Phd2-3 and MPhd2-3. The binding data for amphipathic peptides to LPS are shown in Figure 8B. Polymyxin B has strong binding compared to HBD2-3 as well as LL37 and melittin. The pore forming peptides alamethicin, gramicidin A, and gramicidin S have weak affinity as compared to polymyxin B and their affinity saturated around 0.5 of occupancy factor.
Figure 8.

Displacement of LPS E. coli K-235- bound DC by peptides. The displacement results in de-quenching of DC. The fraction of LPS-bound DC (probe occupancy) was calculated as described in the Experimental section. [LPS E. coli K-235]: 280 μg/mL; [DC]: 12 μM. Buffer: 10 mM phosphate buffer, pH 7.4. 2–5 μL aliquots of peptides were successively added to the cuvette containing the DC: LPS complex. Excitation: 340 nm; emission: 420–600 nm. Band passes: 2.5 nm/5 nm. (A) HBD2-3, non-myristoylated (Phd1-3) and myristoylated (MPhd1-3) defensin analogues. (B) Melittin, LL37 and non-ribosomally synthesized peptides.
The CD spectra of non-myristoylated and myristoylated peptides recorded in buffer and in the presence of LPS E. coli K-235 at a ratio (1:5), indicated that no structural changes were observed for Phd1-3 and MPhd1-3 with LPS (Figure S1).
Transmission electron microscopy (TEM) images of LPS E. coli K-235 obtained in the absence and in the presence of peptides are shown in Figure 9. LPS in aqueous solution shows aggregated structures. This result also indicates the formation of large inhomogeneous aggregates of LPS. In contrast, TEM images confirmed the disaggregation of the assembly of LPS to small dense spherical particles of LPS molecules in the presence of peptides at both (w/w 1:1) and (w/w 1:5) peptide/LPS ratios.
Figure 9.

TEM images of LPS E. coli K-235 control and peptide in the presence of LPS at a ratio of (w/w 1:1) and (w/w 1:5). Scale bar is 0.2 μm.
Discussion
Many therapeutic antibiotics have bactericidal effects but they cannot effectively prevent septic shock caused by LPS because of a lack of ability to neutralize LPS.41,42 Hence, there has been interest in examining whether cationic AMPs, particularly that are a part of host-defense arsenal, can interact with LPS and make them unavailable to cause septic shock. AMPs interact initially with the outer membrane of bacteria. The arrangement of amino acids in the AMP sequence, which determines the structure of the peptide, favors interaction with pathogen surfaces made up of proteins, glycan chains, or surface lipids.43 HBDs are host-defense antibacterial peptides for protection against invading microbes. Defensins kill bacteria by mechanisms different from therapeutic antibiotics. Their initial site of action is the bacterial cell surface, where they interact with LPS in Gram-negative bacteria and peptidoglycan in Gram-positive bacteria.44 This interaction does not result in detergent-like solubilization of the bacterial surface but a pathway is created for an approach to the bacterial “plasma” or cytoplasmic membrane where they exert their activity to kill bacteria. Defensins are also internalized in bacteria wherein they cause destabilization of bacterial metabolism.44,45 Defensins have bactericidal activities and some of them lose their activities in the physiological concentration of NaCl.2−4 They also have LPS-neutralizing activity depending on the LPS, from different species of bacteria.10 In the present study, we have examined the effect of E. coli LPS O26:B6 and E. coli LPS K-235 which differ in length of the polysaccharide chain,23,46 on the bactericidal and hemolytic activities of full length human defensins and their active analogues to delineate the contributions of the peptide sequence and structure for their interaction with LPS. The interaction of nonspecific membrane lytic linear amphipathic peptides and the cyclic peptide gramicidin S with LPS was also examined.
LPS effectively inhibits the antibacterial activity of Phd1-3, MPhd1-3, and HBD2-3 against E. coli and S. aureus. Relative binding affinities of HBD2-3, defensin analogues, amphipathic peptides, and LPS binding peptides, polymyxin B examined in terms of their ability to displace DC from LPS, indicate that the HBD2-3, defensin analogues, and other peptides bind to LPS with lower affinity compared to polymyxin B. However, the binding affinity of peptides was not reflected in their inhibition of antibacterial activity in the presence of LPS. In addition, the binding did not result in structural changes in the defensin analogues, indicating that the interaction is electrostatic and the peptide chain is not intimately associated with the LPS. The importance of the cationic nature of the peptides for LPS interaction is evident from the lack of inhibition of antibacterial activity of alamethicin that has one negative charge, gramicidin S that has two positive charges and gramicidin A which has no charge. The chemical structure of LPS varies among different species of bacteria, with each species expressing structurally heterogeneous LPS molecules.47 The amphiphilic nature of LPS results in the formation of oligomers above a critical concentration (CMC). CMC of LPS E. coli 026:B6 is 14 μg/mL.48 CMC is considered to be directly proportional to the polysaccharide chain length. E. coli K-235 will have low CMC compared to E. coli 026:B6. There are reports that both monomeric and aggregated forms of LPS exhibited pro-inflammatory responses.49,50 EM studies indicate the association of defensin analogues and other peptides polymyxin B, alamethicin, gramicidin A, cyclic gramicidin S, melittin, and LL37 with LPS results in the disintegration of LPS aggregates. The disaggregation of LPS is evident both at higher and lower ratios of the peptide to LPS. Studies on defensins have shown that certain aromatic amino acids such as Trp, Tyr, and Arg play a dominant role in stabilizing the peptide-pathogen surface complexes.43 It is possible that the cationic defensin peptides and LPS are intimately associated, indicating that free peptide is excluded at a higher ratio of the peptide to LPS (w/w 1:5), due to which the peptides are unable to interact with the bacterial cell surface, which is the first step in bacterial killing. This interaction also displayed their activities in limulus amebocyte lysate assay which results in the loss of LPS ability to cause coagulation of haemolymph. Hydrophobic interactions are the main driving force for the insertion of non-ribosomally synthesized peptides such as polymyxin B, alamethicin, gramicidin A, and gramicidin S. Polymyxin B and gramicidin S can also neutralize LPS. The mechanism of Gram-negative bacterial killing by gramicidin S and polymyxin B involves initial interaction with LPS resulting in a self-promoted uptake.51 It is likely that the peptides rapidly insert into membranes and do not interact with the bacterial cell surface. While the LPS structure appears to be disintegrated by these peptides but they may not be bound to the debris as they can kill bacteria.
Association of MPhd1, MPhd3 and melittin as well as LL37 with LPS also prevents their hemolytic activity indicating the intimate association of these peptides with LPS after disintegration. In the case of alamethicin, gramicidin A, and gramicidin S no inhibition of hemolysis is observed. Alamethicin, gramicidin A, and gramicidin S form highly ordered structures and form channels in membranes.52−54 Gramicidin A and alamethicin can interact with the fatty acyl chains of LPS, perturb the LPS, and then associate with the bacterial inner membrane which is the site of action. When this type of mechanism is operational, LPS does not inhibit their ability to kill bacteria as well as to lyse RBCs. Our results suggest that evaluating the interaction of antibacterial and hemolytic peptides with LPS is a compelling way of elucidating the mechanism of bacterial killing or hemolysis.
In nature, defensins may have evolved with twin roles of inhibiting bacterial killing and neutralizing LPS activity. Because bacterial killing would be the early event, subsequent binding of defensins with LPS would not affect the role of defensins in bacterial killing. Whereas, once LPS is removed from the pathogen, the disaggregated LPS–peptide complex causes neutralization of LPS. Our present study suggests a possible dual role even for defensin analogues in host defense. This study also suggests that evaluating the interaction of antibacterial and hemolytic peptides with LPS could be of help in elucidating the mechanism of bacterial killing or hemolysis.
Experimental Section
Materials
Fluorenylmethoxycarbonyl (F-moc)-protected amino acids used were from Novabiochem AG, Switzerland and Advanced Chemtech, Louisville, KY, USA. Coupling reagents used in peptide synthesis were N-hydroxybenzotriazole hydrate (HOBt) (Advanced Chemtech), 2-(1-H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (Advanced Chemtech), and N,N-diisopropylethylamine (Sigma). Myristic acid used for acylation of peptides. LPS E. coli K-235 and LPS E. coli 026:B6 were from Sigma. HBD2-3 were purchased from Peptides International (Louisville, KY). LL37, melittin, alamethicin, gramicidin A, and gramicidin S were from Sigma. UV spectroscopic grade methanol and acetonitrile were from Spectrochem Pvt Ltd., India.
Peptide Synthesis
The synthesis of the resin-bound C-terminal segment of HBD1-3, Phd1-3 with a free amino group at the N-terminus, and corresponding N-terminal myristoylated MPhd1-3 have been described earlier.34,36 Briefly, the peptides were synthesized on the HMPA resin using F-moc chemistry. Both non-myristoylated and myristoylated peptides were cleaved from the resin using trifluoroacetic acid containing thioanisole, m-cresol, and ethanedithiol (10:1:1:0.5 v/v). They were purified by high-performance liquid chromatography (HPLC) on a reversed phase C-18 column [Agilent Extent C-18 (4.6 mm × 250 mm)] as described earlier. The molecular masses of HPLC-purified Phd1-3 and MPhd1-3 were confirmed on a matrix-assisted laser desorption ionization (MALDI) mass spectrometer (AB4800 MALDI-TOF/TOF) from Applied Biosystems (PerSeptive Biosystems, Foster City, CA) in the in-house proteomics facility (Figure S2).
Effects of LPS on Antibacterial Activity
Antibacterial activity of the peptides against bacterial strains of wild-type E. coli K-12 strain MG1655 (CGSC-7740) subsequently referred to as E. coli and S. aureus (ATCC 8530) in the presence of LPS was determined as described earlier.34,36,38 Bacteria was grown overnight in LB medium at 37 °C. After 20 h, 0.2 mL from this suspension was subcultured for 3 h in 20 mL of LB broth to obtain a mid-log-phase culture. Cells were harvested by centrifugation, washed thoroughly with 10 mM phosphate buffer, pH 7.4 (PB), to remove LB medium. Cells were resuspended in the same buffer, and the concentration was adjusted to 106 colony-forming units (cfu)/mL. Peptides Phd1-3, MPhd1-3, and HBD2, 3 were diluted to their MBC (MBC is the concentration of the peptides at which no viable colonies formed) as determined earlier.34,36,38 The protocol used was similar to the one used for defensins55,56 because their activity in the presence of the salt is attenuated. MBC concentrations were mixed with LPS E. coli K-235 or LPS E. coli 026:B6 at different ratios (w/w 1:1 or w/w 1:5). LL37, melittin, alamethicin, gramicidin A, gramicidin S, and polymyxin B diluted to their MBC and mixed with LPS E. coli K-235 at a fixed ratio (w/w 1:1 or w/w 1:5). After incubation with LPS for 30 min, 106 cfu/mL cells were added and incubated for 2 h at 37 °C in sterile 96-well plates in a final volume of 100 μL. Suitably diluted aliquots were spread on LB agar plates. The plates were incubated at 37 °C for 18 h. The cfu were counted, and percentage killing of bacteria relative to the cfu in untreated controls was calculated which was determined from the average of the three independent experiments done in duplicates.
Effects of LPS on Hemolytic Activity
Red blood cells were isolated from heparinized rat blood by centrifugation and washed thrice with 10 mM phosphate buffer, pH 7.4 containing 0.15 M NaCl just before the assays were performed. LPS E. coli K-235 at a ratio (w/w 1:5) was mixed with peptides MPhd1, 3, and LL37, melittin, alamethicin, gramicidin A, and gramicidin S at different concentrations in duplicates in Eppendorf tubes and aliquots of cell suspension (107/mL) were added and incubated at 37 °C for 30 min with gentle mixing. The samples were centrifuged and the absorbance of the supernatant was measured at 540 nm. The lysis obtained with 1% Triton X-100 was considered as 100%. The percentage lysis was calculated. The experiments were repeated thrice with duplicate samples.
LPS Binding Using Fluorescence Spectroscopy
Fluorescence measurements were carried out on a Hitachi F-7000 fluorescence spectrophotometer to study the binding affinity of HBD2-3 Phd1-3, MPhd1-3, LL37, melittin, polymyxin B, alamethicin, gramicidin A, and gramicidin S with LPS E. coli K-235. DC a fluorescent probe specifically binds to LPS and its displacement from LPS by the peptides results in dequenching of its fluorescence.57 The samples were excited at 340 nm, and the emission spectrum was recorded from 420 to 600 nm. The excitation and emission slits were set to 2.5 and 5 nm, respectively. All measurements were carried out at 25 °C. The fluorescence spectrum of DC (12 μM) was recorded and fluorimetric titrations were carried out with increasing concentration of LPS in 10 mM phosphate buffer pH 7.4 and maximum fluorescence intensity was observed at (280 μg/mL) LPS concentration. Increasing concentrations of the displacer was added and dequenching fluorescence intensity was recorded. Initially, the displacement experiment was done using polymyxin B as a positive control. Subsequently, the experiment with other peptides was carried out in a similar manner. Occupancy factor (F – F0)/(Fmax – F0) was calculated, where F0 is the fluorescence intensity of DC alone, Fmax is the intensity in the presence of the maximum LPS concentration and F are the intensities of the DC–LPS mixture at different displacer (peptide) concentrations. The estimation of occupancy in this manner implies a value of 1 at zero displacer concentration (F = Fmax) and 0 when DC is completely displaced with an excess of the displacing ligand (F = F0).
LPS Neutralization Assay
The peptides’ ability to neutralize LPS activity was assayed by using a commercially available semi-quantitative LAL E-TOXATE kit.39 The protocol explained in the manufacturer’s instructions was followed. The LPS in Gram-negative bacteria, activates a pro-enzyme in the LAL. The peptides were dissolved in the pyrogen-free water supplied with the kit, and the pH was adjusted to 7.0 with 1 N HCl which is prepared in pyrogen-free water. Peptides were incubated with 0.2 endotoxin units (EU) of LPS E. coli K-235 at a peptide-LPS ratio (w/w 1:5) in a volume of 50 μL for 30 min, and 50 μL of LAL reagent was added to the peptide–EU complex and further incubated for 60 min at 37 °C without shaking. After an incubation of 60 min for the reaction, gel formation was recorded.
Transmission Electron Microscopy
Defensins analogues Phd1-3, MPhd1-3, and amphipathic peptides polymyxin B, LL37, melittin, alamethicin, gramicidin A, and gramicidin S were incubated with LPS E. coli K-235 (0.1 mg/mL) at ratios (w/w 1:1) and (w/w 1:5) for 30 min in 10 mM phosphate buffer pH 7.4. Then, 1 μL aliquot was placed on a formvar coated copper grid, stained with 1% uranyl acetate, dried, and imaged in a JEOL JEM 2100 electron microscope at 120 kV. An LPS control sample without incubation with peptides was prepared in the same manner.
Acknowledgments
R.N. acknowledges the National Academy of Sciences, India for NASI Platinum Jubilee Senior Scientist Fellowship. R.N. is the recipient of JC Bose Fellowship from the Department of Science and Technology, India.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03770.
CD spectra of non-myristoylated and myristoylated peptides recorded in buffer and in the presence of LPS E. coli K-235 at a ratio (1:5) and MALDI-TOF MS spectra of N-terminal myristoylated peptides MPhd1-3(PDF)
Author Contributions
V.K., T.M.B. performed experiments. H.A. performed TEM experiment and all authors evaluated the data. V.K. and R.N. designed the experiments and wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Sawai M. V.; Jia H. P.; Liu L.; Aseyev V.; Wiencek J. M.; GanzMcCray T. Jr.; Kearney W. R.; Tack B. F.; Tack B. F. The NMR structure of human beta-defensin-2 reveals a novel alpha-helical segment. Biochemistry 2001, 40, 3810–3816. 10.1021/bi002519d. [DOI] [PubMed] [Google Scholar]
- Harder J.; Bartels J.; Christophers E.; Schröder J.-M. Isolation and characterization of human beta-defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. 10.1074/jbc.m008557200. [DOI] [PubMed] [Google Scholar]
- Bowdish D. M. E.; Davidson D. J.; Hancock R. E. Immunomodulatory properties of defensins and cathelicidins. Curr. Top. Microbiol. Immunol. 2006, 306, 27–66. 10.1007/3-540-29916-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover D. M.; Wu Z.; Tucker K.; Lu W.; Lubkowski J. Antimicrobial characterization of human beta-defensin 3 derivatives. Antimicrob. Agents Chemother. 2003, 47, 2804–2809. 10.1128/aac.47.9.2804-2809.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selsted M. E.; Ouellette A. J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- Schneider J. J.; Unholzer A.; Schaller M.; Schäfer-Korting M.; Korting H. C. Human Defensins. J. Mol. Med. 2005, 83, 587–595. 10.1007/s00109-005-0657-1. [DOI] [PubMed] [Google Scholar]
- Pazgier M.; Hoover D. M.; Yang D.; Lu W.; Lubkowski J. Human beta-defensins. Cell. Mol. Life Sci. 2006, 63, 1294–1313. 10.1007/s00018-005-5540-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- Reddick L. E.; Alto N. M. Bacteria fighting back: how pathogens target and subvert the host innate immune system. Mol. Cell 2014, 54, 321–328. 10.1016/j.molcel.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-H.; Jun H.-K.; Lee H.-R.; Chung C.-P.; Choi B.-K. Antibacterial and lipopolysaccharide (LPS)-neutralising activity of human cationic antimicrobial peptides against periodontopathogens. Int. J. Antimicrob. Agents 2010, 35, 138–145. 10.1016/j.ijantimicag.2009.09.024. [DOI] [PubMed] [Google Scholar]
- Wang C.; Shen M.; Zhang N.; Wang S.; Xu Y.; Chen S.; Chen F.; Yang K.; He T.; Wang A.; Su Y.; Cheng T.; Zhao J.; Wang J. Reduction Impairs the Antibacterial Activity but Benefits the LPS Neutralization Ability of Human Enteric Defensin 5. Sci. Rep. 2016, 6, 22875. 10.1038/srep22875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.-H.; Baek D.-H. Antibacterial and neutralizing effect of human beta-defensins on Enterococcus faecalis and Enterococcus faecalis lipoteichoic acid. J. Endod. 2012, 38, 351–356. 10.1016/j.joen.2011.12.026. [DOI] [PubMed] [Google Scholar]
- Scott M. G.; Vreugdenhil A. C. E.; Buurman W. A.; Hancock R. E. W.; Gold M. R. Cutting Edge: Cationic Antimicrobial Peptides Block the Binding of Lipopolysaccharide (LPS) to LPS Binding Protein. J. Immunol. 2000, 164, 549–553. 10.4049/jimmunol.164.2.549. [DOI] [PubMed] [Google Scholar]
- Rosenfeld Y.; Papo N.; Shai Y. Endotoxin (lipopolysaccharide) neutralization by innate immunity host-defense peptides. Peptide properties and plausible modes of action. J. Biol. Chem. 2006, 281, 1636–1643. 10.1074/jbc.m504327200. [DOI] [PubMed] [Google Scholar]
- Giacometti A.; Cirioni O.; Ghiselli R.; Mocchegiani F.; Orlando F.; Silvestri C.; Bozzi A.; Di Giulio A.; Luzi C.; Mangoni M. L.; Barra D.; Saba V.; Scalise G.; Rinaldi A. C. Interaction of antimicrobial peptide temporin L with lipopolysaccharide in vitro and in experimental rat models of septic shock caused by gram-negative bacteria. Biochim. Biophys. Acta 2006, 50, 2478–2486. 10.1128/aac.01553-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaconis Y.; Kowalski I.; Howe J.; Brauser A.; Richter W.; Razquin-Olazarán I.; Iñigo-Pestaña M.; Garidel P.; Rössle M.; Martinez de Tejada G.; Gutsmann T.; Brandenburg K. Biophysical mechanisms of endotoxin neutralization by cationic amphiphilic peptides. Biophys. J. 2011, 100, 2652–2661. 10.1016/j.bpj.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhunia A.; Mohanram H.; Domadia P. N.; Torres J.; Bhattacharjya S. Designed beta-boomerang antiendotoxic and antimicrobial peptides: structures and activities in lipopolysaccharide. J. Biol. Chem. 2009, 284, 21991–22004. 10.1074/jbc.m109.013573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silhavy T. J.; Kahne D.; Walker S. The bacterial cell envelope. Cold Spring Harbor Perspect. Biol. 2010, 2, a000414. 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerle A.; Kidric J.; Jerala R. Enhancement of antibacterial and lipopolysaccharide binding activities of a human lactoferrin peptide fragment by the addition of acyl chain. J. Antimicrob. Chemother. 2003, 51, 1159–1165. 10.1093/jac/dkg219. [DOI] [PubMed] [Google Scholar]
- Rosenfeld Y.; Shai Y. Lipopolysaccharide (Endotoxin)-host defense antibacterial peptides interactions: role in bacterial resistance and prevention of sepsis. Biochim. Biophys. Acta 2006, 1758, 1513–1522. 10.1016/j.bbamem.2006.05.017. [DOI] [PubMed] [Google Scholar]
- Papo N.; Shai Y. A molecular mechanism for lipopolysaccharide protection of Gram-negative bacteria from antimicrobial peptides. J. Biol. Chem. 2005, 280, 10378–10387. 10.1074/jbc.m412865200. [DOI] [PubMed] [Google Scholar]
- Rosenfeld Y.; Barra D.; Simmaco M.; Shai Y.; Mangoni M. L. A synergism between temporins toward Gram-negative bacteria overcomes resistance imposed by the lipopolysaccharide protective layer. J. Biol. Chem. 2006, 281, 28565–28574. 10.1074/jbc.m606031200. [DOI] [PubMed] [Google Scholar]
- Mangoni M. L.; Epand R. F.; Rosenfeld Y.; Peleg A.; Barra D.; Epand R. M.; Shai Y. Lipopolysaccharide, a key molecule involved in the synergism between temporins in inhibiting bacterial growth and in endotoxin neutralization. J. Biol. Chem. 2008, 283, 22907–22917. 10.1074/jbc.m800495200. [DOI] [PubMed] [Google Scholar]
- Mangoni M. L.; Shai Y. Temporins and their synergism against Gram-negative bacteria and in lipopolysaccharide detoxification. Biochim. Biophys. Acta 2009, 1788, 1610–1619. 10.1016/j.bbamem.2009.04.021. [DOI] [PubMed] [Google Scholar]
- Pulido D.; Nogués M. V.; Boix E.; Torrent M. Lipopolysaccharide neutralization by antimicrobial peptides: a gambit in the innate host defense strategy. J. Innate Immun. 2012, 4, 327–336. 10.1159/000336713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tack B. F.; Sawai M. V.; Kearney W. R.; Robertson A. D.; Sherman M. A.; Wang W.; Hong T.; Boo L. M.; Wu H.; Waring A. J.; Lehrer R. I. SMAP-29 has two LPS-binding sites and a central hinge. Eur. J. Biochem. . 2002, 269, 1181–1189. 10.1046/j.0014-2956.2002.02751.x. [DOI] [PubMed] [Google Scholar]
- Bhunia A.; Mohanram H.; Bhattacharjya S. Lipopolysaccharide bound structures of the active fragments of fowlicidin-1, a cathelicidin family of antimicrobial and antiendotoxic peptide from chicken, determined by transferred nuclear Overhauser effect spectroscopy. Biopolymers 2009, 92, 9–22. 10.1002/bip.21104. [DOI] [PubMed] [Google Scholar]
- Bhunia A.; Ramamoorthy A.; Bhattacharjya S. Helical hairpin structure of a potent antimicrobial peptide MSI-594 in lipopolysaccharide micelles by NMR spectroscopy. Chemistry 2009, 15, 2036–2040. 10.1002/chem.200802635. [DOI] [PubMed] [Google Scholar]
- Bhunia A.; Domadia P. N.; Torres J.; Hallock K. J.; Ramamoorthy A.; Bhattacharjya S. NMR structure of pardaxin, a pore-forming antimicrobial peptide, in lipopolysaccharide micelles: mechanism of outer membrane permeabilization. J. Biol. Chem. 2010, 285, 3883–3895. 10.1074/jbc.m109.065672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhunia A.; Saravanan R.; Mohanram H.; Mangoni M. L.; Bhattacharjya S. NMR Structures and Interactions of Temporin-1Tl and Temporin-1Tb with Lipopolysaccharide Micelles. J. Biol. Chem. 2011, 286, 24394–24406. 10.1074/jbc.m110.189662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram N.; Nagaraj R. Interaction of antimicrobial peptides with biological and model membranes: structural and charge requirements for activity. Biochim. Biophys. Acta 1999, 1462, 29–54. 10.1016/s0005-2736(99)00199-6. [DOI] [PubMed] [Google Scholar]
- Saberwal G.; Nagaraj R. Cell-lytic and antibacterial peptides that act by perturbing the barrier function of membranes: facets of their conformational features, structure-function correlations and membrane-perturbing abilities. Biochim. Biophys. Acta 1994, 1197, 109–131. 10.1016/0304-4157(94)90002-7. [DOI] [PubMed] [Google Scholar]
- Krishnakumari V.; Sharadadevi A.; Singh S.; Nagaraj R. Single disulfide and linear analogues corresponding to the carboxy-terminal segment of bovine beta-defensin-2: effects of introducing the beta-hairpin nucleating sequence d-pro-gly on antibacterial activity and Biophysical properties. Biochemistry 2003, 42, 9307–9315. 10.1021/bi034403y. [DOI] [PubMed] [Google Scholar]
- Krishnakumari V.; Singh S.; Nagaraj R. Antibacterial activities of synthetic peptides corresponding to the carboxy-terminal region of human beta-defensins 1-3. Peptides 2006, 27, 2607–2613. 10.1016/j.peptides.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Krishnakumari V.; Nagaraj R. N-Terminal fatty acylation of peptides spanning the cationic C-terminal segment of bovine beta-defensin-2 results in salt-resistant antibacterial activity. Biophys. Chem. 2015, 199, 25–33. 10.1016/j.bpc.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Krishnakumari V.; Guru A.; Adicherla H.; Nagaraj R. Effects of increasing hydrophobicity by N-terminal myristoylation on the antibacterial and hemolytic activities of the C-terminal cationic segments of human-beta-defensins 1-3. Chem. Biol. Drug Des. 2018, 92, 1504–1513. 10.1111/cbdd.13317. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Chapple D. S. Peptide antibiotics. Antimicrob. Agents. Chemother. 1999, 43, 1317–1323. 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumari V.; Packiyanathan K. K.; Nagaraj R. Human-beta-defensins-1-3 and analogs do not require proton motive force for antibacterial activity against Escherichia coli. FEMS Microbiol. Lett. 2013, 348, 52–57. 10.1111/1574-6968.12242. [DOI] [PubMed] [Google Scholar]
- Wachtel R. E.; Tsuji K. Comparison of limulus amebocyte lysates and correlation with the United States Pharmacopeial pyrogen test. Appl. Environ. Microbiol. 1977, 33, 1265–1269. 10.1128/aem.33.6.1265-1269.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov T.; Thompson P. E.; Nation R. L.; Li J. Structure–Activity Relationships of Polymyxin Antibiotics. J. Med. Chem. 2010, 53, 1898–1916. 10.1021/jm900999h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues M.; Santos N.; Castanho M. Antimicrobial peptide rBPI21: a translational overview from bench to clinical studies. Curr. Protein Pept. Sci. 2012, 13, 611–619. 10.2174/138920312804142101. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Wang X.; Wang J. Recent Advances in Antibacterial and Antiendotoxic Peptides or Proteins from Marine Resources. Mar. Drugs 2018, 16, 57–74. 10.3390/md16020057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Eckert T.; Lutteke T.; Hanstein S.; Scheidig A.; Bonvin A. M.; Nifantiev N. E.; Kozar T.; Schauer R.; Enani M. A.; Siebert H. C. Structure-Function Relationships of Antimicrobial Peptides and Proteins with Respect to Contact Molecules on Pathogen Surfaces. Curr. Top. Med. Chem. 2015, 16, 89–98. 10.2174/1568026615666150703120753. [DOI] [PubMed] [Google Scholar]
- Pachón-Ibáñez M. E.; Smani Y.; Pachón J.; Sánchez-Céspedes J. Perspectives for clinical use of engineered human host defense antimicrobial peptides. FEMS Microbiol. Rev. 2017, 41, 323–342. 10.1093/femsre/fux012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew B.; Nagaraj R. Antimicrobial activity of human alpha-defensin 5 and its linear analogs: N-terminal fatty acylation results in enhanced antimicrobial activity of the linear analogs. Peptides 2015, 71, 128–140. 10.1016/j.peptides.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Fung F. M.; Su M.; Feng H. T.; Li S. F. Y. Extraction, separation and characterization of endotoxins in water samples using solid phase extraction and capillary electrophoresis-laser induced fluorescence. Sci. Rep. 2017, 7, 10774–10783. 10.1038/s41598-017-11232-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S. G. Bacterial lipopolysaccharides-themes and variations. Prog. Lipid Res. 1996, 35, 283–343. 10.1016/s0163-7827(96)00004-5. [DOI] [PubMed] [Google Scholar]
- Santos N. C.; Silva A. C.; Castanho M. A. R. B.; Martins-Silva J.; Saldanha C. Evaluation of lipopolysaccharide aggregation by light scattering spectroscopy. Chembiochem 2003, 4, 96–100. 10.1002/cbic.200390020. [DOI] [PubMed] [Google Scholar]
- Sasaki H.; White S. H. Aggregation Behavior of an Ultra-Pure Lipopolysaccharide that Stimulates TLR-4 Receptors. Biophys. J. 2008, 95, 986–993. 10.1529/biophysj.108.129197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller M.; Lindner B.; Kusumoto S.; Fukase K.; Schromm A. B.; Seydel U. Aggregates are the biologically active units of endotoxin. J. Biol. Chem. 2004, 279, 26307–26313. 10.1074/jbc.m401231200. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Dhillon P.; Yan H.; Farmer S.; Hancock R. E. W. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. 10.1128/aac.44.12.3317-3321.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. E.; Vodyanoy I.; Balasubramanian T. M.; Marshall G. R. Alamethicin. A rich model for channel behavior. Biophys. J. 1984, 45, 233–247. 10.1016/s0006-3495(84)84151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H.-A.; Bamberg E. Influence of membrane thickness and ion concentration on the properties of the gramicidin A channel Autocorrelation, spectral power density, relaxation and single-channel studies. Biochim. Biophys. Acta 1977, 464, 127–141. 10.1016/0005-2736(77)90376-5. [DOI] [PubMed] [Google Scholar]
- Wu M.; Maier E.; Benz R.; Hancock R. E. W. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. 10.1021/bi9826299. [DOI] [PubMed] [Google Scholar]
- Jung S.; Mysliwy J.; Spudy B.; Lorenzen I.; Reiss K.; Gelhaus C.; Podschun R.; Leippe M.; Grötzinger J. Human beta-defensin 2 and beta-defensin 3 chimeric peptides reveal the structural basis of the pathogen specificity of their parent molecules. Antimicrob. Agents Chemother. 2011, 55, 954–960. 10.1128/aac.00872-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericksen B.; Wu Z.; Lu W.; Lehrer R. I. Antibacterial activity and specificity of the six human {alpha}-defensins. Antimicrob. Agents Chemother. 2005, 49, 269–275. 10.1128/aac.49.1.269-275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S. A.; Awasthi S. K.; Balaram P. The role of polar and facial amphipathic character in determining lipopolysaccharide-binding properties in synthetic cationic peptides. J. Endotoxin Res. 2000, 6, 249–256. 10.1177/09680519000060030601. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.